

Abstract
Objective:
To describe leptomeningeal blood-barrier impairment reflected by MRI gadolinium-enhanced lesions in patients with aquaporin-4 immunoglobulin G (AQP4-IgG)–positive neuromyelitis optica spectrum disorder (NMOSD).
Methods:
A retrospective case series of 11 AQP4-IgG–positive NMOSD patients with leptomeningeal enhancement (LME) were collected from 5 centers. External neuroradiologists, blinded to the clinical details, evaluated MRIs.
Results:
LME was demonstrated on postcontrast T1-weighted and fluid-attenuated inversion recovery images as a sign of leptomeningeal blood-barrier disruption and transient leakage of contrast agent into the subarachnoid space in 11 patients, 6 in the brain and 6 in the spinal cord. The patterns of LME were linear or extensive and were accompanied by periependymal enhancement in 5 cases and intraparenchymal enhancement in all cases. The location of LME in the spinal cord was adjacent to intraparenchymal contrast enhancement with involvement of a median number of 12 (range 5–17) vertebral segments. At the time of LME on MRI, all patients had a clinical attack such as encephalopathy (36%) and/or myelopathy (70%) with median interval between symptom onset and LME of 12 days (range 2–30). LME occurred in association with an initial area postrema attack (44%), signs of systemic infection (33%), or AQP4-IgG in CSF (22%) followed by clinical progression. LME was found at initial clinical presentation in 5 cases and at clinical relapses leading to a diagnosis of NMOSD in 6 cases.
Conclusion:
This study suggests that altered leptomeningeal blood barrier may be accompanied by intraparenchymal blood-brain barrier breakdown in patients with AQP4-IgG–positive NMOSD during relapses.
Neuromyelitis optica spectrum disorder (NMOSD) is an inflammatory, autoimmune disease of the CNS that follows a relapsing course in the majority of patients. NMOSD-related neurologic disability frequently occurs as a consequence of cumulative attacks.1 The inflammation of NMOSD is often associated with blood-brain barrier (BBB) leakage, reflected by MRI gadolinium (Gd)-enhanced lesions.2–4 Serum-derived aquaporin-4 immunoglobulin G (AQP4-IgG) antibody is believed to play an important and potentially pivotal role in NMOSD pathogenesis1; however, the mechanism whereby AQP4-IgG contributes to BBB leakage remains poorly characterized. The autoantigen AQP4 is densely localized on ependymal cells and astrocyte end-feet, forming the glia limitans of the BBB.5 The lack of a basement membrane and expression of tight junctions in ependymal cells may allow AQP4-IgG in the CSF to get access to its target.5–7 AQP4-IgG may exert pathologic effects by binding selectively to AQP4 on CNS astrocytes, inducing complement-dependent astrocyte damage and inflammation.8
MRI-detected leptomeningeal enhancement (LME), which reflects the leptomeningeal blood-barrier disruption, has been reported in patients with NMOSD.2 LME refers to an enhancement pattern that follows the pial surface of the CNS and fills the subarachnoid spaces of sulci and cisterns. The LME pattern depends on the extent and intensity of the involved inflammatory area and may provide useful clues to the diagnosis of, e.g., neurosarcoidosis.9
In this retrospective case study, we investigated whether compromised integrity of the intraparenchymal BBB occurs concurrently with LME and whether such an alteration of BBB is associated with disease activity in patients with AQP4-IgG–positive NMOSD.
METHODS
Study design and population.
The study was designed as a retrospective case series with clinical and radiologic follow-up and collected by a multicenter study group in 5 centers (Japan, South Korea, United States, France, and Denmark). Patients were included if they met the following criteria: (1) AQP4-IgG–positive NMOSD, qualifying for the 2006 Wingerchuk criteria10 or/and the International Panel for NMO Diagnosis (IPND) 201511 and (2) LME as visualized by MRI following the standard protocols.12,13 Patients were excluded if they had (1) systemic inflammatory disease, e.g., sarcoidosis or (2) neoplastic disease. The information was obtained by review of medical records and reevaluation of previous MRIs of the CNS by local neuroradiologists. In addition, 2 expert neuroradiologists not involved in patient management evaluated images and were blinded to the clinical details (H.P.S. and J.W.).
Neuroimaging.
Because the study was retrospective, several types of 1.5T MRI scanners were used. T2-weighted (T2W) and T1-weighted (T1W) images with or without Gd, diffusion-weighted imaging, and fluid-attenuated inversion recovery (FLAIR) sequences were available for analysis for all brain scans. T2W and T1W with or without Gd and short tau inversion recovery (STIR) sequences were available for spinal cord images. Precontrast and postcontrast images were evaluated to determine the presence of abnormal contrast enhancement on T1W and/or FLAIR sequences according to the criteria previously reported.12–14 If performed, postcontrast FLAIR sequences obtained at least 10 minutes after a single dose of contrast were also used for evaluation.
