

ABSTRACT
Human cerebral malaria (HCM) is a serious complication of Plasmodium falciparum infection. The most severe outcomes for patients include coma, permanent neurological deficits, and death. Recently, a large-scale magnetic resonance imaging (MRI) study in humans identified brain swelling as the most prominent predictor of fatal HCM. Therefore, in this study, we sought to define the mechanism controlling brain edema through the use of the murine experimental cerebral malaria (ECM) model. Specifically, we investigated the ability of CD8 T cells to initiate brain edema during ECM. We determined that areas of blood-brain barrier (BBB) permeability colocalized with a reduction of the cerebral endothelial cell tight-junction proteins claudin-5 and occludin. Furthermore, through small-animal MRI, we analyzed edema and vascular leakage. Using gadolinium-enhanced T1-weighted MRI, we determined that vascular permeability is not homogeneous but rather confined to specific regions of the brain. Our findings show that BBB permeability was localized within the brainstem, olfactory bulb, and lateral ventricle. Concurrently with the initiation of vascular permeability, T2-weighted MRI revealed edema and brain swelling. Importantly, ablation of the cytolytic effector molecule perforin fully protected against vascular permeability and edema. Furthermore, perforin production specifically by CD8 T cells was required to cause fatal edema during ECM. We propose that CD8 T cells initiate BBB breakdown through perforin-mediated disruption of tight junctions. In turn, leakage from the vasculature into the parenchyma causes brain swelling and edema. This results in a breakdown of homeostatic maintenance that likely contributes to ECM pathology.
KEYWORDS: blood-brain barrier, CD8 T cell, experimental cerebral malaria, perforin
INTRODUCTION
Malaria is a serious global health issue with approximately 200 million new cases occurring annually. Despite improvements in comprehensive prevention measures each year, there were still an estimated 438,000 deaths from malaria in 2015 (1). Approximately 70% of fatal malaria cases occur in children less than 5 years old (1). While uncomplicated Plasmodium falciparum infection is highly treatable, the most severe complication, cerebral malaria, presents a significant problem for patients. Human cerebral malaria (HCM) is characterized by disruption of the blood-brain barrier (BBB), coma, seizures, and death (2, 3). Patients that survive HCM are prone to persisting cognitive and neurological deficits after they have recovered from the infection (4, 5). While antimalarial drugs have shown efficacy in killing parasites, treatments to improve the outcome of HCM are lacking (6). Furthermore, the cellular mechanisms regulating HCM disease progression remain poorly understood. The difficulties associated with studying human patients during infection have limited our understanding of HCM and have created a need for additional preclinical research.
Part of the failure to adequately understand HCM is the limited ability to collect human data pertaining to neurological function. Recently, Seydel et al. carried out an expansive magnetic resonance imaging (MRI) study analyzing HCM patients in Malawi (7). Their study characterized physiological changes occurring in brain structure during cerebral malaria in a large cohort of patients. The findings, obtained using T2-weighted MRI, determined that brain swelling is the strongest prognostic indicator that differentiated fatal cases of cerebral malaria from less severe outcomes. Furthermore, their study suggested relieving cranial pressure by reducing brain swelling as a potential route for therapeutic intervention to treat cerebral malaria.
Experimental cerebral malaria (ECM) using Plasmodium berghei ANKA infection of C57BL/6 mice provides a setting for determining mechanistic details that is not feasible with human studies (8–10). The ECM system models many features of HCM. In both humans and mice, parasitic infection initiates BBB disruption leading to fatal neurological dysfunction (11). The mechanism of disease onset involves a complex network of known effector cells and proteins. Studies have demonstrated that an influx of CD8 T cells, macrophages, and neutrophils into the brain occurs during ECM (12). In addition to immune infiltration, nitric oxide availability appears to play a role during ECM (13, 14). Dysfunction of endothelial nitric oxide synthase (eNOS) and neuronal NOS (nNOS) contributes to decreased nitric oxide bioavailability, which in turn mediates cerebrovascular pathology (15).
Previous studies have determined that T cells traffic to the brain and accumulate during ECM (16). CD4 T cells play an important role in generating the population of effector CD8 T cells in the brain. These T cells are required for disease, and when either CD4 or CD8 T cells are depleted, ECM is completely prevented (17–19). Additionally, CD8 T cells promote pathology through the cytotoxic effector molecules perforin and granzyme B. Ablation of the perforin and granzyme B genes in knockout mice confers resistance to fatal ECM (20–22).
