

ABSTRACT
The strategies utilized by pathogens to exit host cells are an area of pathogenesis which has received surprisingly little attention, considering the necessity of this step for infections to propagate. Even less is known about how exit through these pathways affects downstream host-pathogen interactions and the generation of an immune response. Chlamydia trachomatis exits host epithelial cells through two equally active mechanisms: lysis and extrusion. Studies have characterized the outcome of interactions between host innate immune cells, such as dendritic cells and macrophages, and free, extracellular Chlamydia bacteria, such as those resulting from lysis. Exit via extrusion generates a distinct, host-membrane-bound compartment of Chlamydia separate from the original infected cell. In this study, we assessed the effect of containment within extrusions upon the interaction between Chlamydia and host dendritic cells. Extrusion dramatically affected the outcome of Chlamydia-dendritic cell interactions for both the bacterium and the host cell. Dendritic cells rapidly underwent apoptosis in response to engulfment of an extrusion, while uptake of an equivalent dose of free Chlamydia had no such effect. Containment within an extrusion also prolonged bacterial survival within dendritic cells and altered the initial innate immune signaling by the dendritic cell.
KEYWORDS: Chlamydia, apoptosis, dendritic cell, extrusion
INTRODUCTION
Chlamydia trachomatis is a highly successful Gram-negative bacterial pathogen, being the leading bacterial cause of sexually transmitted infections and the leading cause of infectious blindness globally (1–3). In the absence of diagnosis and treatment, Chlamydia infections can lead to severe long-term outcomes, such as chronic pelvic pain, infertility, and ectopic pregnancy (4, 5). During infection, innate and adaptive immune responses are mounted against Chlamydia; however, approximately 50% of infections last for a year or more (6). Even after the resolution of infection, only partial protective immunity is achieved and reinfection is common (7–9). This suggests that Chlamydia is adept at establishing and sustaining infection in the face of immune recognition.
Due to Chlamydia's obligate intracellular nature and a historically limited genetic toolbox, key attributes of the host-Chlamydia interaction are still unknown. Chlamydia infection is initiated by the uptake of metabolically inactive elementary bodies (EB) into mucosal epithelial cells, which transition into reticulate bodies (RB) within a vacuole called an inclusion. Here, the metabolically active RB replicates robustly to generate hundreds of Chlamydia bacteria that undergo transition back to EB (10). Chlamydia inhibits apoptosis of the host epithelial cell, ensuring its ability to complete this replication cycle before exit from the cell (11–13). Bacterial exit from host cells is a crucial but underscrutinized stage of the life cycle of this and other intracellular pathogens. Chlamydia possesses two distinct, equally prevalent exit mechanisms: lysis and extrusion (14, 15). Lysis proceeds through the sequential destruction of the inclusion and nuclear and plasma membranes, culminating in the release of individual bacteria into the extracellular space (14). Sloughing of infected epithelial cells, sometimes driven by polymorphonuclear leukocytes (PMNs), has been observed in vivo and may precede lysis (16). In contrast, extrusion begins with the contraction of the plasma and inclusion membranes, resulting in the detachment of a separate compartment of Chlamydia enclosed in a double host membrane (14, 17). This is a controlled process dependent on both bacterial and host factors, which leaves the host cell intact and often residually infected (14, 17). The role of extrusion during human infection is unknown, but it occurs in approximately 50% of infected cells in vitro and has been observed in an animal model of in vivo infection (14, 17, 18). The potential infectious advantages of extrusion for immune evasion, bacterial survival, and dissemination are striking.
C. trachomatis preferentially infects columnar epithelial cells of the genital tract. Upon exiting these cells via lysis or extrusion, C. trachomatis encounters innate immune cells, such as PMNs, macrophages, and dendritic cells (DCs). PMNs are rapidly recruited in response to Chlamydia infection and interact directly with infected cells at the epithelial surface (16, 19, 20). DCs and macrophages are found in the epithelial submucosa of the genital tract during Chlamydia infection, and DCs are known to directly engulf bacteria across mucosal epithelial layers (21–23). These cells readily phagocytose Chlamydia bacteria and generate a cytokine response to pathogen-associated molecular patterns (PAMPs) detected on the bacteria (24, 25). Survival of C. trachomatis within macrophages and DCs is dramatically reduced compared to that in epithelial cells (26). Previous work from our group has shown that murine bone marrow macrophages phagocytose extrusions and that containment within extrusions enhances the survival of Chlamydia within these cells (27). DCs are a critical intermediary between innate and adaptive immunity, possessing high phagocytic capacity and potent activity as antigen-presenting cells. Depending on the environment sensed by DCs, T cells are differentially polarized for distinct responses (28, 29). During Chlamydia infection, detection of the bacteria and subsequent interleukin-12 (IL-12) production by DCs is essential to the initiation of Th1 responses necessary for protection (30–32). It is unknown how containment within an extrusion affects the interaction of Chlamydia with these crucial immunity-stimulating cells.
In this study, we demonstrate that bone marrow-derived DCs (BMDCs) engulf extrusions and that encasement within an extrusion prolongs the survival of Chlamydia within these cells. These findings mirror our previous results in macrophages (27), indicating a similar extrusion-mediated benefit to Chlamydia during interactions with both of these cell types. A marked and provocative contrast, however, was that extrusion engulfment resulted in dramatically different host cell outcomes for DCs and macrophages. DC uptake of extrusions specifically triggered rapid apoptosis, which was inhibited by blocking activation of caspase 3 and caspase 7. Importantly, containment within an extrusion also significantly modified the transcriptional upregulation of biologically relevant cytokines by DCs in response to Chlamydia.
RESULTS
Dendritic cells engulf extrusions.
Extrusions are comprised of a double layer of host membrane encasing large numbers of Chlamydia bacteria. The inner membrane originates from the inclusion membrane which surrounds the Chlamydia bacteria within an infected host epithelial cell, while the outer extrusion membrane is generated from the epithelial cell plasma membrane. Phosphatidylserine (PS) is externalized to the extrusion surface over time (27), a process which we hypothesize may allow the extrusion to mimic an apoptotic cell. We have previously shown that macrophages engulf extrusions in a partially PS-dependent manner (27). During infection, DCs are likely among the first host cells to encounter Chlamydia bacteria. Uptake of extrusions by DCs could impact the function of these potent activators of adaptive immunity, for example, by altering the antigens presented or the kinetics of T cell activation.