Standard protocol approvals, registrations, and patient consents.
All patients provided oral and written informed consent for publication.
RESULTS
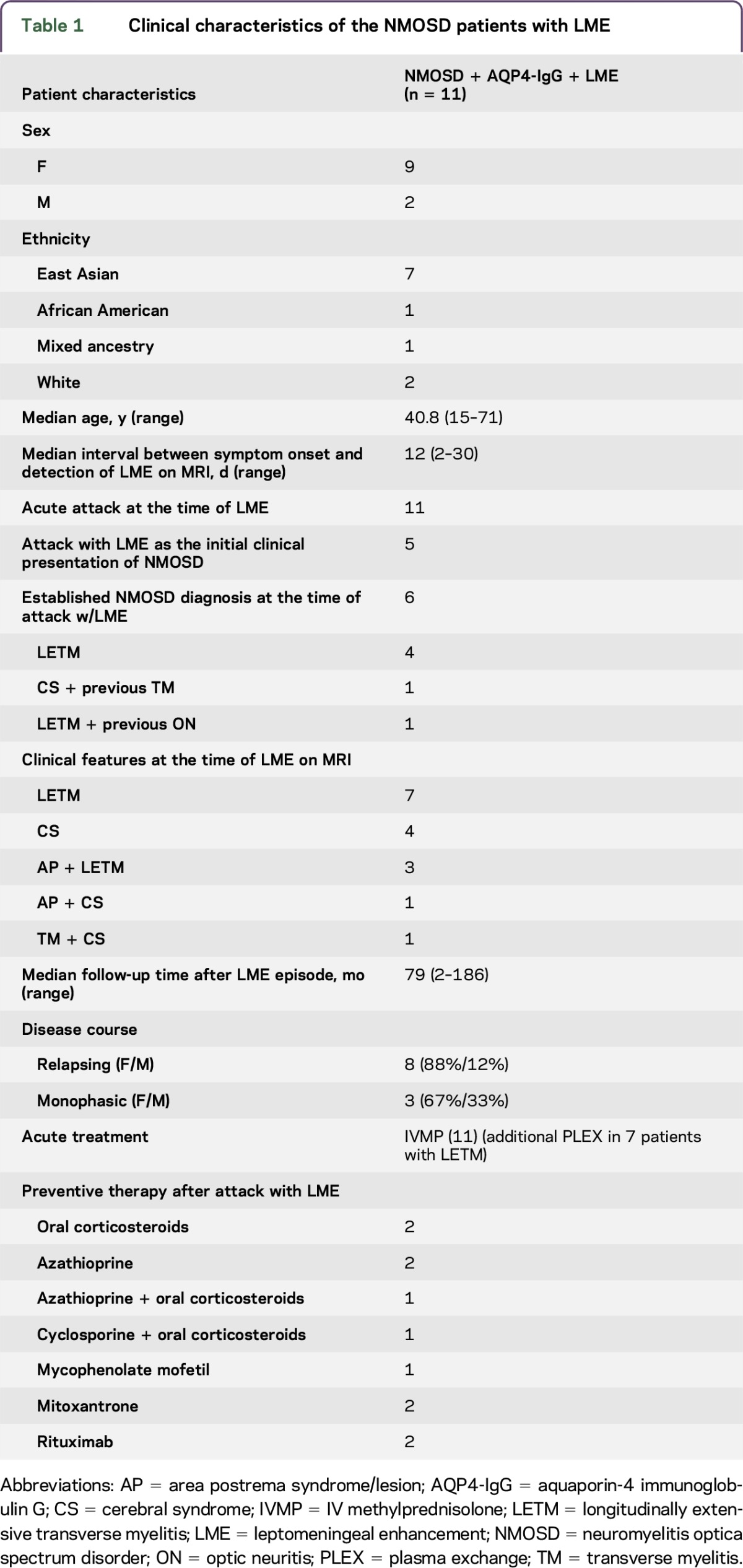
Retrospective MRI and clinical data from 11 AQP4-IgG–positive (measured either in a cell-based assay or ELISA15) NMOSD patients with leptomeningeal enhancement (LME) were obtained for review (table 1). The female: male ratio was 9:2, and the median age at onset of disease was 40.8 years (range 15–71 years). Seven patients were East Asian, 1 African American, 1 of mixed ancestry, and 2 whites.
Table 1.
Clinical characteristics of the NMOSD patients with LME
Radiologic characteristics.
LME localization.
Contrast enhancement in the leptomeningeal space was evaluated by precontrast and postcontrast T1W and/or FLAIR images and observed in all 11 patients, 6 in the brain and 6 in the spinal cord; 1 patient had both brain and spinal cord LME.