Although CD8 T cells appear to be a critical proximal effector cell type in mediating ECM pathology, the mechanism underlying their activation is poorly understood. It is clear that following activation, parasite-specific CD8 T cells promote BBB breakdown (23–25). Understanding the interactions between the various infiltrating immune cells and the components of the BBB is paramount for identifying therapies for cerebral malaria. The BBB is made up of a heterogeneous population of cells. The primary barrier is formed by an extensive network of tight-junction proteins between adjacent endothelial cells forming a unique boundary that restricts the blood-derived factors within the endothelial lumen from freely crossing into the central nervous system (CNS) parenchyma (26). Pericytes that tightly wrap the endothelial cells, astrocyte end-feet that protrude toward the vessel, and neuronal processes all contribute to the maintenance of BBB integrity (27). The complexity of this barrier makes it difficult to determine which cell types are directly involved in BBB disruption during ECM.
To identify critical factors related to edema and brain swelling, we developed an MRI platform to visualize the brains in live animals during ECM. Using gadolinium-enhanced T1-weighted MRI to measure vascular leakage and T2-weighted MRI to measure brain swelling and edema, we observed the physiological changes occurring during disease progression. With this technology, we visualized the distinct spatial localization of vascular permeability within the brain. Pairing MRI with immunofluorescence microscopy allowed us to define protein alterations that colocalize with BBB permeability. Specifically, we investigated the cerebral endothelial tight-junction proteins. These observations build upon current understanding and help to identify important physiological changes occurring within the brain during ECM.
RESULTS
Perforin-deficient mice are protected from ECM despite brain infiltration of CD8 T cells equivalent to that in susceptible mice.
This study expands upon the previously identified role CD8 T cells play in mediating pathology during ECM. We infected mice with red blood cells parasitized with Plasmodium berghei ANKA to cause ECM. C57BL/6 mice became symptomatic abruptly at 6 days postinfection (dpi). Mice began experiencing a loss of balance, respiratory distress, and ataxia and then ultimately and rapidly proceeded to a moribund state. Deletion of the effector molecule perforin resulted in complete protection from ECM (Fig. 1A). One possible explanation for mice failing to succumb to pathology could be a defect in the ability of CD8 T cells to respond to infection. However, although we saw that the mice were protected from ECM, CD8 T cells still accumulated within the brain at the same frequency and total number as they did in susceptible C57BL/6 mice (Fig. 1B to D). It is also necessary to rule out the possibility that the perforin knockout (pfp−/−) mice respond uniquely to infection. We examined the peripheral infection in pfp−/− and wild-type (WT) C57BL/6 mice and saw no differences in the circulating parasite loads between the groups over the course of infection (Fig. 1E). These data demonstrate that although mice are infected with P. berghei ANKA parasites and CD8 T cells traffic normally to the brain, pathology during ECM requires perforin.
FIG 1.

CD8 T cell trafficking to the brain and parasitemia are independent of perforin during experimental cerebral malaria. (A) Survival curve showing fatal disease progression in C57BL/6 mice 6 days after infection with 106 parasitized erythrocytes containing Plasmodium berghei ANKA compared with perforin-deficient mice (pfp−/−) that are fully protected from ECM (P < 0.0001). Survival curve statistical analysis was done by log rank test, with 10 mice, with results pooled from two independent experiments. ****, P < 0.0001. (B) Representative plot illustrating gating strategy for quantifying brain-infiltrating immune cells. (C and D) CD8 T cells infiltrating the brain at 6 dpi are at the same frequencies (C) and total numbers (D) in ECM-protected pfp−/− and WT C57BL/6 mice. Ten mice per group were used, and results were pooled from two independent experiments. (E) Cumulative percentages of parasitemia postinfection show no difference between pfp−/− and WT C57BL/6 mice. Giemsa-stained peripheral blood smears were analyzed at ×100 magnification from P. berghei ANKA-infected mice. Error bars represent standard errors of the mean (SEM), and P values were determined by an unpaired two-tailed t test.
CNS vascular permeability colocalizes with disruption of BBB tight junctions.