We tested whether DCs are able to engulf extrusions by incubating BMDCs with extrusions prepared from McCoy cells infected with green fluorescent protein (GFP)-expressing Chlamydia trachomatis lymphogranuloma venereum (LGV) L2 (termed GFP-Chlamydia). Cells were washed to remove extracellular bacteria and then fixed and stained with phalloidin to elucidate cortical actin structures. Cells were imaged by fluorescence microscopy, and z-stacked images were evaluated to determine whether GFP-Chlamydia bacteria were contained within the planes of the dendritic cell. GFP-Chlamydia bacteria were visible in clusters within extrusion-exposed DCs immediately after uptake (Fig. 1A). Approximately 5% of DCs contained extrusions. In contrast, bacteria were rarely visible in DCs exposed to free Chlamydia bacteria, and these bacteria were seen as distinct, single bacteria rather than the dense clusters typical of extrusions (Fig. 1B). These data indicate that DCs engulf extrusions. At 2 to 5 h postexposure (hpe), GFP-Chlamydia bacteria were frequently dispersed throughout the cells (Fig. 1C and D), as revealed by fluorescence microscopy and a significant increase in the interbacterial distance between Chlamydia within individual extrusion-containing DCs and that in bacteria immediately after uptake (1 hpe). Inclusion-like structures were not observed within extrusion-exposed DCs, as indicated by bacterial distribution and a lack of localized staining for inclusion membrane proteins (data not shown). These data may indicate that extrusion membranes break down within DCs, either releasing Chlamydia bacteria into the cytosol or separating the extrusion into multiple vacuoles within the cell. Notably, this dispersal of bacteria suggests that trafficking after extrusion uptake differs markedly from the homotypic fusion that occurs when multiple free C. trachomatis bacteria infect a cell.
FIG 1.

Extrusions are engulfed by DCs. C57BL/6 BMDCs were exposed to Chlamydia trachomatis LGV L2 extrusions (A and C) or free C. trachomatis bacteria (B). One hour later, the cells were washed and then fixed and stained at the indicated time points with Alexa Fluor 647 phalloidin (purple), DAPI (blue), and anti-GFP (green) to detect GFP-expressing C. trachomatis. (A) Extrusion-exposed DCs at 1 hpe. (B) Free-C. trachomatis-exposed DCs at 1 hpe. (C) Extrusion-exposed DCs at 5 hpe. Arrows indicate extrusion clusters; arrowheads indicate individual bacteria. (D) Mean distance between bacteria within extrusion-exposed DCs, indicating scatter over time. The graph represents at least 25 cells (data points) in each group; horizontal lines represent means and SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Extrusions enhance survival of Chlamydia trachomatis inside DCs.
Chlamydia trachomatis survival in DCs is low, in contrast to the robust growth seen in its main target, the epithelial cell (26). We have previously shown that containment within extrusions increases the survival of Chlamydia in macrophages, prolonging the period during which viable bacteria can be recovered from these cells (27). As innate phagocytic cells and potent stimulators of cell-mediated adaptive immunity, DCs are likely to play a key role in generating protective host responses to Chlamydia (28, 30, 31, 33). To determine how extrusions might improve the survival of Chlamydia within DCs, we exposed DCs to intact extrusions or to an equivalent number of free bacteria generated by sonication of an aliquot of extrusions (this control is referred to as free Chlamydia hereinafter). Sonication did not decrease the viability or infectivity of Chlamydia from extrusions (see Fig. S1 in the supplemental material). At 1 to 5 h after infection, we lysed the DCs and quantified viable bacteria by an inclusion-forming unit (IFU) assay. We observed an increased recovery of viable bacteria from extrusion-exposed DCs in comparison to that in DCs exposed to free bacteria at 1 to 5 h after infection (Fig. 2). These data suggest that containment within extrusions enhances survival of Chlamydia in DCs, similarly to what is seen in macrophages.
FIG 2.

Extrusions prolong survival of Chlamydia bacteria in dendritic cells. C57BL/6 BMDCs were exposed to C. trachomatis LGV L2 extrusions or dose-matched free bacteria. At the indicated hours postexposure, DCs were lysed by sonication and used to infect McCoy cell monolayers for quantification of IFUs. Data are means ± SEM of results of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Extrusion engulfment alters transcriptional upregulation of cytokine production in DCs.
Interactions between host cells and bacterial pathogens activate signaling pathways in response to bacterial PAMPs, culminating in the activation of bactericidal mechanisms and the production of cytokines (34–36). DCs generate a robust response to non-extrusion-contained (or free) Chlamydia, including IL-12p70, tumor necrosis factor alpha (TNF-α), IL-8, IL-6, IL-18, and IL-1β (24, 25, 30). The response of DCs to extrusions has not been explored and is important because containment within these novel pathogenic structures may substantially alter how DCs sense and respond to Chlamydia. Extrusions externalize PS, which may enable them to mimic apoptotic cells, a strategy that Leishmania amazonensis employs to profoundly modify the activation of an effective DC response during infection (37, 38). Phagocytosis of apoptotic bodies by DCs hinders maturation and alters cytokine expression profiles, including inducing immune tolerance through production of IL-10, transformation growth factor beta (TGF-β), and the surface ligand programmed death ligand 1 (PD-L1) (39–42). Containment within extrusions may also alter how Chlamydia bacteria are detected by DCs, including interactions at the cell surface or in endosomal or cytosolic sensing pathways.
We exposed DCs to extrusions or an equivalent dose of free Chlamydia bacteria from sonication-disrupted extrusions and then quantified the transcriptional response at 3 hpe. As described above, sonication of extrusions did not decrease the viability or infectivity of Chlamydia (Fig. S1). We estimated that a DC which has engulfed an extrusion is exposed to approximately 20 times as many bacteria as DCs exposed to sonication-disrupted extrusions. This estimate is based on our microscopy data, which revealed that only ∼5% of DCs engulf an extrusion, whereas bacteria in the sonication-disrupted group would be equally distributed to all DCs in that sample. To control for the effect of this difference in dose, we also exposed DCs to a dose titration of Chlamydia EBs at 1 times, 10 times, and 100 times the concentration found in sonication-disrupted extrusions (100 times was the highest concentration that was feasible to obtain). To obtain a pure population of extrusion-containing DCs, we sorted the extrusion-exposed population (Fig. 3A, blue) for GFP-positive, 7-aminoactinomycin D-negative (GFP+ 7-AAD−) cells, using uninfected cells (Fig. 3A, red) to set the gate. Our sort data indicated that 1.3% of extrusion-exposed DCs contained extrusions, which is approximately consistent with the 5% extrusion-containing DCs observed by fluorescence microscopy (data not shown). To confirm that our sort had enriched for extrusion-containing DCs, we quantified chlamydial 16S mRNA (Fig. 3B). After sorting, the extrusion sample had a 45-fold-higher 16S mRNA level than the same sample prior to sorting, indicating a successful sort. Chlamydial 16S mRNA levels were approximately similar between extrusion-containing DCs and DCs exposed to 100 times the EB dose, indicating that a similar bacterial dose was achieved in this control (Fig. 3B). We confirmed by microscopy that a high Chlamydia burden was engulfed by DCs at this EB dose, visualizing bacterial load immediately after uptake in DCs exposed to 100 times the EB dose (Fig. S2). EBs comprise the vast majority of Chlamydia bacteria contained within extrusions (27), so this EB control utilizes Chlamydia bacteria in a biologically similar state to those encountered by an extrusion-containing DC.