In the brain, T1W contrast-enhanced images revealed a linear or thick and extensive leptomeningeal and pial enhancement with localization in both cerebral hemispheres (2 patients), bilateral medial surfaces of the cerebral hemispheres (1), bilateral frontal, medial parietal lobe (1), right parietal lobe (1), and bilateral parieto-occipital lobes (1) (figure 1, A.a and b, C.a and b). FLAIR (figure 1, A.c, B.c, C.c) and FLAIR postcontrast (figure 1B.a) sequences revealed similar leptomeningeal signal intensity changes.
Figure 1. Leptomeningeal enhancement accompanied by intraparenchymal enhancement during attacks in patients with NMOSD.

Contrast-enhanced cerebral T1W (A.a, A.b, B.b, C.a, and C.b), FLAIR sequences (A.c, B.a, B.c, and C.c) and FLAIR post–contrast-enhanced image (B.a) MRI from 3 patients with AQP4-IgG–positive NMOSD (A, B, and C) during attacks, showing leptomeningeal enhancement (A.a, A.b, B.a, C.a, and C.b), marked with arrows. Parenchymal lesions are seen including cloud-like enhancement (B.c, A.c, and C.c). Notably, enhancement occurred at the ependymal surface of the lateral ventricles (B.a). AQP4-IgG = aquaporin-4 immunoglobulin G; FLAIR = fluid-attenuated inversion recovery; NMOSD = neuromyelitis optica spectrum disorder; T1W = T1 weighted.
In the spinal cord, T1W contrast-enhanced images showed LME in 6 patients, conus to cauda equina (2), multifocal spinal cord (1), and thoracic segments (3) (figure 2, A.a and c, B.a and C, figure 3B, figure 4, A and D, figure e-1, H1 and H3 at Neurology.org/nn).
Figure 2. LETM in association with leptomeningeal enhancement.

Sagittal (A.a and B.a) and axial (A.c and B.c) T1W contrast-enhanced images and sagittal T2W (A.b and B.b) and axial images (A.d and B.d) of spinal cord MRI from 2 (A and B) AQP4-IgG–positive patients with NMOSD. Images were obtained during acute LETM attacks with leptomeningeal and intraparenchymal (A.a and A.c at th10; B.a and B.c at th12) T1W contrast enhancement. The spinal cord lesions were also demonstrated on T2W and STIR images (A.b, A.d, B.b, and B.d). AQP4-IgG = aquaporin-4 immunoglobulin G; LETM = longitudinally extensive transverse myelitis; NMOSD = neuromyelitis optica spectrum disorder; STIR = short tau inversion recovery; T1W = T1 weighted; T2W = T2 weighted.
Figure 3. An area postrema lesion in association with leptomeningeal enhancement and intraparenchymal BBB breakdown.

Spinal cord MRI (A–C) and brain MRI (D–G) from an AQP4-IgG–positive patient with NMOSD who presented with recurrent myelopathy of 1 month duration and area postrema lesion, which progressed into LETM. Images were obtained during acute attacks. An area postrema lesion (A) in association with subacute progression to LETM with leptomeningeal and intraparenchymal T1W contrast enhancement (arrows, B). The spinal cord lesions were also demonstrated on T2W and STIR images (C). Cerebral MRI during another attack with periependymal enhancement on T1W images at the lateral (D) and third ventricles (F). FLAIR images showing lesions along lateral ventricle (E), thalamus, and hypothalamus (G). (B) Used with permission of the publisher from Flanagan EP, Weinshenker BG. Neuromyelitis optica spectrum disorders. Curr Neurol Neurosci Rep 2014;14:483. Copyright © 2014, Springer Science+Business Media New York. AQP4-IgG = aquaporin-4 immunoglobulin G; BBB = blood-brain barrier; FLAIR = fluid-attenuated inversion recovery; LETM = longitudinally extensive transverse myelitis; NMOSD = neuromyelitis optica spectrum disorder; T1W = T1 weighted; T2W = T2 weighted.
Figure 4. Leptomeningeal enhancement in association with the presence of AQP4-IgG in CSF and LETM.

Sagittal contrast-enhanced T1W (A and B), sagittal (C), axial (D), and axial (E) T2W MRIs of the spinal cord of an AQP4-IgG–positive NMOSD patient during attacks, showing leptomeningeal and intraparenchymal T1W contrast enhancement (A, B, and D). The spinal cord lesions were also demonstrated on T2W and STIR images (C and E). The patient had a subacute onset with atypical chest pain as the initial symptom, which after 7 days progressed to paraplegia and LETM on MRI. This patient was positive for AQP4-IgG in CSF. AQP4-IgG = aquaporin-4 immunoglobulin G; LETM = longitudinally extensive transverse myelitis; NMOSD = neuromyelitis optica spectrum disorder; STIR = short tau inversion recovery; T1W = T1 weighted; T2W = T2 weighted.
