

ABSTRACT
Studies of the human pathogen group A Streptococcus (GAS) define the carrier phenotype to be an increased ability to adhere to and persist on epithelial surfaces and a decreased ability to cause disease. We tested the hypothesis that a single amino acid change (Arg135Gly) in a highly conserved sensor kinase (LiaS) of a poorly defined GAS regulatory system contributes to a carrier phenotype through increased pilus production. When introduced into an emm serotype-matched invasive strain, the carrier allele (the gene encoding the LiaS protein with an arginine-to-glycine change at position 135 [liaSR135G]) recapitulated a carrier phenotype defined by an increased ability to adhere to mucosal surfaces and a decreased ability to cause disease. Gene transcript analyses revealed that the liaS mutation significantly altered transcription of the genes encoding pilus in the presence of bacitracin. Elimination of pilus production in the isogenic carrier mutant decreased its ability to colonize the mouse nasopharynx and to adhere to and be internalized by cultured human epithelial cells and restored the virulence phenotype in a mouse model of necrotizing fasciitis. We also observed significantly reduced survival of the isogenic carrier mutant compared to that of the parental invasive strain after exposure to human neutrophils. Elimination of pilus in the isogenic carrier mutant increased the level of survival after exposure to human neutrophils to that for the parental invasive strain. Together, our data demonstrate that the carrier mutation (liaSR135G) affects pilus expression. Our data suggest new mechanisms of pilus gene regulation in GAS and that the invasiveness associated with pilus gene regulation in GAS differs from the enhanced invasiveness associated with increased pilus production in other bacterial pathogens.
KEYWORDS: group A Streptococcus, carrier, sensor kinase, pilus, colonization, asymptomatic carriage
INTRODUCTION
Colonization of a susceptible host is a critical first step in the infection cycle of all bacterial pathogens. Upon contact with a host, a complex interplay between host immune and clearance mechanisms and the ability of a pathogen to subvert host defenses ensues. The result is a continuum ranging from asymptomatic carriage (henceforth referred to simply as carriage) to the breach of host barriers, dissemination, and a multitude of disease manifestations (1). Currently, the vast majority of research and, hence, our understanding of infectious disease pathogenesis are based on the ability of a bacterial pathogen to cause disease. However, the rates of carriage of many bacterial pathogens, such as Neisseria meningitidis (2–4), Streptococcus pneumoniae (5), Staphylococcus aureus (6), Streptococcus agalactiae (group B Streptococcus [GBS]) (7–9), and Streptococcus pyogenes (group A Streptococcus [GAS]) (10), far exceed the rates of disease caused by these pathogens. Thus, study of the bacterial molecular mechanisms that contribute to carriage is critical to identifying enhanced strategies to prevent disease.
GAS is an ideal model organism for the study of bacterial carriage. The diseases caused by GAS range from relatively benign superficial diseases (e.g., impetigo, pharyngitis) to severe and life-threatening conditions (e.g., toxic shock syndrome, necrotizing fasciitis). In the United States, the annual rate of occurrence of serious invasive disease caused by GAS is very low at approximately 3 cases/100,000 population (11). GAS is also carried in the throats of healthy individuals in the absence of symptoms. The rate of GAS carriage among children ranges from 5 to 15% (10), a rate that far exceeds that of any disease caused by GAS. The mechanisms by which GAS causes disease have been intensely investigated and have been the subject of several reviews (12). On the other hand, the GAS carrier state has been described as an enigma for decades, with few efforts being made to explore the underlying bacterial or host mechanisms of GAS carriage (13, 14). Improving our understanding of the bacterial molecular mechanisms contributing to GAS carriage may enhance our understanding of the basic biology of this important human pathogen and facilitate the development of an effective vaccine.
Control of gene expression by regulatory systems along with mutational adaptation may favor bacterial persistence after disease resolution. Comparison of the whole genomes of carriage and disease-causing strains of GAS has identified mutations unique to carrier strains that contribute to GAS carriage (15–18). A recurring theme that has emerged from GAS comparative genomic studies is that mutations affecting bacterial cell surface molecules alter the host-pathogen interaction and are critical to the development of carriage and a carrier phenotype. Gene regulation in GAS, similar to that in other bacteria, uses several two-component systems (TCS) that consist of a membrane-anchored histidine kinase (HK) for sensing environmental stimuli that, upon activation, phosphorylates a cytoplasmic response regulator (RR), leading to altered gene transcription (19). GAS genomes have up to 13 TCS, and the functions of many remain poorly understood (20). Recently, a GAS carrier strain with a mutation leading to a single amino acid change in a sensor kinase (LiaS) of the three-component system (3CS) LiaFSR was identified (17). Importantly, this was the first mutation in a human GAS carrier strain found to alter global gene regulation. The carrier mutation in liaS (causing an arginine-to-glycine change at position 135 of the LiaS protein [liaSR135G]) alters the transcription of genes encoding several virulence factors (17). The relative contribution of individual differential gene expression to the carrier phenotype has not been further explored.
Pili are long filamentous surface structures made by GAS and other bacterial pathogens that have received increased attention, especially in Gram-positive organisms. The GAS pilus was first identified in 2005 (21), and it is now known to be a heteropolymeric structure consisting of a backbone protein (BP) and ancillary proteins (AP) (22) and is the antigenic target for serologic T typing first described by Lancefield (21). Studies suggest that the presence of pilus on the GAS cell surface may simultaneously promote epithelial cell colonization and reduce virulence (23, 24), phenotypes consistent with a carrier state. Regulation of GAS pilus gene expression occurs through stand-alone regulators, including RofA/Nra (25–27) and MsmR (28), and varies by serotype but may also involve GAS TCS. Studies in S. pneumoniae (29) and GBS (30) suggest that the LiaFSR three-component system may directly or indirectly regulate pilus gene expression. However, to date, no studies provide direct evidence of LiaFSR pilus gene regulation in GAS.
In the study described here, we tested the hypothesis that the liaSR135G carrier mutation increases pilus gene transcription and that increased pilus production contributes to the carrier phenotype. In contrast to previous studies, we show that activation of the LiaFSR 3CS by bacitracin results in a significant increase in pilus gene transcription in the isogenic carrier mutant. Elimination of pilus production in the isogenic carrier mutant decreases its ability to adhere to and persist on epithelial surfaces and restores virulence, essentially reversing the carrier phenotype. Our studies are the first to provide direct evidence that the LiaFSR regulatory system alters pilus gene regulation and that increased pilus production contributes to carriage.
RESULTS
Activation of the LiaFSR 3CS significantly increases pilus gene transcription and protein expression in the carrier mutant.
Previous RNA sequencing (RNA-seq) analyses assessing the global gene transcriptional impact of the liaSR135G mutation failed to reveal any significant differences in pilus gene transcription (17). In contrast, deletion of liaS (hk03) in Streptococcus pneumoniae significantly increased pilus gene transcription (29), whereas deletion of liaR in Streptococcus agalactiae significantly decreased pilus gene transcription (30). We hypothesized that the absence of significant changes in pilus gene transcripts in the isogenic carrier mutant was due to the relative inactivity of the LiaFSR three-component system under the previous conditions tested. Activation of LiaFSR- and LiaR-dependent gene activation in response to bacitracin is well established in Bacillus subtilis (31), S. aureus (32), and S. pneumoniae (33). We tested our hypothesis by measuring the bacitracin-induced activation of LiaFSR through gene transcription. Prior to assaying transcript levels, we first assayed the ability of the parental invasive strain MGAS10870 to grow in the presence of bacitracin. MGAS10870 was grown to mid-exponential phase and, upon reaching mid-exponential-phase growth, was treated with bacitracin at concentrations ranging from the MIC (0.025 μg/ml for MGAS10870 and 0.0125 μg/ml for the isogenic mutant MGAS10870 liaSR135G) to over 100-fold the MIC (5.0 μg/ml). The highest concentration of bacitracin resulted in a significant reduction in growth beginning at the 60-min time point (Fig. 1A). No significant differences in growth were observed for the other concentrations used. Additionally, no significant differences in the numbers of CFU between treated and untreated cultures were observed up to bacitracin concentrations of 1.0 μg/ml (Fig. 1A, dashed lines).