FIG 3.

Extrusions alter the DC cytokine response to Chlamydia. C57BL/6 BMDCs were exposed to C. trachomatis LGV L2 extrusions or dose-matched free bacteria. (A) Extrusion-containing DCs were purified by sorting for GFP+ 7-AAD− cells (red indicates an uninfected sample used for gating). (B to F) qRT-PCR quantification of normalized transcript numbers at 3 hpe. Data are from one experiment that is representative of three independent experiments.
Quantitative reverse-transcription PCR (qRT-PCR) analysis of a panel of physiologically relevant cytokines demonstrated elevated beta interferon (IFN-β) transcription in sorted extrusion-containing DCs compared to that in DCs exposed to all doses of free Chlamydia or to uninfected DCs (Fig. 3C). Previous studies indicated that IFN-β production in response to Chlamydia depends on STING (stimulator of interferon genes) in fibroblasts and epithelial cells, a cytosolic sensor of bacterial DNA (43, 44). Therefore, these data suggest that extrusions enhance detection of Chlamydia by the cytosolic sensor STING in DCs and that this response is not simply due to a higher bacterial dose. Extrusion-containing DCs also synthesized elevated transcript levels of the immunosuppressive cytokine IL-10 compared to those of all other groups (Fig. 3D). Transcription of IL-12p40 and IL-12p35 (Fig. 3E and data not shown), cytokines shown to be important for the stimulation of a protective Th1 response to Chlamydia (31), were also elevated in extrusion-containing DCs compared to all other groups. Similarly, the surface molecule PD-L1, which is involved in suppressing T cell responses, was increased in extrusion-containing DCs (Fig. 3F). Transcription of the immunosuppressive cytokine TGF-β (data not shown) was not significantly altered. These data suggest that the DC response to extrusions may differ qualitatively (IFN-β production response specific to extrusion-containing cells) as well as quantitatively (somewhat elevated IL-10, IL-12p40, p35, and PD-L1 levels) from the DC response to an equivalent number of free Chlamydia bacteria. Analysis of sorted unstimulated and infected control samples confirmed that these transcriptional responses are not due to a sorting artifact (data not shown). Therefore, containment within an extrusion alters the cytokine response of DCs to Chlamydia, suggesting that extrusions may interfere with the detection of PAMPs or expose DCs to additional signals that modulate the response.
Engulfment of extrusions causes apoptosis.
In the course of imaging extrusion uptake by DCs, we observed a dramatic change in morphology among extrusion-containing DCs at approximately 5 hpe. These cells specifically became more rounded, and some released membrane blebs, suggesting that engulfment of extrusions was causing cell death. To explore this further, we exposed DCs to extrusions or free bacteria at an equivalent dose and washed them to remove extracellular bacteria. We then stained cells with annexin V to measure phosphatidylserine (PS) on the cell surface, which is a marker of apoptotic cell death (41, 42). Annexin V staining was quantified by fluorescence microscopy. To focus our analysis specifically on the DC response to extrusion uptake within the extrusion-exposed sample, we measured annexin V fluorescence exclusively in DCs that visibly contained an extrusion, based on fluorescence of GFP-Chlamydia bacteria. Extrusion-containing DCs elicited increased annexin V staining at 3 hpe compared to annexin V staining of uninfected or free Chlamydia-exposed DCs, indicating elevated PS externalization (Fig. 4A to C). Annexin V staining was restricted to cells that contained extrusions, not adjacent cells within the same treatment well, indicating that PS externalization occurs downstream of extrusion engulfment and is not driven merely by paracrine signals or surface interaction with an extrusion. Annexin V staining in DCs exposed to free Chlamydia from a frozen seed stock was also overwhelmingly negative, similar to the result from the sonicated free-Chlamydia control (data not shown). To verify that annexin V staining was due to external PS and not an artifact caused by membrane permeability, we also stained with membrane-impermeant LIVE/DEAD fixable viability dye. Most extrusion-containing cells were negative for staining with this dye, indicating that PS was exposed on the surface of DCs (Fig. 4D).
FIG 4.

Extrusion uptake causes phosphatidylserine exposure on DCs. C57BL/6 BMDCs were exposed to C. trachomatis LGV L2 extrusions or dose-matched free bacteria. At 3 hpe, DCs were stained with annexin V-Alexa Fluor 568 or LIVE/DEAD viability dye and imaged. (A and B) Representative images of extrusion-containing (A) or free-C. trachomatis-exposed (B) DCs at 3 hpe. Red, annexin V; blue, DAPI; green, C. trachomatis. A white arrowhead indicates an individual bacterium. Scale bar, 5 μm. (C and D) Quantification of the percentage of cells positive for annexin V (C) or viability dye (D) at 3 hpe. Data are shown as means ± SEM of results of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Annexin V staining suggested that extrusion uptake causes apoptosis in DCs, so we sought to verify this as the mechanism of cell death. We used a FLICA (fluorochrome inhibitor of caspases)-based fluorescent probe to measure the activation of caspases 3 and 7, the apoptosis-associated effector caspases. DCs were exposed to extrusions or an equivalent number of free Chlamydia bacteria and washed to remove extracellular bacteria as before, and then they were stained with the caspase 3/7 probe and fixed for imaging. As before, within the extrusion-exposed sample, we used fluorescence of GFP-Chlamydia to specifically focus our analysis on extrusion-containing DCs. We observed increased caspase 3/7 probe fluorescence in extrusion-containing DCs, compared to the fluorescence of uninfected or free-Chlamydia-infected DCs (Fig. 5A and C). To determine whether DC apoptosis was caused by the high bacterial burden delivered through extrusion uptake, we exposed DCs to higher doses of EBs, from 10 times to 100 times the concentration of bacteria in sonication-disrupted extrusions. Since approximately 1 to 5% of cells contain extrusions, all cells exposed to 100 times the EB dose received a bacterial burden per cell similar to what an extrusion-containing DC would be exposed to. DCs exposed to elevated doses of free Chlamydia showed caspase 3/7 activity equivalent to that of uninfected cells (Fig. 5B and C). These data reveal that extrusion engulfment specifically activates caspase 3 or 7, implicating apoptosis as the mechanism by which extrusion uptake causes DC death.
FIG 5.