Follow-up MRI demonstrated resolution of LME within 1–4 weeks in 9 patients. In one other case, the next MRI was performed after 72 days when the LME had disappeared. One patient did not have a follow-up MRI.
LME in association with parenchymal enhancement.
LME in all cases spatially corresponded to intraparenchymal T1W contrast enhancement or/and FLAIR image abnormalities in the brain (figure 1, A.c, B.c, C.b and c). Periependymal linear T1W contrast enhancement was localized at the lateral ventricles (3 patients) (figure 1B.b, figure 3D) and adjacent to the third and fourth ventricles (2) (figure 3F). Intracerebral cloud-like enhancement (3 patients) (figure 1C.c), enhancement of hypothalamus (2) (figure 3G), and optic chiasm (1) were observed on T1W contrast-enhanced or/and FLAIR sequences. In addition, parenchymal lesions were observed on T2W or/and FLAIR sequences (figure 1, A.c, B.c, C.c, figure 3, A and E).
Spinal cord MRI demonstrated intraparenchymal T1W contrast enhancement in the underlying parenchyma adjacent to the LME (figure 2, A.a and B.a, figure 3B, figure 4B, figure e-1H1), involving cervical, thoracic, and lumbar cords. Spinal cord lesions expanded over a median of 12 segments (range 5–17) as demonstrated on contrast-enhanced T1W (figure 2, A.a and B.a, figure 3B, figure 4B, figure e-1H1) and T2W and STIR images (figure 2, A.b and d, B.b and d, figure 3, A and C, figure 4, C and E, figure e-1, H2 and H4).
CSF abnormalities.
Lumbar puncture was performed after MRI investigation in all patients, and complete CSF analysis was available at the time of the LME for 6 of the 11 patients. CSF examination showed pleocytosis >50 mL (/mm3) in all cases; 2 cases with increased polymorphonuclear leukocytes (PMNLs) had 80% (PMNL 166 and mononuclear leukocyte [MNL] 41/mm3) and 98% (PMNL 212 and MNL 4/mm3) PMNL, respectively. The combination of LME and pleocytosis in all cases, especially in the 2 cases with increased CSF-PMNL and fever, led to extensive virologic and bacteriologic workup, which proved negative both in blood and CSF. The median CSF protein concentration was 169 (range 54–322) (mg/dL), and CSF glucose level was normal in all cases. CSF-restricted oligoclonal bands or elevated IgG CSF/serum ratio were absent, except in 1 patient. AQP4-IgG in CSF was only measured in 2 patients, and it was positive in both patients.
LME and clinical disease activity.
When LME was visible on MRI, all patients had experienced attacks with subacute onset and progressive neurologic deficits over a few weeks (7 patients) or with acute onset and rapid progression within a few days (4 patients). Median interval between symptom onset and MRI scans displaying LME was 12 days (range 2–30 days). Seven patients had longitudinally extensive transverse myelitis (LETM) (4 subacute and 3 acute onset) and 4 had cerebral syndromes (CS) (3 with subacute and 1 with acute onset) as the clinical presentation associated with LME (table e-1).
Spinal cord involvement.
In the LETM group, 3 of the 4 patients with subacute onset initially had experienced an area postrema syndrome/lesion (episodes of nausea, vomiting, and hiccups) (figure 3, A–C) followed a few days later by extremity weakness, which progressed to tetraplegia (1 patient) or paraplegia (2) over 7, 12, and 30 days. MRI of the spinal cord at this time revealed LETM with LME adjacent to LETM. Another patient with a subacute onset of LETM had atypical chest pain as the initial symptom, which after 7 days progressed to paraplegia and MRI of the spinal cord revealed LME and LETM (figure 4, A–E).
In the remaining 3 patients with acute onset of LETM, 2 developed symptom/signs after an upper respiratory tract infection, by 2 and 4 days respectively (without the identification of an infectious agent) and 1 occurred 2 days after an induced abortion. One presented with tetraplegia and 2 with paraplegia. MRI of spinal cord revealed LETM with LME adjacent to the intramedullary lesion (H1-H4).
Brain involvement.
In the CS group, 1 patient initially experienced an area postrema syndrome followed 10 days later by altered consciousness and recurrent generalized seizures. Brain MRI performed 17 days after onset of symptoms revealed LME (figure 1, A.a–c). Another patient experienced dizziness and was treated with corticosteroids. A third patient had nonspecific symptoms such as drowsy mental state, dysarthria, and recurrent episode of myelitis and optic neuritis (ON). LME were in those 2 cases who demonstrated on MRI 24 and 21 days after symptom onset, respectively. The one patient with acute symptom onset had a generalized seizure followed by tetraparesis from TM. LME was in this case demonstrated on MRI 5 days after symptom onset (table e-1).
Acute and long-term immunotherapy.