FIG 1.
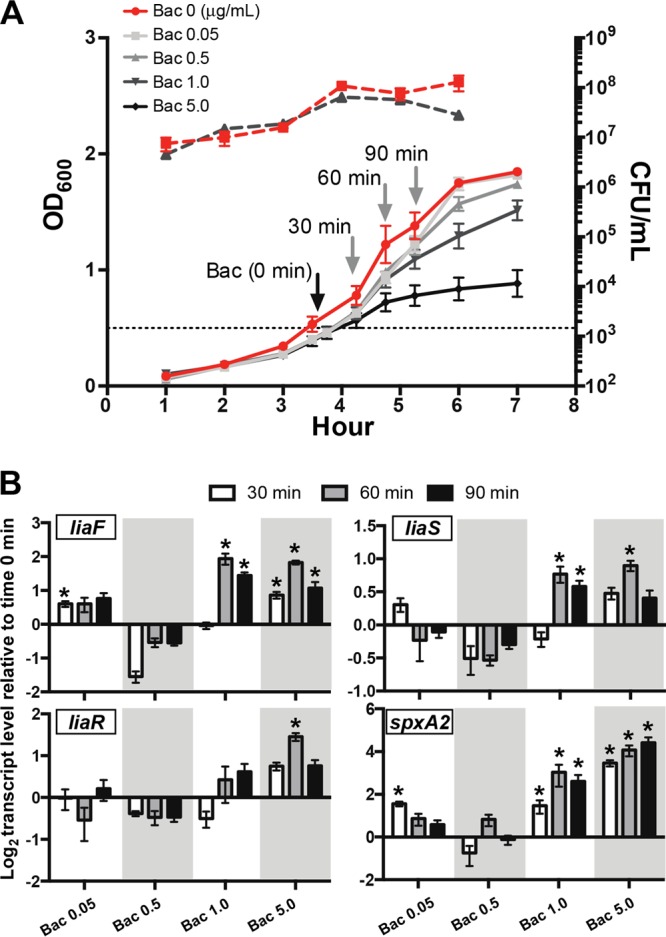
Activation of LiaFSR by bacitracin (Bac) and effect on gene transcription in the serotype M3 invasive strain MGAS10870. (A) MGAS10870 was grown in rich medium (THY), and bacitracin was added when the OD600 was 0.5 (dotted line). Cells were harvested at 30-min intervals (gray arrows) for RNA extraction and quantitative real-time PCR. Dashed lines, number of CFU during growth in the presence of bacitracin (5.0 μg/ml; black dashed line) or without bacitracin (red dashed line). (B) Quantitative real-time PCR of liaF, liaS, liaR, and spxA2 at 30 min (white bars), 60 min (gray bars), and 90 min (black bars) following the addition of bacitracin. *, P < 0.05 (t test, unpaired, Welch's correction) compared to the transcript level at time zero.
We next assayed activation in GAS bacteria harvested at 30-min intervals following the addition of bacitracin using quantitative real-time PCR to measure the transcript levels of liaFSR and compared those levels to the transcript levels from untreated cells. LiaFSR is known to be autoregulated in B. subtilis (31); however, whether autoregulation occurs in GAS is unknown. Significant increases in liaFSR transcript levels following the addition of bacitracin occurred at concentrations of ≥1 μg/ml and ≥60 min posttreatment (Fig. 1B). To provide further evidence of LiaFSR activation, we also assayed the transcript levels of spxA2 (spyM3_1799) following the addition of bacitracin. This gene was assayed because deletion of LiaR in S. agalactiae was found to eliminate spxA transcription (30), and more recently, LiaR has been shown to bind to a 25-bp consensus motif in the promoter of the homologous spxA in Streptococcus mutans (34). Examination of the spxA2 promoter sequence in a serotype M3 GAS strain revealed a similar motif with approximately 85% identity (data not shown). Further, spxA2 was the most highly differentially gene expressed in RNA-seq studies comparing the wild type and its isogenic carrier (liaSR135G) mutant (17). Consistent with spxA2 gene regulation by LiaFSR, we observed significantly increased spxA2 transcript levels following the addition of bacitracin (Fig. 1B, spxA2). The transcription of spxA2 was also concentration and time dependent. However, at the highest concentration used (5.0 μg/ml), transcript levels continued to increase at the 90-min time point, coinciding with a significantly reduced growth rate (Fig. 1B, spxA2). Overall, examination of liaFSR and spxA2 transcript levels revealed a peak at 60 min following treatment with bacitracin at 1.0 μg/ml, suggestive of LiaFSR activation.
Having established the in vitro conditions under which the LiaFSR 3CS was activated, we next tested the hypothesis that the carrier mutation liaSR135G significantly alters pilus gene transcription. The genes necessary for pilus biosynthesis in GAS are contiguously located in a chromosomal region with genes encoding fibronectin binding proteins, collagen binding proteins, and T antigens (FCT) (35). The T antigen is composed of the pilus subunits, including a backbone protein (spyM3_100, tee3) and two ancillary proteins (spyM3_0098 and spyM3_102) (22). Pilus subunits and the associated sortases necessary for assembly are oriented in a predicted operon (Fig. 2A) under the control of a single promoter (Database of Prokaryotic Operons, version 2.0; http://csbl.bmb.uga.edu/DOOR/ [36]). We measured the transcript levels of the genes encoding an ancillary protein (spyM3_0098, cbp) and the backbone protein (spyM3_0100, tee3) from cells of MGAS10870 and the isogenic carrier mutant (MGAS10870 liaSR135G) following treatment with bacitracin (1.0 μg/ml, 60 min) (Fig. 2B). Consistent with our hypothesis, we observed significantly increased pilus gene transcription in the isogenic carrier mutant compared to the parental strain following treatment with bacitracin (Fig. 2C).
FIG 2.

Effect of bacitracin treatment on pilus gene transcript levels. (A) FCT region in serotype M3 GAS. The spectinomycin resistance gene, aad9 (gray block), was used to construct in-frame deletions of the pilus backbone protein (tee3, spyM3_0100). (B) Growth curves for MGAS10870 and MGAS10870 liaSR135G in rich medium with and without bacitracin at 1.0 μg/ml. Bacitracin was added when the OD600 was 0.5, and cells were harvested 60 min after that for RNA extraction and quantitative real-time PCR. (C) Quantitative real-time PCR for determination of the transcript levels of pilus genes (cbp and tee3), pilus gene regulators (nra and msmR), and spxA2. *, P < 0.05 (t test, unpaired, Welch's correction) compared to those in parental strain MGAS10870. (D) Western immunoblotting for Tee3 in the parental invasive strain (MGAS10870), an isogenic carrier mutant, and derived pilus-negative mutants. Cells were treated with bacitracin as described in the legend to panel B, proteins were extracted, and equal amounts of purified cell wall and secreted proteins were probed with an affinity-purified antibody to Tee3 as described in Materials and Methods.
It is possible that modest but significant increases in transcription do not result in increased protein expression in the isogenic carrier mutant compared to the invasive parental strain. We generated an affinity-purified polyclonal antibody against the major pilus backbone protein, Tee3 (SpyM3_0100), to test the hypothesis that the isogenic carrier mutant produced more pilus protein than the parental invasive strain did. Cell wall and secreted proteins were purified after treatment with bacitracin (1.0 μg/ml), as described above for transcript analyses (Fig. 2B). As predicted following Western immunoblotting using the anti-Tee3 antibody, the isogenic carrier mutant showed increased amounts of the Tee3 protein compared to the parental strain in both purified cell wall and secreted protein extracts (2-fold and 1.7-fold increases, respectively; Fig. 2D).
Increased pilus gene transcription in the isogenic carrier mutant is not due to altered gene transcription of known pilus gene regulators.