Extrusion uptake causes activation of caspase 3 and/or 7 in dendritic cells. C57BL/6 BMDCs were exposed to C. trachomatis LGV L2 extrusions or dose-matched free bacteria. At 3 hpe, DCs were stained with the caspase 3/7 probe and imaged. (A and B) Representative images of DCs containing extrusions (A) or DCs exposed to 100 times the free-C. trachomatis EB dose (B) at 3 hpe. Red, caspase 3/7 probe; blue, DAPI; green, C. trachomatis. Scale bar, 5 μm. (C) Quantification of caspase 3/7 probe intensity at 3 hpe. Data are means ± SEM of the results of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Chlamydia are capable of inhibiting the apoptosis of epithelial cells, enabling the bacteria to complete their developmental cycle within these permissive target cells (11, 45, 46). Chlamydia trachomatis has not been shown to inhibit apoptosis in DCs. However, an alternative explanation of our data was that free Chlamydia bacteria prevent DC apoptosis, while extrusion-contained Chlamydia bacteria are unable to do so. In this scenario, coinfecting DCs with free Chlamydia EBs and extrusions should result in minimal apoptosis, due to inhibition of cell death by the free Chlamydia. To test this, we incubated DCs with a dose titration of Chlamydia EBs (1 times, 10 times, and 100 times the bacterial concentration from sonication-disrupted extrusions) either simultaneously with, or 4 h prior to, exposure to extrusions. DCs were subsequently washed to remove extracellular bacteria, incubated with a caspase 3/7 probe, fixed, and assessed by fluorescence microscopy. Apoptosis, as quantified by caspase 3/7 activity, was not diminished in extrusion-containing DCs by coinfection or preinfection with free Chlamydia EBs (Fig. S3). These data suggest that apoptosis is an extrusion-specific outcome, and this extrusion-dependent cell death is predicted to occur in the biologically relevant scenario in which DCs encounter both extrusions and free EBs.
Extrusion-mediated apoptosis can be interrupted by inhibition of caspase 3/7.
Since extrusion uptake causes apoptosis of DCs, we sought to determine whether extrusion-mediated cellular death could be disrupted by inhibition of the apoptosis-associated effector caspases. We incubated DCs with extrusions in the presence of increasing concentrations of DEVD-FMK, an inhibitor of caspases 3 and 7. We used a TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling) assay to quantify apoptosis at an event downstream from activation of the effector caspases. As a negative control, we also exposed DCs to increasing concentrations of WEHD-FMK, a caspase 1 inhibitor. As before, DCs which engulfed extrusions underwent apoptosis at higher rates than uninfected DCs, indicated by elevated levels of TUNEL staining (Fig. 6). Inhibition of caspases 3 and 7, but not caspase 1, decreased TUNEL staining in extrusion-containing DCs in a dose-dependent manner (Fig. 6). These data indicate that extrusion-induced apoptosis is mediated through the apoptosis effector caspases 3 and 7.
FIG 6.
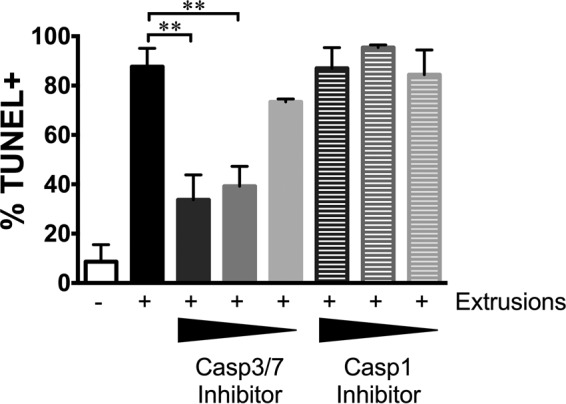
Extrusion-mediated apoptosis requires caspase 3 or 7. C57BL/6 BMDCs were exposed to C. trachomatis LGV L2 extrusions in the presence of specific inhibitors of caspase 3/7 or caspase 1. At 8 hpe, DCs were stained with the Click-iT Plus TUNEL assay and imaged. Wedges indicate increasing inhibitor concentrations. Data are means ± SEM of results of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
DISCUSSION
Our data reveal that containment within an extrusion substantially alters Chlamydia-DC interactions, resulting in dramatically distinct outcomes for the host cell. Engulfment of an extrusion resulted in rapid apoptosis of the DC via a caspase 3/7-dependent mechanism. Since DCs exposed to an equivalent number of free Chlamydia bacteria on a cell-to-cell basis did not undergo apoptosis, the demise of the DC appears to be caused by a distinct property of the extrusion itself, not simply a high bacterial burden. The externalization of phosphatidylserine (PS), a hallmark of apoptosis, on the extrusion surface may disguise extrusions as apoptotic bodies. DCs do not undergo apoptosis following uptake of apoptotic cells or PS-coated liposomes (47–49); however, DC exposure to PS-liposomes predisposes these cells to undergo apoptosis following stimulation with Escherichia coli lipopolysaccharide (LPS) (48). Considering that DCs are likely to encounter Chlamydia LPS during or after extrusion uptake, this experimental setup likely resembles the series of signals encountered by a DC engulfing an extrusion. Our data also demonstrate a profound difference in the responses to extrusion engulfment between DCs and another innate immune phagocyte, bone marrow macrophages, which showed no such cell death after phagocytosis of extrusions (27). Further experiments will be needed to discover the extrusion-specific signal that drives DC apoptosis, as well as to characterize the downstream consequences of extrusion-mediated DC apoptosis during in vivo infection. Protective immunity against Chlamydia is primarily driven by IFN-γ-producing T cells (32, 36, 50), and DCs fulfill a central role in antigen presentation and activation of these protective T cell responses (25, 26, 30). The rapid apoptotic death of DCs after extrusion uptake could directly hinder priming of adaptive immune responses by eliminating a subset of Chlamydia-exposed DCs.
Specific apoptosis of extrusion-containing DCs suggests that containment within an extrusion fundamentally alters the interaction between the host cell and the bacterium. Interactions of free, non-extrusion-contained Chlamydia bacteria with host DCs are well characterized. DCs are adept at activating protective Th1 responses by presenting antigens from free Chlamydia bacteria to T cells (25, 51); however, the context of this encounter is important for activation. Production of IL-12 by DCs in response to Chlamydia is essential to the protective role of these cells during Chlamydia infection (31, 52). In contrast, IL-10 production is correlated with Chlamydia susceptibility in mice (53), and neutralization of IL-10 production by DCs enhances Th1 T cell responses to Chlamydia (54–56). Additionally, the immune-inhibitory ligand PD-L1 was expressed by DCs in a murine model of Chlamydia infection in which PD-L1 expression decreased protective CD8+ T cell responses and bacterial clearance in the genital tract (57). Containment within an extrusion is likely to modify Chlamydia-DC interactions in profound ways, including protecting the bacteria from host bactericidal mechanisms, altering the detection of Chlamydia by pattern recognition receptors, and modifying the ability of host cells to present antigen and activate adaptive immune responses.