In all patients, acute attacks were initially treated with IV methylprednisolone (IVMP), which were administered between the first and the follow-up MRI. The 7 patients with LETM did not clinically respond to IVMP therapy and were subsequently treated by plasma exchange.
Various immunosuppressive drugs as listed in table 1 were used as preventive therapy after the follow-up MRI, which showed resolution of LME; one received azathioprine before the attack and follow-up MRI examination.
LME and NMOSD diagnosis.
LME was found at the initial attack of NMOSD in 5 (3: LETM, 2: area postrema syndrome) of the 11 patients (table 1). In the remaining 6 patients, symptoms at first attack were ON (2 patients), TM (1), AP (1), and CS (2).
A total of 6 patients, including all patients who had LME at the initial attack, qualified for the diagnosis of NMOSD except one, with area postrema syndrome, who received the NMOSD diagnosis 2 years later at testing for AQP4-IgG. Of the 6 patients, 2 had a history of TM and ON, respectively, and received the diagnosis of NMOSD in association with LME and acute attack, 1 with LETM, and 1 with a generalized seizure followed by TM (table 1).
Three patients with previous attacks received the NMOSD diagnosis at median 20 months (range 12–24) after attack with LME on MRI, with disease duration of median 116 months (range 108–132). They were suspected for MS and treated with interferon-β previously, relapses occurred and treatment was discontinued. One patient was diagnosed with NMOSD 2 years before an attack with LME on MRI. She was treated with azathioprine. For all patients, median follow-up after onset of attack and LME on MRI was 79.6 months (range 2–186).
DISCUSSION
In this retrospective case series comprising 11 patients with AQP4-IgG–positive NMOSD, we observed contrast enhancement of the leptomeningeal structures during clinical attacks accompanied by intraparenchymal contrast enhancement spatially corresponding to the LME. LME was observed in association with either an initial area postrema attack, with precedent signs of systemic infection or with detectable AQP4-IgG in CSF, followed by secondary worsening of symptoms/signs and lesions on MRI (figure e-2). These findings suggest that, in some cases, entry of serum-derived AQP4-IgG to the subarachnoid space may be facilitated by primary inflammation at area postrema by systemic infection or by blood-CSF barrier dysfunction (figure e-2). It could be speculated that AQP4-IgG may be deposited at the astrocytic foot processes at pial vessels and cause astrocytic damage. The resultant dysfunction of AQP4 water channels in the pial and subpial surfaces could lead to impairment of leptomeningeal vessel barrier, which may be involved in the development of NMOSD lesions.
In line with this study, pronounced meningeal inflammation has been reported in autopsy findings of patients with NMOSD.16 Our recent experimental study of intrathecal injection of AQP4-IgG may corroborate these findings, as pronounced deposition of AQP4-IgG was observed in the leptomeningeal area, including where pial vessels penetrate the brain parenchyma and form perivascular spaces into the brain parenchyma,17 the so-called paravascular pathway.18 Furthermore, AQP4-IgG initiated perivascular astrocyte-destructive lesions and consequently breakdown of the BBB.17 A significant proportion of subarachnoid CSF circulates through the parenchyma within these paravascular spaces19 which surround penetrating and cerebral surface vessels20–22 and is completely ensheathed by astrocyte end-feet.22 Of interest, experimental data have suggested that the flow of fluid along this paravascular route is mediated by astrocytic AQP4.19
Although direct inferences from an animal study are not possible, our observations suggest that intraparenchymal contrast enhancement lesions may form in anatomical proximity to regions where LME is visible. LME was spatially associated with periependymal enhancement and with intraparenchymal enhancement in the brain, brainstem, hypothalamus, optic chiasm, and spinal cord. These CNS MRI abnormalities in patients with AQP4-IgG–positive NMOSD are believed to be common and typical of the disease with regards to localization and configuration.2,23 Conversely, LME has been reported in few patients only and is presumably more common during NMOSD relapses and in AQP4-IgG–positive patients.23,24 However, the frequency of LME is likely to be underestimated given that most MRIs are performed subsequently at onset of clinical symptoms and thus at a time point where lesion formation is probably completed and LME may have vanished. Timing of MRI may be important for the demonstration of LME, and evaluation of MRI should consider the interval between symptom onset and MRI scan. Last, LME may be suppressed by steroid treatment as immunosuppressive therapy probably decreases BBB permeability.25 It has previously been reported that corticosteroid treatment before MRI may lead to the improvement or resolution of LME9 as part of the treatment response. In this study, patients received treatment with IV corticosteroid before follow-up MRI, and this may have led to the resolution of LME. The temporal evolution of alterations to the BBB permeability is difficult to determine and will require a kinetic study with sequential and frequent MRI images that analyze the dynamic changes of enhancement in newly appearing active lesions. The localization of the LME at the spinal cord in 70% of cases was associated with extremely long acute LETM lesions, and the patients were resistant to steroid therapy. This may on MRI reflect the severity of attacks in the spinal cord at the time of LME. However, such determination of an association between the leptomeningeal blood-barrier disruption and BBB breakdown sites and severity of attacks require larger and prospective studies.