Pilus gene regulation may occur through a number of transcriptional regulators and appears to be M protein serotype and FCT type dependent (25–28). Serotype M3 GAS (FCT type 3) strains may regulate pilus gene expression through regulators that reside within the FCT chromosomal region, including Nra (SpyM3_0097) (26) and MsmR (SpyM3_0103) (28). Additionally, transcriptome analysis following the deletion of the stand-alone regulator Mga suggests that Mga is an activator of pilus in serotype M1 GAS strains (37). One potential explanation for the significant increase in pilus gene transcription in the isogenic carrier mutant relative to the parental strain is alteration of gene transcription in one or more of the known pilus gene regulators. A previous transcriptome analysis of the isogenic carrier strain at two time points failed to identify significant differences in either nra or msmR transcript levels. However, mga transcript levels in the isogenic carrier mutant decreased approximately 2-fold in early exponential phase (17). To explore the potential mechanism of altered pilus gene transcription, we assayed transcripts of nra, msmR, and mga in the isogenic carrier and wild-type parental strains after bacitracin treatment (1.0 μg/ml, 60 min). Consistent with previous RNA-seq data (17), we observed an approximately 2-fold decrease in mga transcription in the isogenic carrier mutant in the absence or presence of bacitracin (Fig. 2C). Interestingly, we observed a significant decrease in the transcript levels of both pilus genes and the pilus gene regulator nra in the parental strain (MGAS10870) after bacitracin exposure (Fig. 2C). No significant differences in the transcript levels of either nra or msmR were observed between the isogenic carrier mutant and the parental strain (Fig. 2C). Nra is a positive regulator of pilus gene transcription in serotype M53 but a negative regulator in serotype M49 (26). Thus, Nra may act as a positive regulator of pilus gene transcription in serotype M3 GAS strains, and the decreased transcription observed in the wild-type MGAS10870 strain may be responsible for the differences in pilus gene expression compared to that in the isogenic carrier strain. Our data indicate that the increased pilus gene transcription conferred by the liaSR135G carrier mutation is not mediated by other known pilus gene regulators.
Deletion of the major pilus subunit gene in the liaSR135G carrier mutant decreases the ability to colonize the mouse nasopharynx and adhere to and be internalized by cultured human epithelial cells.
We have defined the carrier phenotype to be one characterized by an increased ability to adhere to epithelial surfaces and a decreased ability to cause disease, a definition supported by investigation of independent carrier-specific mutations and their contribution to the carrier phenotype (15–18). The liaSR135G carrier mutation causes an increased frequency of mouse colonization in a model of nasopharyngeal infection (17). Further, in serotype M1 GAS strains, elimination of pilus decreases adherence to cultured human epithelial cells (23). Inasmuch as induction of the LiaFSR 3CS by bacitracin in vitro leads to alteration of pilus gene transcription, we hypothesized that the liaSR135G mutation results in increased pilus gene transcription in vivo and contributes to the carrier phenotype, including an increased ability to colonize the mouse nasopharynx. If this is true, elimination of the GAS pilus would lead to decreased recovery following mouse nasopharyngeal infection. To test this hypothesis, we created an in-frame replacement of the major pilus subunit gene (spyM3_0100, tee3) to generate a pilus-negative carrier mutant (MGAS10870 liaSR135G Δtee3) and a wild-type invasive strain (MGAS10870 Δtee3) (Fig. 2A). Both pilus-negative strains lacked bacterial cell surface pilus expression, as determined by Western immunoblotting using an affinity-purified anti-Tee3 polyclonal antibody (Fig. 2D). Mice infected in the nasopharynx with the parental invasive strain (MGAS10870), isogenic carrier mutant (MGAS10870 liaSR135G), pilus-negative invasive strain (MGAS10870 Δtee3), or pilus-negative carrier mutant (MGAS10870 liaSR135G Δtee3) were swabbed daily for 14 days to determine the number of bacterial CFU. Elimination of pilus production significantly reduced the ability of the isogenic carrier strain to colonize the mouse nasopharynx but had only a modest effect on that of the invasive strain (Fig. 3A). Consistent with the findings of previous studies, beginning at day 5 bacteria were significantly more frequently recovered from mice infected with the liaSR135G carrier mutant than from mice infected with the parental strain (Fig. 3A). The pilus-negative liaSR135G mutant was recovered from mice significantly less frequently than the liaSR135G mutant but significantly more frequently than the pilus-negative wild-type invasive strain (Fig. 3A).
FIG 3.

Elimination of pilus in MGAS10870 liaSR135G decreases recovery following mouse nasopharyngeal infection and in vitro adherence to and internalization by human epithelial cells. (A) Percentage of mice from which bacteria were recovered following nasopharyngeal infection with the respective GAS strains. **, P < 0.05 (Kruskal-Wallis repeated-measures ANOVA) relative to mice infected with all other strains after day 5 (dotted line); *, P < 0.05 compared to both the parental and pilus-negative parental strains after day 5. (B) Adherence to cultured human epithelial cells. Assays of adherence to HaCaT and HEp-2 cells were performed in replicates of 12 on at least two occasions. Internalization assays were performed using HEp-2 cells. Percent adherence or percent internalization was calculated by dividing the colony counts following the assay by the input number of bacterial cells and multiplying by 100. P values were determined by the Mann-Whitney U test. NS, not significant.
In addition to decreased recovery following mouse nasopharyngeal infection, we hypothesized that elimination of pilus would also significantly reduce adherence of the carrier mutant to cultured human epithelial cells. Thus, we performed in vitro adherence assays comparing the pilus-negative mutants to their parental counterparts. We observed significantly increased adherence of MGAS10870 liaSR135G to HaCaT cells compared to the parental strain, MGAS10870 (Fig. 3B, HaCaT). We also assayed adherence to HEp-2 cells and observed a phenotype similar to that for adherence to HaCaT cells (Fig. 3B, HEp-2). Elimination of the pilus backbone protein had no significant effect on the adherence of the parental invasive strain to either HaCaT or HEp-2 cells (Fig. 3B). The pilus-negative carrier mutant had significantly reduced adherence to HEp-2 cells and a trend toward reduced adherence to HaCaT cells (Fig. 3B). Interestingly, the pilus-negative carrier mutant maintained significantly increased adherence compared to that of either the parental or the pilus-negative parental strain (Fig. 3B), suggesting that additional molecules are involved in adherence in the carrier mutant.
We also assessed the contribution of the GAS pilus to the ability of the parental and isogenic carrier mutants to be internalized by human epithelial cells. Previous studies suggest that pilus contributes to the ability of GAS to be internalized by epithelial cells (24). Thus, we hypothesized that the increased pilus gene transcription conferred by the carrier mutation would also lead to increased internalization by HEp-2 cells. We performed gentamicin protection assays to determine the level of internalization of the parental, isogenic carrier, and pilus-negative strains. The isogenic carrier mutant had an almost 3-fold increased ability to be internalized by HEp-2 cells compared to the wild-type, parental strain (Fig. 3B). Elimination of pilus did not significantly alter the ability of either the parental or the isogenic carrier mutant to be internalized by HEp-2 cells. However, there was a trend toward significance for the pilus-negative isogenic carrier mutant (P = 0.065). Overall, the data suggest that pilus is a contributor to epithelial cell adherence and internalization in the isogenic carrier mutant but that additional factors also contribute to this phenotype.
Deletion of the pilus major subunit gene partially restores virulence in the liaSR135G carrier mutant in a mouse model of necrotizing fasciitis.