A defining characteristic of ECM is CNS vascular permeability due to disruption of the BBB. To analyze the extent of vascular permeabilization as disease progresses, we intravenously injected fluorescein isothiocyanate (FITC)-albumin 6 dpi and measured the fluorescence intensity within the brain. By immunofluorescence microscopy, we saw that in phosphate-buffered saline (PBS)-injected control mice, intravenously (i.v.) injected FITC-albumin is restricted within the brain vasculature. We observed the tight-junction proteins claudin-5 and occludin connecting cerebral endothelial cells to form a continuous and linearly organized network of microvessels (Fig. 2A). Six days after P. berghei ANKA infection, the continuity of claudin-5 and occludin was lost and FITC-albumin leaked from the microvessel lumen into the perivascular space. The diffuse FITC-albumin leakage specifically colocalized with regions of claudin-5 and occludin disruption (Fig. 2B). The tight-junction proteins that form the proximal seal of the BBB were no longer linear along the wall of the vessels but were broken and disorganized. Importantly, to address the possibility that we missed the plane of intact vasculature during the imaging of these regions of tight-junction disruption, we assessed the volume above and below the area of imaging through creation of three-dimensional (3D) imaging stacks. This confirmed that the tight junctions are disrupted in WT mice during ECM. Interestingly, pfp−/− mice retain tight-junction integrity after P. berghei ANKA infection (Fig. 2C). The intact tight junctions preserve a functional BBB that prevents FITC-albumin from leaking into the brain parenchyma.
FIG 2.

Vascular leakage associates with perforin-dependent tight-junction disruptions during ECM. (A to C) Representative immunofluorescent confocal microscopy images of occludin, claudin-5, and FITC-albumin in mouse brain sections. (A) PBS-injected WT control mice show FITC-albumin restricted within the vasculature. (B) P. berghei ANKA (PbA)-infected C57BL/6 animals show increased FITC-albumin leakage from vessels into the parenchyma and claudin-5 and occludin disruption. (C) P. berghei ANKA-infected pfp−/− mice maintain claudin-5 and occludin tight-junction integrity, and FITC-albumin is again constrained within the microvasculature. Fresh frozen sections were taken from mice 6 dpi. (D) Leakage of intravenously injected FITC-albumin into the CNS was significantly elevated 6 dpi in WT C57BL/6 mice infected with P. berghei ANKA compared to that in PBS-injected controls (P = 0.003) and infected pfp−/− mice (P = 0.002). (E and F) Quantification of claudin-5 (E) and occludin (F) show reductions in the area of tight-junction proteins within the brains of C57BL/6 mice but not pfp−/− mice at 6 dpi compared to those in uninfected controls. (G) Densitometry quantification of Western blots of brain protein lysate shows no detectable elevation of active cleaved caspase-3 in the cortex or olfactory bulb 6 days after P. berghei ANKA infection or PBS injection (P = 0.20). GL261 brain tumor samples show increased cleavage of caspase-3, indicating an active apoptotic pathway. Four mice per experiment were used, and results are representative of three independent experiments. Error bars represent SEM, and P values were determined by analysis of variance (ANOVA) with Bonferroni's post hoc analysis. **, P < 0.01; ***, P < 0.001; ns, nonsignificant.
Total brain fluorescence increases as FITC-albumin is deposited within the brain after BBB disruption. Fluorescence intensity was significantly elevated in the brains of P. berghei ANKA-infected C57BL/6 mice compared to that of control animals. However, in pfp−/− mice, where the BBB remains intact after infection, fluorescence was not elevated relative to that in uninfected mice (Fig. 2D). Quantification of the cerebral tight-junction proteins demonstrated that both claudin-5 and occludin were significantly reduced at 6 dpi in C57BL/6 mice relative to the levels in uninfected controls whereas infected perforin-deficient mice maintained tight-junction levels similar to those of uninfected animals (Fig. 2E and F). Thus, elevated BBB leakage correlates with a perforin-dependent reduction in cerebral tight-junction protein levels during ECM.
Because perforin is a known effector molecule in the lytic granule exocytosis pathway used by CD8 T cells to kill targets, we hypothesized that T cells may directly kill target cells at the BBB. To determine if endothelial cell death was responsible for tight-junction disruption, we immunoblotted brain lysates for detection of cleaved caspase-3, the active form of the protein. However, we did not observe an elevation in the activated cleaved caspase-3 in P. berghei ANKA-infected mice compared to that in uninfected controls in the cortex or the olfactory bulb, a known brain region of vascular permeability during ECM (Fig. 2G). Brain tissue from glioma-bearing mice was used as a positive control for cell death. This implies that perforin-mediated tight-junction disruption may occur by a mechanism independent of the initiation of apoptosis.