In this study, all cases were associated with disease activity at the time of LME on MRI, and LME was accompanied by other characteristic findings such as LETM, area postrema lesion etc.11 In 7 of the 11 patients, the attacks occurred subacutely with a dynamic progression of symptoms/signs. Four of the seven patients had an initial area postrema syndrome. The dynamic formation of an LETM was previously reported with initial involvement of the area postrema during 12 days from the onset of symptoms as visualized on sequential brainstem/spinal cord MRI.26 These findings support the concept that area postrema, a circumventricular area which has incomplete BBB, may serve as entry route to the CNS for circulating AQP4-IgG.20,26,27
In NMOSD, the presence of AQP4-IgG in CSF has been reported to be associated with high titers of AQP4-IgG in serum,28–30 and to be strongly correlated with astrocyte damage, as reflected in elevated levels of glial fibrillary acidic protein in the CSF and BBB disruption during disease attacks.30,31 In this small retrospective study, titers of AQP4-IgG in serum were not determined and AQP4-IgG in CSF was only measured in 2 patients, both of whom had positive results.
Infections may result in events that alter the BBB and thus permit access of AQP4-IgG to the CNS.32,33 Three cases with LETM were preceded acutely by symptoms suggestive of systemic infection. Cases of infectious or parainfectious events before the onset of neurologic symptoms and NMOSD have been reported, adding to the etiological diversity in NMOSD.32–36 It is possible that infection may act as a precipitating factor in the development of NMOSD with the presence of AQP4-IgG.
We observed that leptomeningeal blood-barrier integrity was compromised during relapses and was accompanied by intraparenchymal BBB breakdown in patients with AQP4-IgG–positive NMOSD. Although our data are too preliminary to allow for a firm statement on the putative mechanism of lesion formation in human NMOSD, they suggest in conjunction with previous animal experiments that the question of how pathogenic AQP4-IgG gains access to its CNS target deserves further attention. Moreover, future imaging studies may inform us about the relevance of LME for attack severity and clinical course in NMOSD.
Supplementary Material
GLOSSARY
- AP
area postrema syndrome/lesion
- AQP4
aquaporin-4
- BBB
blood-brain barrier
- CS
cerebral syndromes
- DWI
diffusion-weighted imaging
- FLAIR
fluid-attenuated inversion recovery
- Gd
gadolinium
- IVMP
IV methylprednisolone
- LETM
longitudinally extensive transverse myelitis
- LME
leptomeningeal enhancement
- MNL
mononuclear leukocyte
- NMOSD
neuromyelitis optica spectrum disorder
- ON
optic neuritis
- PLEX
plasma exchange
- PMNL
polymorphonuclear leukocyte
- STIR
short tau inversion recovery
- T1W
T1 weighted
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
N.A.: study concept and design, acquisition of data, interpretation of results, drafting, and revising of the manuscript. E.P.F., K.F., and H.J.K.: acquisition of data, interpretation of results, revising of the manuscript, and approving the final version. H.P.S. and J.W.: MRI reevaluation, interpretation of results, revising of the manuscript, and approving the final version. H.K., S.H.K., and E.M.: acquisition of data, revising of the manuscript, and approving the final version. R.M. and S.J.P.: acquisition of data, interpretation of results, revising of the manuscript, and approving the final version. F.P.: study design, interpretation of results, revising of the manuscript, and approving the final version. B.G.W.: study design, acquisition of data, interpretation of results, revising of the manuscript, and approving the final version.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
N. Asgari received research support from The Vejle Hospital Research Fund of the Region of Southern Denmark, Lundbeck Research Foundation, and The University of Southern Denmark. E.O. Flanagan reports no disclosures. K. Fujihara served on the scientific advisory board for Bayer Schering Pharma, Biogen Idec, Mitsubishi Tanabe Pharma Corporation, Novartis Pharma, Chugai Pharmaceutical, Ono Pharmaceutical, Nihon Pharmaceutical, Alexion Pharmaceuticals, and MedImmune and Medical Review; received travel funding and/or speaker honoraria from Bayer Schering Pharma, Biogen Idec, Eisai Inc, Mitsubishi Tanable Pharma Corporation, Novartis Pharma, Astellas Pharma Inc, Takeda Pharmaceutical Company Limited, Asahi Kasei Medical Co, Daiichi Sankyo, and Nihon Pharmaceutical; served on the editorial board for Clinical and Experimental Neuroimmunology; an advisory board member for Sri Lanka Journal of Neurology; received research support from Bayer Schering Pharma, Biogen Idec Japan, Asahi Kasei Medical, The Chemo-Sero-Therapeutic Research