Analysis of DC transcription following extrusion uptake corroborates this, indicating increased transcription of IFN-β, and to a lesser degree IL-10, IL-12p40, and PD-L1, in extrusion-containing DCs compared to that in free-Chlamydia-exposed DCs. Correlation of protein expression levels would have been informative but was impossible due to the rapid kinetics of extrusion-induced DC cell death and the prohibitive number of extrusion-exposed DCs that would need to be sorted to obtain sufficient extrusion-containing DCs for protein analysis. PS-mediated mimicry of apoptotic cells by extrusions may facilitate phagocytosis and modify the DC response (41, 42). Uptake of apoptotic cells by macrophages and DCs alters cytokine production, increasing levels of the immunosuppressive cytokines IL-10 and TGF-β while diminishing the proinflammatory cytokine response and DC maturation (39, 40, 47, 58). Similarly, engulfment of PS-coated liposomes has been shown to diminish proinflammatory responses and maturation in both human and mouse DCs while increasing IL-10 production by mouse DCs (48, 49). Furthermore, expression of immunosuppressive cytokines and subsequent apoptosis by extrusion-containing DCs may potentiate a positive-feedback loop, where extrusion-containing apoptotic DCs are then engulfed by additional macrophages and DCs, which in turn generate immunosuppressive signals in response to apoptotic cell uptake. Interestingly, the intracellular parasite L. amazonensis has been shown to expose PS on its surface, a strategy that hinders macrophage and DC maturation, diminishing downstream adaptive immune responses (37, 38). Extrusions may provide a similar benefit to Chlamydia by altering the context in which Chlamydia antigens are presented to T cells and in turn modifying protective T cell responses against this pathogen.
In this study, we show that extrusions are engulfed by BMDCs. Based on the surface exposure of PS on extrusions, as well as previous data from our laboratory indicating that macrophages phagocytose extrusions (27), it was expected that DCs would perform this task similarly. Our data also reveal that extrusion-contained Chlamydia bacteria benefit from a survival advantage within DCs, increasing the recovery of viable and infectious Chlamydia bacteria up to 5 h after extrusion uptake. This finding complements earlier work in our laboratory that demonstrated extrusion-prolonged survival of Chlamydia within macrophages (27). The viability and infectivity of C. trachomatis, an obligate intracellular bacterium which preferentially infects epithelial cells, is very limited extracellularly or in phagocytes such as macrophages or DCs (25, 26). During infection of the female genital tract, Chlamydia initially infects the cervix. The spread of infection to the upper genital tract is associated with development of disease sequelae. The mechanisms by which nonmotile Chlamydia spreads to other cells and tissues are not well understood, though it has been suggested that this occurs by canalicular spread, possibly after dislodgement of infected cells by PMNs (16, 59). Several studies indicate that inhibition or knockout of matrix metalloproteinase 9 (MMP-9) hindered Chlamydia ascension to the upper genital tract in a mouse model (60, 61). Mice lacking MMP-9 demonstrated substantially decreased acute inflammation in the genital tract, suggesting that cells in the inflammatory infiltrate (including PMNs and DCs) may play a role in ascension. We propose that extrusion may contribute to this spread, by extending the extracellular viability of Chlamydia (27) and allowing it to hitch a ride in host cells, as has been shown for other bacterial species and for Chlamydia pneumoniae (62–64).
Our data are consistent with a model in which Chlamydia utilizes extrusion to (i) enhance the spread of bacteria to distant sites within extrusions and extrusion-containing phagocytes, (ii) modulate the innate immune response by stimulating production of type 1 interferons and potentially other cytokines, and (iii) diminish activation of the adaptive immune response by killing the subset of DCs that engulfs extrusions before antigen presentation can occur. Our study utilized Chlamydia trachomatis LGV in bone marrow DCs for reasons of feasibility; extending the current study to include other Chlamydia serovars and mucosal DCs will be the focus of future efforts. Upcoming studies directly addressing the effect of extrusions on immune responses in vivo will illuminate the role played by this novel exit pathway during infection. More broadly, these studies will begin to reveal the ways in which distinct mechanisms of pathogen cellular exit can effect pathogenesis.
MATERIALS AND METHODS
Reagents.
Antibodies were obtained from the following sources: mouse anti-Chlamydia–fluorescein isothiocyanate (FITC) conjugate (Meridian Diagnostics), mouse anti-Chlamydia trachomatis LPS donated by Bob Suchland (University of Washington), donkey anti-goat Alexa Fluor 488 (ThermoFisher). Phalloidin Alexa Fluor 647 was obtained from ThermoFisher. Annexin V Alexa Fluor 594, LIVE/DEAD fixable violet viability dye, the Image-iT live caspase 3/7 assay, and the Click-iT TUNEL assay were from Molecular Probes. Caspase 3/7 (DEVD-FMK) and caspase 1 (WEHD-FMK) inhibitors were from R&D Systems.
Cell culture and infection.
McCoy cells were obtained from the ATCC and routinely cultured in RPMI 1640 medium with 10% fetal bovine serum (FBS; HyClone) and 2 mM l-glutamine (HyClone) at 37°C in 5% CO2. Chlamydia trachomatis serovar L2 (LGV 434) was propagated in McCoy cells as previously described (65). Infections were performed by washing cells with Hanks' balanced salt solution (HBSS; HyClone), incubating cells with Chlamydia EBs diluted in HBSS for 2 h at room temperature, and then washing them with HBSS and adding fresh medium. Cells were incubated at 37°C for 48 h, at which point stocks of EBs were produced by lysing infected McCoy cells in sucrose phosphate buffer (5 mM glutamine, 0.2 M sucrose, 0.2 M phosphate) by mechanical disruption using glass beads. Lysates were centrifuged to remove cellular debris, and aliquoted supernatants were frozen at −80°C.
BMDCs from C57BL/6 mice were generated as described previously (66). Briefly, mouse bone marrow cells were isolated from the tibia and femur and incubated in a CO2 incubator in RPMI 1640 medium supplemented with 10 ng/ml of granulocyte macrophage colony-stimulating factor (GM-CSF; ThermoFisher), 2 mM l-glutamine (HyClone), 1× penicillin-streptomycin (HyClone), 1× nonessential amino acids (HyClone), 1 mM sodium pyruvate (HyClone), and 10 mM HEPES (HyClone). Fresh medium was added on days 3, 6, and 8. DCs were harvested at day 10. The purity of DC cultures used for experiments was >80%, as determined by flow cytometry analysis of surface CD11c staining.
Extrusion isolation and sonication.
At 48 h postinfection, cells were washed once with HBSS and then incubated in fresh medium for 2 h at 37°C in 5% CO2. Culture supernatants were centrifuged at room temperature in a Sorvall Legend XTR centrifuge at 90 × g for 5 min to remove cell debris and then at 300 × g for 5 min to enrich extrusions. The extrusion pellet was resuspended in BMDC medium. Part of the extrusion population was disrupted with a hand sonicator set at 20 A, for three cycles for 10 s on ice, to generate an equivalent-dose free bacterial control.
DC infection.
BMDCs were plated in 8-well chamber slides (Nunc), 24-well plates, or 6-well plates and incubated overnight at 37°C. Medium was aspirated the following day, and intact or sonicated extrusions were added before incubation at 37°C for 1 h. Cells were washed twice with HBSS to remove extracellular bacteria, fresh medium was added, and cells were returned to 37°C.