BBB disruption occurs in distinct brain regions during ECM.
We have established that BBB disruption and tight-junction protein alterations occur in the brains of P. berghei ANKA-infected mice. To further examine this phenotype, we used gadolinium-enhanced T1-weighted MRI to analyze the localization of vascular leakage during ECM. Gadolinium is used as a contrast agent and can be visualized as a hyperintense signal when it leaks from vasculature in areas of BBB disruption. Six days after P. berghei ANKA infection, we observed gadolinium leakage in C57BL/6 mice (Fig. 3A) but not in pfp−/− mice (Fig. 3C). We then compiled serial sections to create a 3D rendering of the total volume of gadolinium leakage throughout the whole brain (Fig. 3B and D). The 3D volume (illustrated in red) depicts the extensive vascular leakage within brains of P. berghei ANKA-infected WT mice (Fig. 3B). There was an increase in the area of observable gadolinium leakage in C57BL/6 mice (Fig. 3A and B) compared with the scans of pfp−/− animals, which were devoid of gadolinium leakage (Fig. 3C and D).
FIG 3.

Vascular permeability during ECM is localized to specific vital brain regions. (A to F) Representative T1-weighted gadolinium-enhanced MRI axial sections comparing levels of vascular permeability in the CNS of C57BL/6 (A and E) and pfp−/− mice (C and F). 3D volumetric renderings show gadolinium leakage (red) in C57BL/6 mice (B) that is elevated compared to that in pfp−/− mouse brain scans (D). Midsagittal sections show gadolinium enhancement in the olfactory bulb (arrow a), ventricles (arrow b), and brainstem (arrow c) of C57BL/6 mice (E) but not in pfp−/− mice (F). MRIs were analyzed with Analyze 10.0 (Biomedical Imaging Resource, Mayo Clinic) to render 3D masks of gadolinium leakage volume. All images are representative of three independent experiments of 5 mice per group.
We then analyzed the specific location of gadolinium enhancement. Midsagittal sections illustrate precisely which functional brain regions have BBB disruption in these mice. We determined that vascular leakage is not homogeneous across the entire brain but rather is restricted to specific brain regions (Fig. 3E). Our results indicate BBB disruption and vascular leakage into the olfactory bulb (Fig. 3E, arrow a) and the lateral ventricle (Fig. 3E, arrow b). Importantly, we also observed extensive gadolinium leakage in the brainstems of P. berghei ANKA-infected C57BL/6 mice (Fig. 3E, arrow c). No areas of gadolinium leakage appeared on T1-weighted MRI scans of pfp−/− mice (Fig. 3F). This demonstrates that loss of perforin protects the mice from vascular leakage in these vital brain regions. These MRI results indicate that blocking CD8 T cell effector function protects mice from BBB disruption, thus preventing ECM.
Perforin is required for the development of brain edema during ECM.
Following the identification of the specific localization of BBB permeability during ECM, we next sought to analyze the effect vascular leakage has on brain homeostasis. We hypothesized that the excess fluid entering the brain would result in increased intracranial pressure. To test this, we used T2-weighted MRI to visualize abnormalities within the brain tissue of mice during ECM, as hyperintensity of brain tissue over background levels on these scans is indicative of edema. C57BL/6 mice presented with increased free fluid in the brain 6 dpi compared to that in healthy control animals (Fig. 4A and B). Perforin-deficient mouse brains were devoid of edema, and these mice had brain scans similar to those of healthy uninfected control animals (Fig. 4C). 3D volumetric analysis of T2-weighed MRIs further confirmed that P. berghei ANKA-infected C57BL/6 mice exhibit a significant increase in volume of hyperintense regions compared to both uninfected C57BL/6 controls (P = 0.0015) and P. berghei ANKA-infected perforin knockout mice (P = 0.0016) (Fig. 4D). Sagittal sections of these brains revealed that this abnormal hyperintensity is most extreme surrounding the brainstems of P. berghei ANKA-infected animals (Fig. 4E, asterisks) and is located at the meningeal layer sitting beneath the skull and enveloping the brain tissue. This layer of hyperintensity was not seen in infected pfp−/− animals (Fig. 4F). These data confirm that brain edema during ECM occurs in conjunction with BBB disruption and is regulated by a perforin-dependent mechanism.
FIG 4.