Institute, Teva Pharmaceutical, Mitsubishi Tanabe Pharma, Teijin Pharma, Chugai Pharmaceutical, Ono Pharmaceutical, Nihon Pharmaceutical, Genzyme Japan, Grants-in-Aid for Scientific Research from the Ministry of Education, Science and Technology of Japan, and Ministry of Health, Welfare and Labor of Japan. H.J. Kim received travel funding and/or speaker honoraria from Bayer Schering Pharma, Biogen Idec, Genzyme, MedImmune, Merck, Novartis, Teva-Handok, and UCB; served as an associate editor for Journal of Clinical Neurology, co-editor of Multiple Sclerosis Journal—Experimental, Translational and Clinical; consulted for Bayer Schering Pharma, Biogen Idec, Genzyme, HanAll Biopharma, MedImmune, Merck, Novartis, Teva-Handock, and UCB; received research support from Genzyme, Kael-GemVax, Merck Serono, Teva-Handock, UCB, and The Ministry of Science, ICT and Future Planning. H.P. Skejoe reports no disclosures. J. Wuerfel served on the scientific advisory board for Biogen, Novartis, Genzyme, Teva, and Roche; received travel funding and/or speaker honoraria from Novartis, Bayer, Biogen, and Novartis; was CEO at MIAC AG; received support from German Ministry of Education and Research, German Ministry of Economy, EXIST program, and ZIM program. H. Kuroda received research support from Ministry of Education, Culture, Sports, Science and Technology. S.H. Kim reports no disclosures. E. Maillart served on the scientific advisory board for Genzyme, Roche, Novartis, and Merck; received travel funding and/or speaker honoraria from Biogen, Teva Pharmaceuticals, Genzyme, Roche, and Novartis. R. Marignier served on the scientific advisory board for MedImmune. S.J. Pittock holds patents that relate to functional AQP4/NMO-IgG assays and NMO-IgG as a cancer marker; has consulted for Alexion Pharma, MedImmune, and Chugai Pharma (all consulting compensation is paid to Mayo Clinic); received research support from Alexion, RO1, and Guthy-Jackson Charitable Foundation Grant. F. Paul served on the scientific advisory board for Novartis OCTIMS study and MedImmune; received travel funding and/or speaker honoraria from Bayer, Novartis, Biogen Idec, Teva, Sanofi-Aventis/Genzyme, Merck Serono, Alexion-Chugai, MedImmune, and Shite; is an associate editor for Neurology® Neuroimmunology & Neuroinflammation, an academic editor for PLoS One; consulted for Sanofi Genzyme, Biogen Idec, MedImmune, Shite, and Alexion; received research support from Bayer, Novartis, Biogen Idec, Teva, Sanofi-Aventis/Genzyme, Alexion and Merck Serono, German Research Council, Werth Stiftung of the City of Cologne, German Ministry of Education and Research, Arthur Arnstein Stiftung Berlin, Arthur Arnstein Foundation Berlin, Guthy-Jackson Charitable Foundation, and National Multiple Sclerosis Society of the United States. B.G. Weinshenker served on the scientific advisory board for Novartis, Biogen Idec, and Mitsubishi; served on the editorial board for Canadian Journal of Neurological Sciences, Turkish Journal of Neurology, and Neurology; holds a patent for and receives royalties from NMO-IgG for diagnosis of neuromyelitis optica; is an adjudication committee member for MedImmune and Alexion; and received research support from Guthy-Jackson Charitable Foundation. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007;6:805–815. [DOI] [PubMed] [Google Scholar]
- 2.Kim HJ, Paul F, Lana-Peixoto MA, et al. . MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology 2015;84:1165–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim W, Park MS, Lee SH, et al. . Characteristic brain magnetic resonance imaging abnormalities in central nervous system aquaporin-4 autoimmunity. Mult Scler 2010;16:1229–1236. [DOI] [PubMed] [Google Scholar]
- 4.Asgari N, Skejoe HP, Lillevang ST, Steenstrup T, Stenager E, Kyvik KO. Modifications of longitudinally extensive transverse myelitis and brainstem lesions in the course of neuromyelitis optica (NMO): a population-based, descriptive study. BMC Neurol 2013;13:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci 1997;17:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rash JE, Yasumura T, Hudson CS, Agre P, Nielsen S. Direct immunogold labeling of aquaporin-4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proc Natl Acad Sci USA 1998;95:11981–11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jarius S, Paul F, Franciotta D, et al. . Mechanisms of disease: aquaporin-4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol 2008;4:202–214. [DOI] [PubMed] [Google Scholar]
- 8.Lucchinetti CF, Mandler RN, McGavern D, et al. . A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain 2002;125:1450–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moravan M, Segal BM. Treatment of CNS sarcoidosis with infliximab and mycophenolate mofetil. Neurology 2009;72:337–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485–1489. [DOI] [PubMed] [Google Scholar]