Flow-assisted cellular sorting.
DCs were collected into sorting buffer (phosphate-buffered saline [PBS] with 1% FBS, 10 mM HEPES, 10 μg/ml DNase [Sigma]) and stained with 7-AAD (BD Biosciences) at 5 μl per 2 × 105 cells. Stained cells were sorted on a BD FACSAria cell sorter to obtain live, extrusion-containing (GFP+ 7-AAD−) cells. Uninfected cells were used as a gating control. Data were analyzed with FlowJo X Software.
RNA isolation, cDNA synthesis, and qRT-PCR.
DCs were centrifuged at room temperature in an Eppendorf 5418 microcentrifuge at 500 × g for 5 min after collection and then resuspended in buffer RA1 plus TCEP [Tris(2-carboxyethyl)phosphine hydrochloride] for RNA isolation according to the manufacturer's guidelines (Clontech). Isolated RNA was transcribed into cDNA using an iScript cDNA synthesis kit (Bio-Rad). Select targets were quantified in cDNA using SsoAdvanced universal SYBR green supermix (Bio-Rad) on an Applied Biosystems StepOne Plus real-time PCR system. Transcript levels were normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) with unstimulated DCs as the reference sample, using the Pfaffl method (67).
Immunofluorescence, staining, and imaging.
Cells were fixed in 3.7% formaldehyde, permeabilized in 0.5% Triton X-100, and blocked in 1% bovine serum albumin (BSA)–PBS for immunofluorescence staining. For annexin V, viability, caspase 3 or caspase 7 activity, and TUNEL staining, cells were fixed and stained according to the manufacturer's instructions. Cells were imaged on a Nikon Eclipse Ti-E inverted microscope with a 60× objective, and images were processed in Volocity (PerkinElmer). The percentage of cells positive for annexin V and viability dye staining was determined manually based on the intensity and the distribution of fluorescence intensity, using positive and negative controls to confirm accuracy. Interbacterial distance was calculated in Volocity by quantifying the median distance between individual bacteria within an extrusion-containing DC and then determining the mean of this measurement for 25 or more cells at each time point.
Statistical analysis.
Data were analyzed and graphed using GraphPad Prism software. Graphs indicate means ± standard errors of the mean (SEM). A threshold for positive staining was generated for the TUNEL assay based on the mean plus three times the standard deviation of unstimulated samples. Statistical significance was assessed by one-way analysis of variance (ANOVA), followed by Tukey's test, or by Student's t test for data sets with only two groups, and is indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Supplementary Material
ACKNOWLEDGMENTS
We thank Meghan Zuck for helpful discussions regarding this study and Bob Suchland for antibodies.
A.M.S. and K.H. conceived and designed the experiments, A.M.S. performed the experiments, A.M.S. and K.H. analyzed the data, and A.M.S. and K.H. generated the figures and wrote the paper.
This work was supported by grant R01AI095603 from the National Institutes of Health to K.H. and grant T32 AI07140 from the National Institutes of Health to A.S.
The authors have no conflicts of interest associated with this study. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00046-17.
REFERENCES
- 1.Vasilevsky S, Greub G, Nardelli-Haefliger D, Baud D. 2014. Genital Chlamydia trachomatis: understanding the roles of innate and adaptive immunity in vaccine research. Clin Microbiol Rev 27:346–370. doi: 10.1128/CMR.00105-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Newman L, Rowley J, Vander Hoorn S, Wijesooriya NS, Unemo M, Low N, Stevens G, Gottlieb S, Kiarie J, Temmerman M. 2015. Global estimates of the prevalence and incidence of four curable sexually transmitted infections in 2012 based on systematic review and global reporting. PLoS One 10:e0143304. doi: 10.1371/journal.pone.0143304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burton MJ, Mabey DCW. 2009. The global burden of trachoma: a review. PLoS Negl Trop Dis 3:e460. doi: 10.1371/journal.pntd.0000460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunham RC, Rey-Ladino J. 2005. Immunology of Chlamydia infection: implications for a Chlamydia trachomatis vaccine. Nat Rev Immunol 5:149–161. doi: 10.1038/nri1551. [DOI] [PubMed] [Google Scholar]
- 5.Hafner LM, Wilson DP, Timms P. 2014. Development status and future prospects for a vaccine against Chlamydia trachomatis infection. Vaccine 32:1563–1571. doi: 10.1016/j.vaccine.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 6.Geisler WM. 2010. Duration of untreated, uncomplicated Chlamydia trachomatis genital infection and factors associated with chlamydia resolution: a review of human studies. J Infect Dis 201(Suppl 2):S104–S113. doi: 10.1086/652402. [DOI] [PubMed] [Google Scholar]
- 7.Batteiger BE, Xu F, Johnson RE, Rekart ML. 2010. Protective immunity to Chlamydia trachomatis genital infection: evidence from human studies. J Infect Dis 201(Suppl):S178–S189. doi: 10.1086/652400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katz BP, Batteiger BE, Jones RB. 1987. Effect of prior sexually transmitted disease on the isolation of Chlamydia trachomatis. Sex Transm Dis 14:160–164. doi: 10.1097/00007435-198707000-00008. [DOI] [PubMed] [Google Scholar]
- 9.Rank RGG, Whittum-Hudson JAA. 2010. Protective immunity to chlamydial genital infection: evidence from animal studies. J Infect Dis 201(Suppl 2):S168–S177. doi: 10.1086/652399. [DOI] [PubMed] [Google Scholar]
- 10.Abdelrahman YM, Belland RJ. 2005. The chlamydial developmental cycle. FEMS Microbiol Rev 29:949–959. doi: 10.1016/j.femsre.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Dean D, Powers VC. 2001. Persistent Chlamydia trachomatis infections resist apoptotic stimuli. Infect Immun 69:2442–2447. doi: 10.1128/IAI.69.4.2442-2447.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer SF, Vier J, Kirschnek S, Klos A, Hess S, Ying S, Häcker G. 2004. Chlamydia inhibit host cell apoptosis by degradation of proapoptotic BH3-only proteins. J Exp Med 200:905–916. doi: 10.1084/jem.20040402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verbeke P, Welter-Stahl L, Ying S, Hansen J, Häcker G, Darville T, Ojcius DM. 2006. Recruitment of BAD by the Chlamydia trachomatis vacuole correlates with host-cell survival. PLoS Pathog 2:e45. doi: 10.1371/journal.ppat.0020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hybiske K, Stephens RS. 2007. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc Natl Acad Sci U S A 104:11430–11435. doi: 10.1073/pnas.0703218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hybiske K, Stephens RS. 2008. Exit strategies of intracellular pathogens. Nat Rev Microbiol 6:99–110. doi: 10.1038/nrmicro1821. [DOI] [PubMed] [Google Scholar]