It has been shown that deletion of pilus in serotype M1 GAS strains results in increased virulence in mouse models of invasive GAS infection (23, 24). Thus, we next hypothesized that if increased pilus expression is a contributor to the carrier phenotype (i.e., decreased virulence) conferred by the liaSR135G mutation, elimination of pilus would restore virulence in isogenic carrier mutant MGAS10870 liaSR135G. Using strains identical to those used for mouse nasopharyngeal infection, mice were infected intramuscularly and followed for survival. No significant difference in survival was observed between mice infected with the isogenic liaSR135G carrier mutant strain and mice infected with the pilus-negative liaSR135G mutant (MGAS10870 liaSR135G Δtee3) (Fig. 4A). However, consistent with an inverse relationship between pilus expression and virulence, we observed significantly greater mortality in mice infected with the MGAS10870 Δtee3 mutant strain than mice infected with the carrier liaSR135G mutant strain (Fig. 4A). Examination of mouse limbs at 72 h postinfection revealed that the pilus-negative liaSR135G mutant caused an infection that appeared similar to that caused by the wild type, with larger areas of devitalized tissue and necrosis that were indistinguishable from those caused by the parental invasive strain (Fig. 4B). In contrast, the MGAS10870 liaSR135G strain caused a small isolated area of infection surrounded by healthy tissue (Fig. 4B), similar to previous observations (17). Using a validated pathology score (38), the isogenic carrier mutant (MGAS10870 liaSR135G) produced significantly less tissue damage than the wild-type or pilus-negative strain (Fig. 4C). Thus, consistent with our hypothesis, elimination of pilus partially restored virulence in the liaSR135G mutant strain.
FIG 4.
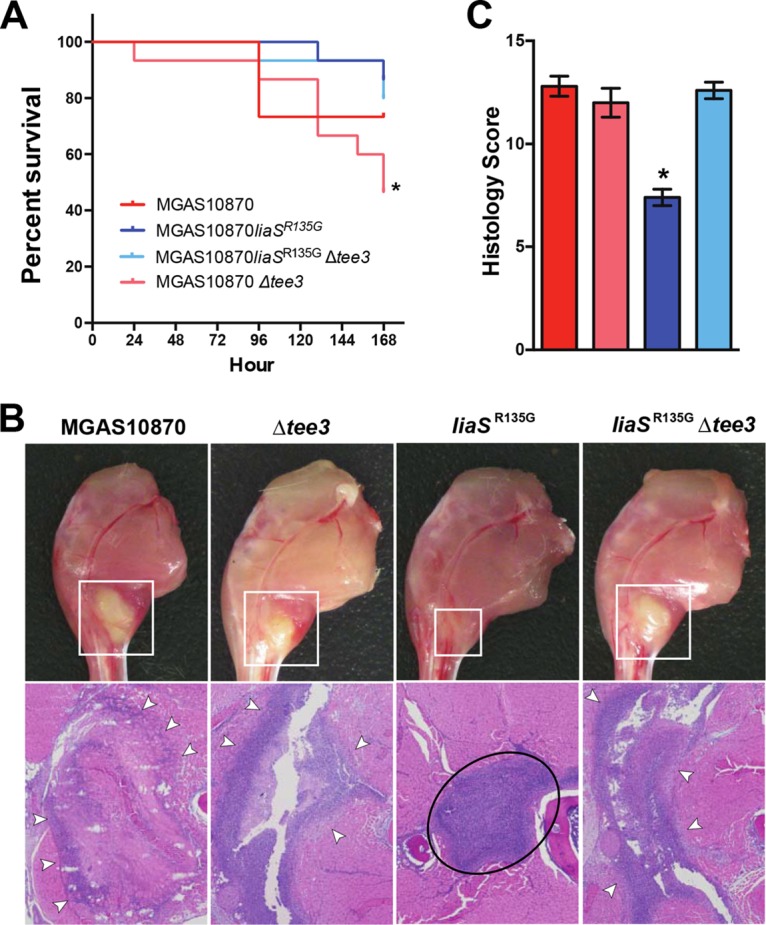
Deletion of the pilus backbone gene in MGAS10870 liaSR135G partially restores virulence and reverses the carrier phenotype. (A) Kaplan-Meier survival curve of mice following intramuscular infection with MGAS10870 (parental strain), MGAS10870 Δtee3 (parental strain, pilus negative), MGAS10870 liaSR135G (carrier mutant), or MGAS10870 liaSR135G Δtee3 (carrier mutant, pilus negative). *, P < 0.05 (log-rank test) relative to both the pilus-positive and pilus-negative carrier mutant. (B) Visual inspection and histopathology of infected mouse limbs at 72 and 24 h postinfection, respectively. (Top row) White boxes, areas of abscess-like formation centered at the inoculation site of the limbs; (bottom row) white arrowheads, areas of marked neutrophil infiltration and necrotic fascial and muscle tissue that extend beyond the border of the field of view; black oval, a well-circumscribed abscess that is limited to the fascial plane and surrounded by healthy muscle tissue (magnification, ×4). (C) Pathology score for wild-type, isogenic carrier, and pilus-negative derivatives based on microscopic examination at 24 h postinfection. *, P < 0.05 (Wilcoxon rank sum) compared to all other strains.
The isogenic carrier mutant had decreased survival after exposure to human neutrophils, a phenotype reversed upon elimination of pilus production.
Previously, an inverse relationship between pilus expression and survival in human blood was reported for serotype M1 GAS strains (23). The liaSR135G carrier mutation is known to confer a decreased ability to grow in human blood ex vivo (17). We hypothesized that increased pilus transcription and increased pilus expression in the isogenic carrier mutant contribute to this phenotype. Further, we hypothesized that, as reported for serotype M1 GAS strains (23), decreased survival of the isogenic carrier mutant in human blood would be due to increased pilus expression. To test this hypothesis, we performed GAS survival assays following exposure to purified human neutrophils. Consistent with our previous observations in whole blood ex vivo (17), the isogenic carrier mutant had significantly reduced survival compared to the parental invasive strain (Fig. 5). The pilus-negative carrier mutant strain (MGAS10870 liaSR135G Δtee3) had significantly increased survival compared to the pilus-positive carrier mutant (Fig. 5). In contrast, elimination of pilus in the invasive strain did not result in significantly increased survival (Fig. 5).
FIG 5.
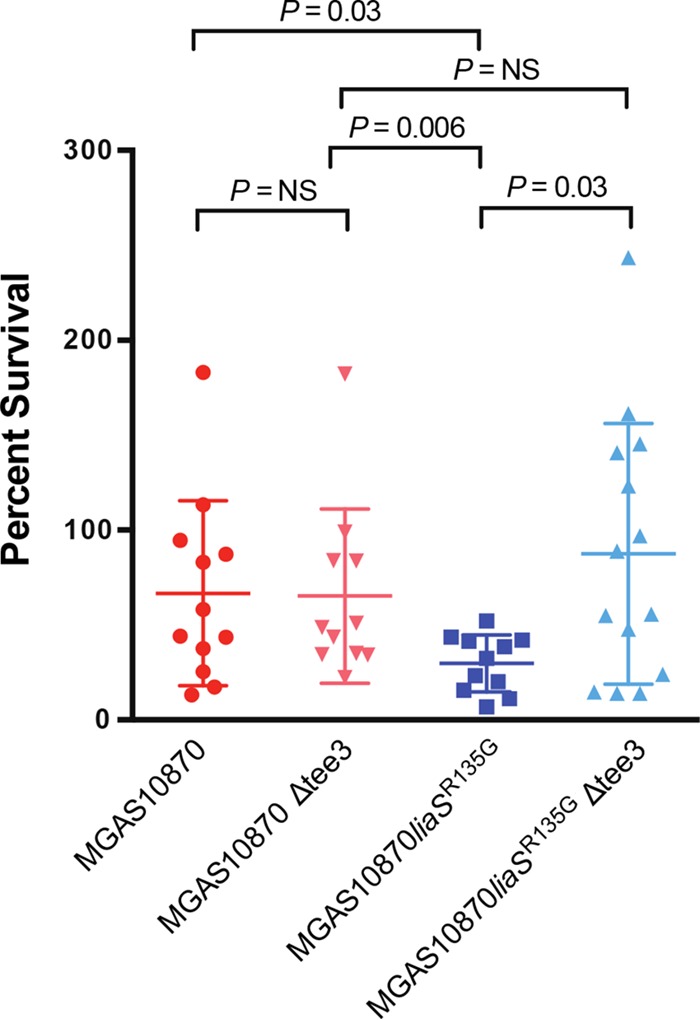
Elimination of pilus in MGAS10870 liaSR135G increases survival in purified human neutrophils. The survival of MGAS10870 (parental strain), MGAS10870 Δtee3 (parental strain, pilus negative), MGAS10870 liaSR135G (carrier mutant), or MGAS10870 liaSR135G Δtee3 (carrier mutant, pilus negative) was assessed 3 h following interaction with human neutrophils. Percent survival was calculated with the equation (CFU+PMN at 3 h/CFU+PMN at 0 h) × 100. P values were determined by the Mann-Whitney U test.