- 11.Wingerchuk DM, Banwell B, Bennett JL, et al. . International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85:177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fukui MB, Meltzer CC, Kanal E, Smirniotopoulos JG. MR imaging of the meninges: part II: neoplastic disease. Radiology 1996;201:605–612. [DOI] [PubMed] [Google Scholar]
- 13.Meltzer CC, Fukui MB, Kanal E, Smirniotopoulos JG. MR imaging of the meninges: part I: normal anatomic features and nonneoplastic disease. Radiology 1996;201:297–308. [DOI] [PubMed] [Google Scholar]
- 14.Eisele P, Griebe M, Szabo K, et al. . Investigation of leptomeningeal enhancement in MS: a postcontrast FLAIR MRI study. Neurology 2015;84:770–775. [DOI] [PubMed] [Google Scholar]
- 15.Zekeridou A, Lennon VA. Aquaporin-4 autoimmunity. Neurol Neuroimmunol Neuroinflamm 2015;2:e110 doi: 10.1212/NXI.0000000000000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saji E, Arakawa M, Yanagawa K, et al. . Cognitive impairment and cortical degeneration in neuromyelitis optica. Ann Neurol 2013;73:65–76. [DOI] [PubMed] [Google Scholar]
- 17.Asgari N, Berg CT, Morch MT, Khorooshi R, Owens T. Cerebrospinal fluid aquaporin-4-immunoglobulin G disrupts blood brain barrier. Ann Clin Trans Neurol 2015;2:857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iliff JJ, Lee H, Yu M, et al. . Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Invest 2013;123:1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iliff JJ, Wang M, Liao Y, et al. . A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Trans Med 2012;4:147ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Broadwell RD, Sofroniew MV. Serum proteins bypass the blood-brain fluid barriers for extracellular entry to the central nervous system. Exp Neurol 1993;120:245–263. [DOI] [PubMed] [Google Scholar]
- 21.Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature 2010;468:232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rangroo Thrane V, Thrane AS, Plog BA, et al. . Paravascular microcirculation facilitates rapid lipid transport and astrocyte signaling in the brain. Scientific Rep 2013;3:2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pekcevik Y, Orman G, Lee IH, Mealy MA, Levy M, Izbudak I. What do we know about brain contrast enhancement patterns in neuromyelitis optica? Clin Imaging 2016;40:573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tahara M, Ito R, Tanaka K, Tanaka M. Cortical and leptomeningeal involvement in three cases of neuromyelitis optica. Eur J Neurol 2012;19:e47–e48. [DOI] [PubMed] [Google Scholar]
- 25.Barkhof F, Tas MW, Frequin ST, et al. . Limited duration of the effect of methylprednisolone on changes on MRI in multiple sclerosis. Neuroradiology 1994;36:382–387. [DOI] [PubMed] [Google Scholar]
- 26.Asgari N, Skejoe HP, Lennon VA. Evolution of longitudinally extensive transverse myelitis in an aquaporin-4 IgG-positive patient. Neurology 2013;81:95–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Apiwattanakul M, Popescu BF, Matiello M, et al. . Intractable vomiting as the initial presentation of neuromyelitis optica. Ann Neurol 2010;68:757–761. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi T, Fujihara K, Nakashima I, et al. . Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain 2007;130:1235–1243. [DOI] [PubMed] [Google Scholar]
- 29.Jarius S, Franciotta D, Paul F, et al. . Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: frequency, origin, and diagnostic relevance. J Neuroinflammation 2010;7:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato DK, Callegaro D, de Haidar Jorge FM, et al. . Cerebrospinal fluid aquaporin-4 antibody levels in neuromyelitis optica attacks. Ann Neurol 2014;76:305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takano R, Misu T, Takahashi T, Sato S, Fujihara K, Itoyama Y. Astrocytic damage is far more severe than demyelination in NMO: a clinical CSF biomarker study. Neurology 2010;75:208–216. [DOI] [PubMed] [Google Scholar]
- 32.Graber DJ, Levy M, Kerr D, Wade WF. Neuromyelitis optica pathogenesis and aquaporin 4. J Neuroinflammation 2008;5:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sellner J, Hemmer B, Muhlau M. The clinical spectrum and immunobiology of parainfectious neuromyelitis optica (Devic) syndromes. J Autoimmun 2010;34:371–379. [DOI] [PubMed] [Google Scholar]
- 34.Jarius S, Ruprecht K, Wildemann B, et al. . Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation 2012;9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology 1999;53:1107–1114. [DOI] [PubMed] [Google Scholar]
- 36.Jarius S, Wildemann B, Paul F. Neuromyelitis optica: clinical features, immunopathogenesis and treatment. Clin Exp Immunol 2014;176:149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.