- 16.Rank RG, Whittimore J, Bowlin AK, Wyrick PB. 2011. In vivo ultrastructural analysis of the intimate relationship between polymorphonuclear leukocytes and the chlamydial developmental cycle. Infect Immun 79:3291–3301. doi: 10.1128/IAI.00200-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chin E, Kirker K, Zuck M, James G, Hybiske K. 2012. Actin recruitment to the Chlamydia inclusion is spatiotemporally regulated by a mechanism that requires host and bacterial factors. PLoS One 7:e46949. doi: 10.1371/journal.pone.0046949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doughri A, Storz J, Altera KP. 1972. Mode of entry and release of chlamydiae in infections of intestinal epithelial cells. J Infect Dis 126:652–657. doi: 10.1093/infdis/126.6.652. [DOI] [PubMed] [Google Scholar]
- 19.Darville T, Andrews CW, Laffoon KK, Shymasani W, Kishen LR, Rank RG. 1997. Mouse strain-dependent variation in the course and outcome of chlamydial genital tract infection is associated with differences in host response. Infect Immun 65:3065–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barteneva N, Theodor I, Peterson EM, de la Maza LM. 1996. Role of neutrophils in controlling early stages of a Chlamydia trachomatis infection. Infect Immun 64:4830–4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agrawal T, Vats V, Wallace PK, Singh A, Salhan S, Mittal A. 2009. Recruitment of myeloid and plasmacytoid dendritic cells in cervical mucosa during Chlamydia trachomatis infection. Clin Microbiol Infect 15:50–59. doi: 10.1111/j.1469-0691.2008.02113.x. [DOI] [PubMed] [Google Scholar]
- 22.Parr MB, Kepple L, Parr EL. 1991. Langerhans cells phagocytose vaginal epithelial cells undergoing apoptosis during the murine estrous cycle. Biol Reprod 45:252–260. doi: 10.1095/biolreprod45.2.252. [DOI] [PubMed] [Google Scholar]
- 23.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, Granucci F, Kraehenbuhl JP, Ricciardi-Castagnoli P. 2001. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol 2:361–367. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 24.Gervassi A, Alderson MR, Suchland R, Maisonneuve JF, Grabstein KH, Probst P. 2004. Differential regulation of inflammatory cytokine secretion by human dendritic cells upon Chlamydia trachomatis infection. Infect Immun 72:7231–7239. doi: 10.1128/IAI.72.12.7231-7239.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matyszak MK, Young JL, Gaston JS. 2002. Uptake and processing of Chlamydia trachomatis by human dendritic cells. Eur J Immunol 32:742–751. doi:. [DOI] [PubMed] [Google Scholar]
- 26.Steele LN, Balsara ZR, Starnbach MN. 2004. Hematopoietic cells are required to initiate a Chlamydia trachomatis-specific CD8+ T cell response. J Immunol 173:6327–6337. doi: 10.4049/jimmunol.173.10.6327. [DOI] [PubMed] [Google Scholar]
- 27.Zuck M, Ellis T, Venida A, Hybiske K. 21 November 2016. Extrusions are phagocytosed and promote Chlamydia survival within macrophages. Cell Microbiol doi: 10.1111/cmi.12683. [DOI] [PubMed] [Google Scholar]
- 28.Kaiko GE, Horvat JC, Beagley KW, Hansbro PM. 2008. Immunological decision-making: how does the immune system decide to mount a helper T-cell response? Immunology 123:326–338. doi: 10.1111/j.1365-2567.2007.02719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kapsenberg ML. 2003. Dendritic-cell control of pathogen-driven T-cell polarization. Nat Rev Immunol 3:984–993. doi: 10.1038/nri1246. [DOI] [PubMed] [Google Scholar]
- 30.Su H, Messer R, Whitmire W, Fischer E, Portis JC, Caldwell HD. 1998. Vaccination against chlamydial genital tract infection after immunization with dendritic cells pulsed ex vivo with nonviable Chlamydiae. J Exp Med 188:809–818. doi: 10.1084/jem.188.5.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu H, Zhong G, Lu H. 1999. Interleukin-12 production is required for chlamydial antigen-pulsed dendritic cells to induce protection against live Chlamydia trachomatis infection. Infect Immun 67:1763–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gondek DC, Olive AJ, Stary G, Starnbach MN. 2012. CD4+ T cells are necessary and sufficient to confer protection against Chlamydia trachomatis infection in the murine upper genital tract. J Immunol 189:2441–2449. doi: 10.4049/jimmunol.1103032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steinman RM, Hemmi H. 2006. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol 311:17–58. [DOI] [PubMed] [Google Scholar]
- 34.Iwasaki A, Medzhitov R. 2015. Control of adaptive immunity by the innate immune system. Nat Immunol 16:343–353. doi: 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hafner L, Beagley K, Timms P, Hafner L. 2008. Chlamydia trachomatis infection: host immune responses and potential vaccines. Mucosal Immunol 1:116–130. doi: 10.1038/mi.2007.19. [DOI] [PubMed] [Google Scholar]
- 36.Darville T, Hiltke TJ. 2010. Pathogenesis of genital tract disease due to Chlamydia trachomatis. J Infect Dis 201(Suppl):S114–S125. doi: 10.1086/652397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wanderley JLM, Thorpe PE, Barcinski MA, Soong L. 2013. Phosphatidylserine exposure on the surface of Leishmania amazonensis amastigotes modulates in vivo infection and dendritic cell function. Parasite Immunol 35:109–119. doi: 10.1111/pim.12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wanderley JLM, Moreira MEC, Benjamin A, Bonomo AC, Barcinski MA. 2006. Mimicry of apoptotic cells by exposing phosphatidylserine participates in the establishment of amastigotes of Leishmania (L) amazonensis in mammalian hosts. J Immunol 176:1834–1839. doi: 10.4049/jimmunol.176.3.1834. [DOI] [PubMed] [Google Scholar]
- 39.Kushwah R, Oliver JR, Zhang J, Siminovitch KA, Hu J. 2009. Apoptotic dendritic cells induce tolerance in mice through suppression of dendritic cell maturation and induction of antigen-specific regulatory T cells. J Immunol 183:7104–7118. doi: 10.4049/jimmunol.0900824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kushwah R, Hu J. 2010. Dendritic cell apoptosis: regulation of tolerance versus immunity. J Immunol 185:795–802. doi: 10.4049/jimmunol.1000325. [DOI] [PubMed] [Google Scholar]