Elevated brain edema during ECM correlates with regions of BBB disruption. (A to C) Axial sections from representative T2-weighted MRIs of murine brains during ECM. T2 hyperintensity is seen in ventricles of all samples but also in the cortex of P. berghei ANKA (PbA)-infected C57BL/6 mice. (D) Quantification of hyperintensity volumes illustrates brain edema during ECM in C57BL/6 mice. Hyperintensity in P. berghei ANKA-infected C57BL/6 mice is significantly increased compared to that in both uninfected C57BL/6 (P = 0.0015) and infected pfp−/− (P = 0.0016) animals. *, P < 0.05. (E and F) Sagittal sections of brains 6 dpi demonstrate regions of meningeal hyperintensity (asterisks) in C57BL/6 animals (E) that do not appear in pfp−/− mice (F). Images were analyzed using Analyze 10.0 (Biomedical Imaging Resource, Mayo Clinic). Five mice per group were used, and data are representative of two individual experiments. Error bars represent SEM, and P values were determined using ANOVA with Bonferroni's post hoc analysis.
Perforin production from CD8 T cells alone causes edema during ECM.
To determine the cellular source of perforin production contributing to the development of brain edema, we performed an adoptive transfer of purified WT CD8 T cells from infected donors into perforin-deficient mice. Animals receiving wild-type CD8 T cells developed ECM. In contrast, no mice receiving pfp−/− CD8 T cells developed symptoms or succumbed to disease (Fig. 5A). Next, we performed T2-weighted MRI scans to assess edema in the brains of the recipient animals. Mice that received perforin-deficient CD8 T cells displayed no edema within the brain parenchyma. However, when animals were given wild-type CD8 T cells, edema was visible in both the cortex and meningeal layer (Fig. 5B). Volumetric analysis of the brains of these animals revealed significantly elevated hyperintensity when perforin-expressing CD8 T cells were transferred in comparison to the intensity in the controls (Fig. 5C). These data illustrate that CD8 T cell-produced perforin is necessary to initiate fatal brain edema during ECM.
FIG 5.
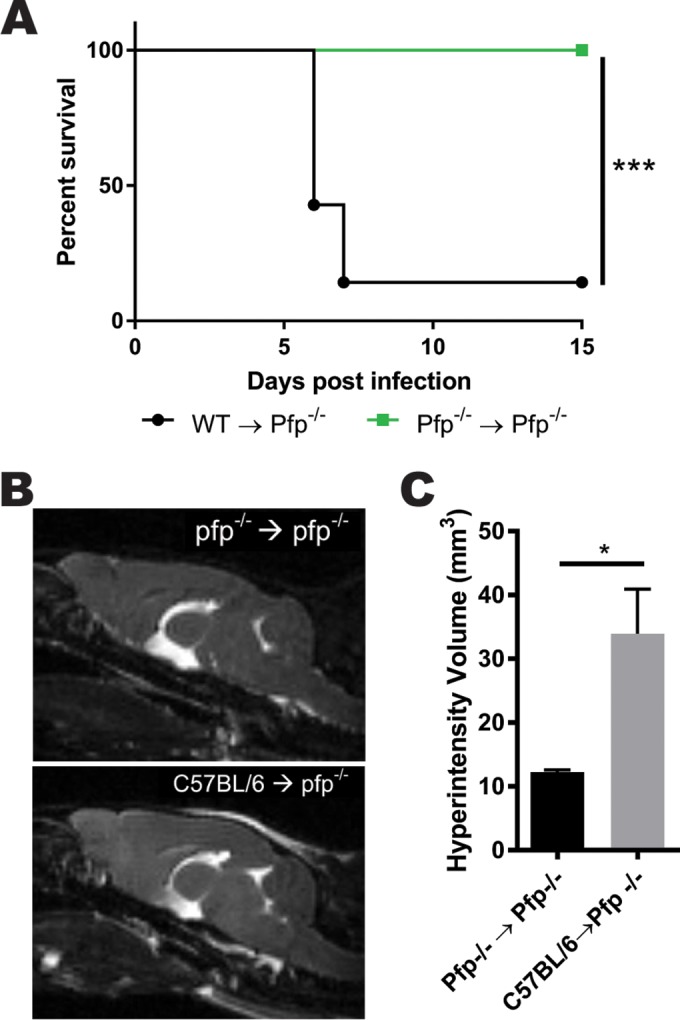
Perforin secretion is required for CD8 T cells to cause brain edema during ECM. (A) Survival curve demonstrating that 6 days after adoptive transfer of purified CD8 T cells from WT C57BL/6 mice into pfp−/− recipients, animals develop ECM. In contrast, the control group of pfp−/− animals receiving pfp−/− CD8 T cells remains protected from disease (P = 0.0008). Survival was analyzed using a log rank test with 8 animals, and results were pooled from two independent experiments. ***, P < 0.001. (B) Representative T2-weighted MRIs of sagittal sections from mice 6 days after adoptive transfer and P. berghei ANKA infection. (C) Volumetric quantification of hyperintensity from T2-weighted MRI illustrates brain edema during ECM in pfp−/− mice with adoptively transferred C57BL/6 CD8 T cells but not in pfp−/− donors (P = 0.0213). Error bars represent SEM, and the P value was obtained using an unpaired t test. *, P < 0.05.