DISCUSSION
Neither the bacterial nor the host molecular mechanisms that contribute to the persistence of bacterial pathogens in the upper respiratory tract of humans are well understood. Few studies have directly examined the bacterial molecular genetic factors that lead to the persistence of GAS in humans (15, 16, 18). Our findings substantially enhance understanding of the molecular mechanism underlying the carrier mutation (liaSR135G) and its contribution to the asymptomatic carrier phenotype in GAS. We demonstrated that the carrier mutation altered pilus gene regulation and that increased pilus gene expression contributed to increased adherence and internalization by epithelial cells and decreased virulence (i.e., the carrier phenotype). This is the first report of pilus as a major contributor to a carrier phenotype in GAS and differs strikingly from the enhanced invasiveness reported for piliated pneumococci (39, 40) and GBS (41, 42).
Our discovery that a GAS carrier mutation alters expression of bacterial surface structures and contributes to the GAS carrier phenotype is in concert with previous findings. We reported a mutation unique to carrier strains in the promoter of the stand-alone regulator Mga that led to decreased transcription of mga and Mga-regulated genes, including emm, encoding M protein (18). We also identified a carrier mutation that restored the function of the GAS surface protein SclA (streptococcal collagen-like protein A) (16). In addition, we discovered that mutations eliminating GAS capsule production were the most frequently identified mutations in GAS carrier strains (15). In the aggregate, our studies have revealed multiple independent pathways to a carrier phenotype defined by increased persistence, decreased virulence, and decreased inflammation, a phenotype that stands in stark contrast to an invasive disease phenotype. A recurring theme has emerged, in that independent genetic changes altering cell surface molecules and, potentially, the host-microbe interaction are critical to the carrier phenotype.
Little is known regarding GAS gene regulation by the three-component system LiaFSR. We previously showed that the liaSR135G carrier mutation significantly altered the transcript levels of approximately 7% of the genes in serotype M3 GAS strains (17). Here, we show that gene regulation by LiaFSR and LiaSR135G is affected following activation by bacitracin. Specifically, we show that, consistent with the findings of studies with GBS (30) and S. pneumoniae (29), the GAS LiaFSR exerts some level of control over pilus gene transcription. Targeted gene transcription of known pilus gene regulators identified nra to be one potential mechanism for the increased pilus gene transcription in the isogenic carrier mutant. However, the association was indirect, as the only significant differences in nra transcription following treatment with bacitracin were in the parental invasive strain (in which nra transcription was decreased 3.6-fold), suggesting that nra acts as a positive regulator of pilus gene transcription, similar to the findings for serotype M53 GAS (26). We also observed a 1.9-fold decrease in mga transcription in the isogenic carrier mutant following treatment with bacitracin. Mga-deficient mutants of serotype M1 GAS have decreased pilus gene expression (37), suggesting that Mga is also an activator of pilus gene expression.
It is unknown if the LiaFSR three-component system directly interacts with the pilus operon or known pilus gene regulators. Serotype M1 GAS strains lacking LiaS have a reduced ability to grow under acidic conditions (43). Recently, site-directed mutagenesis suggested that LiaS may contribute to extracellular acidification and may be activated under acidic conditions, and this directly affected biofilm formation (44). Further, GAS pili contribute to biofilm formation, and, at least for a subset of FCT types, a lower environmental pH enhances pilus gene transcription (45). More studies are needed to further define the precise mechanism of pilus gene regulation by LiaFSR and the mechanism by which the liaSR135G carrier mutation alters pilus gene transcription.
Our data strongly support the possibility that pilus expression confers a distinct advantage to the development of a carrier phenotype. In studies of serotype M1 GAS strains, mutants lacking pili had a reduced ability to adhere to pharyngeal cells (46), human tonsillar epithelium, and primary human keratinocytes (47). Pilus-defective mutants of serotype M1 GAS were also shown to have a decreased ability to adhere to cultured human epithelial cells and reduced virulence in murine models of invasive disease (23, 24). Consistent with the findings of previous studies, our data demonstrate that increased pilus expression contributes to the increased adherence and decreased virulence of the isogenic carrier mutant. Moreover, our findings echo those of others showing an inverse relationship between expression of bacterial adhesins and virulence (48–50). The pili of several Gram-positive pathogens, including GAS, are under investigation as potential vaccine targets (21, 51). Our findings that pili influence virulence but also carriage and persistence have direct implications in vaccine development.
The current study shows that the LiaFSR 3CS is directly involved in GAS pathogenesis and the host-pathogen interaction. No previous studies have directly linked the LiaFSR regulatory system to immune evasion. The liaSR135G single amino acid change results in a significantly decreased ability to survive exposure to human neutrophils. This finding begins to define a mechanism for the observation that the carrier mutation decreases survival in whole human blood ex vivo (17). Previous studies of GAS-neutrophil interactions identified the two-component regulatory system Ihk/Irr to be critical to GAS survival (52), and irr-negative mutants were more easily killed by neutrophils (53). RNA-seq analysis of the liaSR135G mutant at two separate time points in vitro did not show any significant changes in irr and ihk transcription (17). Further investigation is needed to determine the precise mechanism by which the LiaFSR 3CS contributes to immune evasion.
Our study has several limitations. We performed targeted gene transcript analysis. It is possible that additional genes are affected by the LiaSR135G carrier amino acid substitution upon activation by bacitracin or other cell wall-active agents. It is also possible that activation of the LiaFSR three-component system by bacitracin remains incomplete and other LiaFSR-activating factors may further contribute to gene regulatory differences. Further, elimination of pilus in the isogenic carrier strain did not fully restore the invasive phenotype. Previous transcriptome studies comparing the parental and isogenic carrier strains showed that several virulence factors are affected (17). Alteration of pilus gene expression contributes to the overall carrier phenotype, but it is likely that the complete phenotype imparted by the liaSR135G carrier mutation is multifactorial.
In summary, our study adds substantially to the existing framework of the bacterial molecular mechanisms that contribute to carriage of GAS and other bacterial pathogens. The single amino acid change in LiaS alters the GAS phenotype in favor of asymptomatic carriage. Rather than alter expression of a single factor, the liaSR135G mutation likely alters expression of multiple surface/secreted factors, such as pilus, that contribute to the carrier phenotype (Fig. 6). It is reasonable to hypothesize that further study of the gene regulatory changes associated with the liaSR135G carrier mutation will reveal new mechanisms contributing to the carrier phenotype.
FIG 6.

Schematic hypothetical model of the mechanism by which the carrier single amino acid replacement (LiaSR135G) leads to altered pilus expression and the carrier phenotype. LiaS (blue) is a histidine kinase with two transmembrane-spanning domains. Activation signals for LiaS are unknown, but sensing is believed to involve both LiaS and the accessory membrane protein LiaF (black). Upon activation, LiaS undergoes autophosphorylation at the single conserved histidine (H). Following phosphorylation (circled P), the cognate response regulator, LiaR (yellow), is phosphorylated by phosphotransfer from LiaS. However, one hypothesis is that the carrier mutation decreases the autophosphorylation of LiaS, thereby decreasing phosphotransfer to LiaR and altering gene transcription. Decreased activation of LiaS and LiaR leads to alterations in global gene transcription, including the transcription of genes encoding pilus proteins. Ultimately, increased expression of pilus favors increased adherence, decreased immune evasion/inflammation, and decreased virulence, key characteristics of the carrier phenotype and persistence. WT, wild type.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The strains used in this study are listed in Table S1 in the supplemental material. MGAS10870 is a serotype M3 GAS invasive strain isolated in 2002 in Ontario, Canada, and its genome has been sequenced to high quality (54). Strain MGAS10870 liaSR135G is a previously described isogenic mutant differing from the parental strain, MGAS10870, only by the carrier liaS allele (17). All GAS strains were grown in Todd-Hewitt broth containing 0.2% (wt/vol) yeast extract (THY; Difco Laboratories), on THY agar, or on Trypticase soy agar containing 5% sheep blood agar (SBA; Becton Dickinson).