- 41.Savill J, Fadok V. 2000. Corpse clearance defines the meaning of cell death. Nature 407:784–788. doi: 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- 42.Ravichandran KS, Lorenz U. 2007. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol 7:964–974. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Y, Yeruva L, Marinov A, Prantner D, Wyrick PB, Lupashin V, Nagarajan UM. 2014. The DNA sensor, cyclic GMP-AMP synthase, is essential for induction of IFN-β during Chlamydia trachomatis infection. J Immunol 193:2394–2404. doi: 10.4049/jimmunol.1302718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barker JR, Koestler BJ, Carpenter VK, Burdette DL, Waters CM, Vance RE, Valdivia RH. 2013. STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during Chlamydia trachomatis infection. mBio 4:e00018-13. doi: 10.1128/mBio.00018-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, Greenberg AH, Zhong G. 1998. Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J Exp Med 187:487–496. doi: 10.1084/jem.187.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fischer SF, Schwarz C, Vier J, Häcker G. 2001. Characterization of antiapoptotic activities of Chlamydia pneumoniae in human cells. Infect Immun 69:7121–7129. doi: 10.1128/IAI.69.11.7121-7129.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. 1997. Immunosuppressive effects of apoptotic cells. Nature 390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 48.Chen X, Doffek K, Sugg SL, Shilyansky J. 2004. Phosphatidylserine regulates the maturation of human dendritic cells. J Immunol 173:2985–2994. doi: 10.4049/jimmunol.173.5.2985. [DOI] [PubMed] [Google Scholar]
- 49.Shi D, Fu M, Fan P, Li W, Chen X, Li C, Qi X, Gao T, Liu Y. 2007. Artificial phosphatidylserine liposome mimics apoptotic cells in inhibiting maturation and immunostimulatory function of murine myeloid dendritic cells in response to 1-chloro-2,4-dinitrobenze in vitro. Arch Dermatol Res 299:327–336. doi: 10.1007/s00403-007-0770-9. [DOI] [PubMed] [Google Scholar]
- 50.Gondek DC, Roan NR, Starnbach MN. 2009. T cell responses in the absence of IFN-gamma exacerbate uterine infection with Chlamydia trachomatis. J Immunol 183:1313–1319. doi: 10.4049/jimmunol.0900295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ojcius DM, Bravo de Alba Y, Kanellopoulos JM, Hawkins RA, Kelly KA, Rank RG, Dautry-Varsat A. 1998. Internalization of Chlamydia by dendritic cells and stimulation of Chlamydia-specific T cells. J Immunol 160:1297–1303. [PubMed] [Google Scholar]
- 52.Marks E, Tam MA, Lycke NY. 2010. The female lower genital tract is a privileged compartment with IL-10 producing dendritic cells and poor Th1 immunity following Chlamydia trachomatis infection. PLoS Pathog 6:e1001179. doi: 10.1371/journal.ppat.1001179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang X, HayGlass KT, Brunham RC. 1996. Genetically determined differences in IL-10 and IFN-gamma responses correlate with clearance of Chlamydia trachomatis mouse pneumonitis infection. J Immunol 156:4338–4344. [PubMed] [Google Scholar]
- 54.Yang X, Gartner J, Zhu L, Wang S, Brunham RC. 1999. IL-10 gene knockout mice show enhanced Th1-like protective immunity and absent granuloma formation following Chlamydia trachomatis lung infection. J Immunol 162:1010–1017. [PubMed] [Google Scholar]
- 55.Igietseme JU, Ananaba GA, Bolier J, Bowers S, Moore T, Belay T, Eko FO, Lyn D, Black CM. 2000. Suppression of endogenous IL-10 gene expression in dendritic cells enhances antigen presentation for specific Th1 induction: potential for cellular vaccine development. J Immunol 164:4212–4219. doi: 10.4049/jimmunol.164.8.4212. [DOI] [PubMed] [Google Scholar]
- 56.He Q, Moore TT, Eko FO, Lyn D, Ananaba GA, Martin A, Singh S, James L, Stiles J, Black CM, Igietseme JU. 2005. Molecular basis for the potency of IL-10-deficient dendritic cells as a highly efficient APC system for activating Th1 response. J Immunol 174:4860–4869. doi: 10.4049/jimmunol.174.8.4860. [DOI] [PubMed] [Google Scholar]
- 57.Fankhauser SC, Starnbach MN. 2014. PD-L1 limits the mucosal CD8+ T cell response to Chlamydia trachomatis. J Immunol 192:1079–1090. doi: 10.4049/jimmunol.1301657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Getts DR, Turley DM, Smith CE, Harp CT, McCarthy D, Feeney EM, Getts MT, Martin AJ, Luo X, Terry RL, King NJC, Miller SD. 2011. Tolerance induced by apoptotic antigen-coupled leukocytes is induced by PD-L1+ and IL-10-producing splenic macrophages and maintained by T regulatory cells. J Immunol 187:2405–2417. doi: 10.4049/jimmunol.1004175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rank RG, Whittimore J, Bowlin AK, Dessus-Babus S, Wyrick PB. 2008. Chlamydiae and polymorphonuclear leukocytes: unlikely allies in the spread of chlamydial infection. FEMS Immunol Med Microbiol 54:104–113. doi: 10.1111/j.1574-695X.2008.00459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Imtiaz MT, Schripsema JH, Sigar IM, Kasimos JN, Ramsey KH. 2006. Inhibition of matrix metalloproteinases protects mice from ascending infection and chronic disease manifestations resulting from urogenital Chlamydia muridarum infection. Infect Immun 74:5513–5521. doi: 10.1128/IAI.00730-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Imtiaz MT, Distelhorst JT, Schripsema JH, Sigar IM, Kasimos JN, Lacy SR, Ramsey KH. 2007. A role for matrix metalloproteinase-9 in pathogenesis of urogenital Chlamydia muridarum infection in mice. Microbes Infect 9:1561–1566. doi: 10.1016/j.micinf.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davis JM, Ramakrishnan L. 2009. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 136:37–49. doi: 10.1016/j.cell.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moazed TC, Kuo CC, Grayston JT, Campbell LA. 1998. Evidence of systemic dissemination of Chlamydia pneumoniae via macrophages in the mouse. J Infect Dis 177:1322–1325. doi: 10.1086/515280. [DOI] [PubMed] [Google Scholar]
- 64.Edelson BT, Bradstreet TR, Hildner K, Carrero JA, Frederick KE, Wumesh KC, Belizaire R, Aoshi T, Schreiber RD, Miller MJ, Murphy TL, Unanue ER, Murphy KM. 2011. CD8α+ dendritic cells are an obligate cellular entry point for productive infection by Listeria monocytogenes. Immunity 35:236–248. doi: 10.1016/j.immuni.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schachter J, Wyrick PB. 1994. Culture and isolation of Chlamydia trachomatis, p 377–390. In Clark VL, Bavoil PM (ed), Bacterial pathogenesis. Part B: interaction of pathogenic bacteria with host cells. California Academic Press, Inc., San Diego, CA. [Google Scholar]
- 66.Lutz MB, Kukutsch N, Ogilvie ALJ, Rößner S, Koch F, Romani N, Schuler G. 1998. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods 223:77–92. doi: 10.1016/S0022-1759(98)00204-X. [DOI] [PubMed] [Google Scholar]
- 67.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.