Generation of pilus-negative mutants in strains MGAS10870 and MGAS10870 liaSR135G.
The plasmids and primers used in this study are listed in Table S2. We used a previously described procedure (55) for in-frame replacement of the gene encoding the pilus backbone protein tee3 (spyM3_0100) with a spectinomycin resistance gene (aad9) in strains MGAS10870 and MGAS10870 liaSR135G. Experimental details are provided in the supplemental material. Mutants were confirmed and shown, using whole-genome sequencing, to lack spurious mutations.
Anti-Tee3 antibody production and Western immunoblot analysis.
Recombinant, purified Tee3 protein was produced using an Impact system (New England BioLabs). Briefly, tee3 (spyM3_0100) was synthesized, optimized for expression in Escherichia coli, and cloned into pTXB1 (GenScript). Protein overexpression and purification were carried out as described in the manufacturer's instructions (New England BioLabs). Purified Tee3 protein was subjected to mass spectrometry for identification prior to use in antibody production. Affinity-purified rabbit polyclonal anti-Tee3 antibody was generated by Covance (Denver, PA). Isolation of bacterial proteins and Western immunoblot analysis were carried out as previously described (18).
Mouse virulence studies.
All mouse experiments were conducted under a protocol approved by the Houston Medical Research Institute Institutional Animal Care and Use Committee. Mouse intranasal and intramuscular infections were carried out as previously described (17). For nasopharyngeal infections, a total of 30, 3- to 4-week-old female CD1 mice (Harlan Laboratories) were intranasally inoculated with 5 × 107 CFU of the appropriate GAS strain in 50 μl phosphate-buffered saline (PBS), after which the mice were swabbed daily for 14 days. For intramuscular infections, mice (n = 15) were infected in the right hind limb with 5 × 106 CFU of the appropriate GAS strain in 100 μl PBS. Mice were evaluated at 24 h postinfection for histopathology. All mice were observed and sacrificed when they reached near mortality, determined using predefined criteria. For histopathology scoring, a pathologist blind to the strain treatment groups reviewed each slide (n = 5 per strain) as previously described (38).
Human neutrophil survival assays.
Human neutrophils (polymorphonuclear leukocytes [PMNs]) were isolated from heparinized venous blood of healthy volunteer donors (n = 14) in accordance with a protocol approved by the Institutional Review Board for Human Subjects at Montana State University. All subjects provided written informed consent. Neutrophils were isolated as previously described (56). Complete procedural details are provided in the supplemental material. Percent survival was determined by dividing the number of bacterial CFU after 3 h exposure by the number of bacterial CFU at time zero.
Cultured human epithelial cell adherence and internalization assays.
Adherence to cultured human epithelial cells was carried out as previously described (15). Complete experimental details can be found in the supplemental material. Percent adherence was calculated by dividing the recovered number of CFU by the original inoculum. GAS internalization by cultured epithelial cells was carried out as previously described (15). Percentage internalization was calculated by dividing the recovered number of CFU by the original inoculum. All assays were performed in replicates of 12.
RNA isolation and qRT-PCR analysis.
GAS strains were grown overnight in THY, diluted 1:50 in fresh, prewarmed THY, and incubated at 37°C. Growth was monitored hourly using spectrophotometry (determination of the optical density at 600 nm [OD600]), and when the strains reached an OD600 of 0.5, bacitracin (Sigma-Aldrich) was added to achieve the final concentration indicated above. Cells were harvested at 30, 60, and 90 min following the addition of bacitracin. RNA was isolated and purified with an RNeasy minikit (Qiagen) as previously described (18). The quantity and quality of RNA were determined using an Agilent 2100 bioanalyzer and an RNA 6000 Nano kit (Agilent Technologies).
TaqMan (Life Technologies) quantitative real-time PCR (qRT-PCR) of cDNA produced using SuperScript III reverse transcriptase (Invitrogen) was performed with an ABI 7500 Fast real-time system. The TaqMan primers and probes used in the analyses are listed in Table S2. The endogenous control gene tufA was used for all TaqMan analyses. Transcript levels were compared between strains using the ΔΔCT threshold cycle (CT) method (User Bulletin No. 2, ABI Prism 7700 sequence detection system; Life Technologies). All reactions were performed in triplicate using RNA purified from at least three biological replicates.
Statistics.
All statistical analyses were performed with Prism (version 6) software (GraphPad Software, Inc.). A Mann-Whitney U test was used to compare survival following exposure to purified human neutrophils and adherence to or internalization by cultured human epithelial cells for the strains. A two-tailed t test (unequal variance) was used to compare gene transcript levels. A log rank test was used to compare survival, and Kruskal-Wallis analysis of variance (ANOVA) was used to compare the rates of nasopharyngeal GAS recovery from mice. The Wilcoxon rank sum test was used to compare histopathology scores. A P value of <0.05 was considered significant for all tests unless otherwise indicated.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by a grant from the Harold Amos Medical Faculty Development Program of the Robert Wood Johnson Foundation (to A.R.F.) and funds from the Fondren Foundation (to J.M.M.).
We have no conflicts of interest to declare.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00949-16.
REFERENCES
- 1.Casadevall A, Pirofski LA. 2003. The damage-response framework of microbial pathogenesis. Nat Rev Microbiol 1:17–24. doi: 10.1038/nrmicro732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cartwright KA, Stuart JM, Jones DM, Noah ND. 1987. The Stonehouse survey: nasopharyngeal carriage of meningococci and Neisseria lactamica. Epidemiol Infect 99:591–601. doi: 10.1017/S0950268800066449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caugant DA, Hoiby EA, Magnus P, Scheel O, Hoel T, Bjune G, Wedege E, Eng J, Froholm LO. 1994. Asymptomatic carriage of Neisseria meningitidis in a randomly sampled population. J Clin Microbiol 32:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Claus H, Maiden MC, Wilson DJ, McCarthy ND, Jolley KA, Urwin R, Hessler F, Frosch M, Vogel U. 2005. Genetic analysis of meningococci carried by children and young adults. J Infect Dis 191:1263–1271. doi: 10.1086/428590. [DOI] [PubMed] [Google Scholar]
- 5.Bogaert D, De Groot R, Hermans PW. 2004. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis 4:144–154. doi: 10.1016/S1473-3099(04)00938-7. [DOI] [PubMed] [Google Scholar]
- 6.Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, Nouwen JL. 2005. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis 5:751–762. doi: 10.1016/S1473-3099(05)70295-4. [DOI] [PubMed] [Google Scholar]
- 7.Campbell JR, Hillier SL, Krohn MA, Ferrieri P, Zaleznik DF, Baker CJ. 2000. Group B streptococcal colonization and serotype-specific immunity in pregnant women at delivery. Obstet Gynecol 96:498–503. [DOI] [PubMed] [Google Scholar]
- 8.Regan JA, Klebanoff MA, Nugent RP. 1991. The epidemiology of group B streptococcal colonization in pregnancy. Vaginal Infections and Prematurity Study Group. Obstet Gynecol 77:604–610. [PubMed] [Google Scholar]
- 9.Yancey MK, Schuchat A, Brown LK, Ventura VL, Markenson GR. 1996. The accuracy of late antenatal screening cultures in predicting genital group B streptococcal colonization at delivery. Obstet Gynecol 88:811–815. doi: 10.1016/0029-7844(96)00320-1. [DOI] [PubMed] [Google Scholar]
- 10.Shaikh N, Leonard E, Martin JM. 2010. Prevalence of streptococcal pharyngitis and streptococcal carriage in children: a meta-analysis. Pediatrics 126:e557–e564. doi: 10.1542/peds.2009-2648. [DOI] [PubMed] [Google Scholar]
- 11.Nelson GE, Pondo T, Toews KA, Farley MM, Lindegren ML, Lynfield R, Aragon D, Zansky SM, Watt JP, Cieslak PR, Angeles K, Harrison LH, Petit S, Beall B, Van Beneden CA. 2016. Epidemiology of invasive group A streptococcal infections in the United States, 2005-2012. Clin Infect Dis 63:478–486. doi: 10.1093/cid/ciw248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walker MJ, Barnett TC, McArthur JD, Cole JN, Gillen CM, Henningham A, Sriprakash KS, Sanderson-Smith ML, Nizet V. 2014. Disease manifestations and pathogenic mechanisms of group A Streptococcus. Clin Microbiol Rev 27:264–301. doi: 10.1128/CMR.00101-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeMuri GP, Wald ER. 2014. The group A streptococcal carrier state reviewed: still an enigma. J Pediatr Infect Dis Soc 3:336–342. doi: 10.1093/jpids/piu030. [DOI] [PubMed] [Google Scholar]
- 14.Kaplan EL. 1980. The group A streptococcal upper respiratory tract carrier state: an enigma. J Pediatr 97:337–345. doi: 10.1016/S0022-3476(80)80178-8. [DOI] [PubMed] [Google Scholar]
- 15.Flores AR, Jewell BE, Olsen RJ, Shelburne SA III, Fittipaldi N, Beres SB, Musser JM. 2014. Asymptomatic carriage of group A Streptococcus is associated with elimination of capsule production. Infect Immun 82:3958–3967. doi: 10.1128/IAI.01788-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flores AR, Jewell BE, Versalovic EM, Olsen RJ, Bachert BA, Lukomski S, Musser JM. 2015. Natural variant of collagen-like protein A in serotype M3 group A Streptococcus increases adherence and decreases invasive potential. Infect Immun 83:1122–1129. doi: 10.1128/IAI.02860-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flores AR, Jewell BE, Yelamanchili D, Olsen RJ, Musser JM. 2015. A single amino acid replacement in the sensor kinase LiaS contributes to a carrier phenotype in group A Streptococcus. Infect Immun 83:4237–4246. doi: 10.1128/IAI.00656-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flores AR, Olsen RJ, Wunsche A, Kumaraswami M, Shelburne SA III, Carroll RK, Musser JM. 2013. Natural variation in the promoter of the gene encoding the Mga regulator alters host-pathogen interactions in group A Streptococcus carrier strains. Infect Immun 81:4128–4138. doi: 10.1128/IAI.00405-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao R, Stock AM. 2009. Biological insights from structures of two-component proteins. Annu Rev Microbiol 63:133–154. doi: 10.1146/annurev.micro.091208.073214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sitkiewicz I, Musser JM. 2006. Expression microarray and mouse virulence analysis of four conserved two-component gene regulatory systems in group A Streptococcus. Infect Immun 74:1339–1351. doi: 10.1128/IAI.74.2.1339-1351.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mora M, Bensi G, Capo S, Falugi F, Zingaretti C, Manetti AG, Maggi T, Taddei AR, Grandi G, Telford JL. 2005. Group A Streptococcus produce pilus-like structures containing protective antigens and Lancefield T antigens. Proc Natl Acad Sci U S A 102:15641–15646. doi: 10.1073/pnas.0507808102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Falugi F, Zingaretti C, Pinto V, Mariani M, Amodeo L, Manetti AG, Capo S, Musser JM, Orefici G, Margarit I, Telford JL, Grandi G, Mora M. 2008. Sequence variation in group A Streptococcus pili and association of pilus backbone types with Lancefield T serotypes. J Infect Dis 198:1834–1841. doi: 10.1086/593176. [DOI] [PubMed] [Google Scholar]
- 23.Crotty Alexander LE, Maisey HC, Timmer AM, Rooijakkers SH, Gallo RL, von Kockritz-Blickwede M, Nizet V. 2010. M1T1 group A streptococcal pili promote epithelial colonization but diminish systemic virulence through neutrophil extracellular entrapment. J Mol Med 88:371–381. doi: 10.1007/s00109-009-0566-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakata M, Koller T, Moritz K, Ribardo D, Jonas L, McIver KS, Sumitomo T, Terao Y, Kawabata S, Podbielski A, Kreikemeyer B. 2009. Mode of expression and functional characterization of FCT-3 pilus region-encoded proteins in Streptococcus pyogenes serotype M49. Infect Immun 77:32–44. doi: 10.1128/IAI.00772-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kreikemeyer B, Nakata M, Koller T, Hildisch H, Kourakos V, Standar K, Kawabata S, Glocker MO, Podbielski A. 2007. The Streptococcus pyogenes serotype M49 Nra-Ralp3 transcriptional regulatory network and its control of virulence factor expression from the novel eno ralp3 epf sagA pathogenicity region. Infect Immun 75:5698–5710. doi: 10.1128/IAI.00175-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo F, Lizano S, Bessen DE. 2008. Heterogeneity in the polarity of Nra regulatory effects on streptococcal pilus gene transcription and virulence. Infect Immun 76:2490–2497. doi: 10.1128/IAI.01567-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molinari G, Rohde M, Talay SR, Chhatwal GS, Beckert S, Podbielski A. 2001. The role played by the group A streptococcal negative regulator Nra on bacterial interactions with epithelial cells. Mol Microbiol 40:99–114. doi: 10.1046/j.1365-2958.2001.02373.x. [DOI] [PubMed] [Google Scholar]
- 28.Nakata M, Podbielski A, Kreikemeyer B. 2005. MsmR, a specific positive regulator of the Streptococcus pyogenes FCT pathogenicity region and cytolysin-mediated translocation system genes. Mol Microbiol 57:786–803. doi: 10.1111/j.1365-2958.2005.04730.x. [DOI] [PubMed] [Google Scholar]
- 29.Rosch JW, Mann B, Thornton J, Sublett J, Tuomanen E. 2008. Convergence of regulatory networks on the pilus locus of Streptococcus pneumoniae. Infect Immun 76:3187–3196. doi: 10.1128/IAI.00054-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klinzing DC, Ishmael N, Dunning Hotopp JC, Tettelin H, Shields KR, Madoff LC, Puopolo KM. 2013. The two-component response regulator LiaR regulates cell wall stress responses, pili expression and virulence in group B Streptococcus. Microbiology 159:1521–1534. doi: 10.1099/mic.0.064444-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kesel S, Mader A, Hofler C, Mascher T, Leisner M. 2013. Immediate and heterogeneous response of the LiaFSR two-component system of Bacillus subtilis to the peptide antibiotic bacitracin. PLoS One 8:e53457. doi: 10.1371/journal.pone.0053457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuroda M, Kuroda H, Oshima T, Takeuchi F, Mori H, Hiramatsu K. 2003. Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol Microbiol 49:807–821. [DOI] [PubMed] [Google Scholar]
- 33.Majchrzykiewicz JA, Kuipers OP, Bijlsma JJ. 2010. Generic and specific adaptive responses of Streptococcus pneumoniae to challenge with three distinct antimicrobial peptides, bacitracin, LL-37, and nisin. Antimicrob Agents Chemother 54:440–451. doi: 10.1128/AAC.00769-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shankar M, Mohapatra SS, Biswas S, Biswas I. 2015. Gene regulation by the LiaSR two-component system in Streptococcus mutans. PLoS One 10:e0128083. doi: 10.1371/journal.pone.0128083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bessen DE, Kalia A. 2002. Genomic localization of a T serotype locus to a recombinatorial zone encoding extracellular matrix-binding proteins in Streptococcus pyogenes. Infect Immun 70:1159–1167. doi: 10.1128/IAI.70.3.1159-1167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mao X, Ma Q, Zhou C, Chen X, Zhang H, Yang J, Mao F, Lai W, Xu Y. 2014. DOOR 2.0: presenting operons and their functions through dynamic and integrated views. Nucleic Acids Res 42:D654–D659. doi: 10.1093/nar/gkt1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ribardo DA, McIver KS. 2006. Defining the Mga regulon: comparative transcriptome analysis reveals both direct and indirect regulation by Mga in the group A Streptococcus. Mol Microbiol 62:491–508. doi: 10.1111/j.1365-2958.2006.05381.x. [DOI] [PubMed] [Google Scholar]
- 38.Olsen RJ, Sitkiewicz I, Ayeras AA, Gonulal VE, Cantu C, Beres SB, Green NM, Lei B, Humbird T, Greaver J, Chang E, Ragasa WP, Montgomery CA, Cartwright J Jr, McGeer A, Low DE, Whitney AR, Cagle PT, Blasdel TL, DeLeo FR, Musser JM. 2010. Decreased necrotizing fasciitis capacity caused by a single nucleotide mutation that alters a multiple gene virulence axis. Proc Natl Acad Sci U S A 107:888–893. doi: 10.1073/pnas.0911811107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barocchi MA, Ries J, Zogaj X, Hemsley C, Albiger B, Kanth A, Dahlberg S, Fernebro J, Moschioni M, Masignani V, Hultenby K, Taddei AR, Beiter K, Wartha F, von Euler A, Covacci A, Holden DW, Normark S, Rappuoli R, Henriques-Normark B. 2006. A pneumococcal pilus influences virulence and host inflammatory responses. Proc Natl Acad Sci U S A 103:2857–2862. doi: 10.1073/pnas.0511017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iovino F, Hammarlof DL, Garriss G, Brovall S, Nannapaneni P, Henriques-Normark B. 2016. Pneumococcal meningitis is promoted by single cocci expressing pilus adhesin RrgA. J Clin Invest 126:2821–2826. doi: 10.1172/JCI84705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banerjee A, Kim BJ, Carmona EM, Cutting AS, Gurney MA, Carlos C, Feuer R, Prasadarao NV, Doran KS. 2011. Bacterial pili exploit integrin machinery to promote immune activation and efficient blood-brain barrier penetration. Nat Commun 2:462. doi: 10.1038/ncomms1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maisey HC, Quach D, Hensler ME, Liu GY, Gallo RL, Nizet V, Doran KS. 2008. A group B streptococcal pilus protein promotes phagocyte resistance and systemic virulence. FASEB J 22:1715–1724. doi: 10.1096/fj.07-093963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ichikawa M, Minami M, Isaka M, Tatsuno I, Hasegawa T. 2011. Analysis of two-component sensor proteins involved in the response to acid stimuli in Streptococcus pyogenes. Microbiology 157:3187–3194. doi: 10.1099/mic.0.050534-0. [DOI] [PubMed] [Google Scholar]
- 44.Isaka M, Tatsuno I, Maeyama J, Matsui H, Zhang Y, Hasegawa T. 2016. The YvqE two-component system controls biofilm formation and acid production in Streptococcus pyogenes. APMIS 124:574–585. doi: 10.1111/apm.12538. [DOI] [PubMed] [Google Scholar]
- 45.Manetti AG, Koller T, Becherelli M, Buccato S, Kreikemeyer B, Podbielski A, Grandi G, Margarit I. 2010. Environmental acidification drives S. pyogenes pilus expression and microcolony formation on epithelial cells in a FCT-dependent manner. PLoS One 5:e13864. doi: 10.1371/journal.pone.0013864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manetti AG, Zingaretti C, Falugi F, Capo S, Bombaci M, Bagnoli F, Gambellini G, Bensi G, Mora M, Edwards AM, Musser JM, Graviss EA, Telford JL, Grandi G, Margarit I. 2007. Streptococcus pyogenes pili promote pharyngeal cell adhesion and biofilm formation. Mol Microbiol 64:968–983. doi: 10.1111/j.1365-2958.2007.05704.x. [DOI] [PubMed] [Google Scholar]
- 47.Abbot EL, Smith WD, Siou GP, Chiriboga C, Smith RJ, Wilson JA, Hirst BH, Kehoe MA. 2007. Pili mediate specific adhesion of Streptococcus pyogenes to human tonsil and skin. Cell Microbiol 9:1822–1833. doi: 10.1111/j.1462-5822.2007.00918.x. [DOI] [PubMed] [Google Scholar]
- 48.McElroy MC, Cain DJ, Tyrrell C, Foster TJ, Haslett C. 2002. Increased virulence of a fibronectin-binding protein mutant of Staphylococcus aureus in a rat model of pneumonia. Infect Immun 70:3865–3873. doi: 10.1128/IAI.70.7.3865-3873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Natanson S, Sela S, Moses AE, Musser JM, Caparon MG, Hanski E. 1995. Distribution of fibronectin-binding proteins among group A streptococci of different M types. J Infect Dis 171:871–878. doi: 10.1093/infdis/171.4.871. [DOI] [PubMed] [Google Scholar]
- 50.Nyberg P, Sakai T, Cho KH, Caparon MG, Fassler R, Bjorck L. 2004. Interactions with fibronectin attenuate the virulence of Streptococcus pyogenes. EMBO J 23:2166–2174. doi: 10.1038/sj.emboj.7600214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nuccitelli A, Cozzi R, Gourlay LJ, Donnarumma D, Necchi F, Norais N, Telford JL, Rappuoli R, Bolognesi M, Maione D, Grandi G, Rinaudo CD. 2011. Structure-based approach to rationally design a chimeric protein for an effective vaccine against group B Streptococcus infections. Proc Natl Acad Sci U S A 108:10278–10283. doi: 10.1073/pnas.1106590108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Voyich JM, Sturdevant DE, Braughton KR, Kobayashi SD, Lei B, Virtaneva K, Dorward DW, Musser JM, DeLeo FR. 2003. Genome-wide protective response used by group A Streptococcus to evade destruction by human polymorphonuclear leukocytes. Proc Natl Acad Sci U S A 100:1996–2001. doi: 10.1073/pnas.0337370100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Voyich JM, Braughton KR, Sturdevant DE, Vuong C, Kobayashi SD, Porcella SF, Otto M, Musser JM, DeLeo FR. 2004. Engagement of the pathogen survival response used by group A Streptococcus to avert destruction by innate host defense. J Immunol 173:1194–1201. doi: 10.4049/jimmunol.173.2.1194. [DOI] [PubMed] [Google Scholar]
- 54.Beres SB, Carroll RK, Shea PR, Sitkiewicz I, Martinez-Gutierrez JC, Low DE, McGeer A, Willey BM, Green K, Tyrrell GJ, Goldman TD, Feldgarden M, Birren BW, Fofanov Y, Boos J, Wheaton WD, Honisch C, Musser JM. 2010. Molecular complexity of successive bacterial epidemics deconvoluted by comparative pathogenomics. Proc Natl Acad Sci U S A 107:4371–4376. doi: 10.1073/pnas.0911295107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lukomski S, Hoe NP, Abdi I, Rurangirwa J, Kordari P, Liu M, Dou SJ, Adams GG, Musser JM. 2000. Nonpolar inactivation of the hypervariable streptococcal inhibitor of complement gene (sic) in serotype M1 Streptococcus pyogenes significantly decreases mouse mucosal colonization. Infect Immun 68:535–542. doi: 10.1128/IAI.68.2.535-542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Voyich JM, Braughton KR, Sturdevant DE, Whitney AR, Said-Salim B, Porcella SF, Long RD, Dorward DW, Gardner DJ, Kreiswirth BN, Musser JM, DeLeo FR. 2005. Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J Immunol 175:3907–3919. doi: 10.4049/jimmunol.175.6.3907. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.