DISCUSSION
CD8 T cells have been previously identified as one of the critical effector cell types mediating ECM. However, the mechanism by which these cells initiate fatal disease pathology is not well understood. Here, we identify a noncytotoxic role for the effector molecule perforin in promoting disease. We show that pfp deficiency prevents the disruption of cerebral endothelial cell tight-junction proteins during P. berghei ANKA infection. The integrity of the tight-junction proteins studied here, claudin-5 and occludin, is critical for the maintenance of a selectively permeable barrier between the endothelial lumen and the CNS (28, 29). This study demonstrates that disruption in the organization of tight junctions has significant global consequences that can lead to organ-wide pathology. Recent work has identified a reduction in claudin-5 during ECM (30). Here, we confirm a significant reduction in claudin-5 and also show that this phenomenon is shared by the tight-junction protein occludin. The reductions in claudin-5 and occludin both depend upon functional CD8 T cells. Furthermore, this work shows that CD8 T cell-mediated disruption of tight junctions results in significant brain edema that is ultimately fatal. This link between fatal brain edema and CD8 T cells provides a novel avenue to pursue as a potential therapeutic target for treatment. Still, it is important to consider the role of CD8 T cells in modulating endothelial tight-junction integrity in the context of their interactions with other key brain-infiltrating immune cells. Recent work has shown that activated endothelial cells are capable of cross-presenting parasitic antigens (25, 31). This would suggest a direct interaction between the CD8 T cells and the cerebral endothelial cells as the most straightforward model.
In our experiments, we observed no signs of increased cell death of brain endothelial cells during ECM. This was interesting, as a primary role of activated CD8 T cells is to employ cytolytic effector functions. However, this observation is in agreement with other recently reported results where apoptosis could not be detected (32, 33). While some studies have reported evidence of apoptosis, this has not been seen at levels great enough to cause the extensive vascular leakage responsible for cerebral malaria (21). Additionally, while in vitro analysis shows the capacity for CD8 T cells to kill, the extent to which these cells kill in vivo is still being identified (31, 34). Rigorous quantification of both endothelial cell density and number of apoptotic cells in the brain suggests it is a rare event (12, 32). Here, we show that vascular leakage and edema are not rare but are diffuse events affecting large regions of the brain (Fig. 3 and 4). Additionally, we see no difference in the activation of the apoptotic pathway, as measured by caspase 3 cleavage (Fig. 2E). Taken together, these data suggest an alternative mechanism of ECM pathology than endothelial cell apoptosis alone.
Although this pathway has yet to be fully delineated, this study demonstrates that perforin plays a critical role in disruption of tight junctions. Previous studies have indicated that the CD8 T cells recruited during ECM are specific to parasitic antigens (25, 35, 36). Therefore, this pathway appears to be initiated when CD8 T cells recognize major histocompatibility complex (MHC) class I molecules loaded with parasitic antigens and mediate BBB disruption downstream through perforin. However, these CD8 T cells do not appear to directly kill the endothelial cells. Alternatively, the T cells initiate a process that ultimately disrupts endothelial tight junctions, weakening the BBB. This work contributes to a growing body of evidence for a noncytotoxic mechanism of CD8 T cell-mediated pathology during ECM.
Noncytolytic roles for perforin and granzyme B have recently been demonstrated in systems other than P. berghei ANKA infection. A report recently identified an extracellular role for granzyme B in facilitating lymphocyte migration through interactions with basement membranes (37). This is an example of a protease functioning as expected, but in an alternative location. Here, granzyme B cleaves extracellular matrix proteins outside a target cell. This supports the hypothesis that the canonical cytolytic effector molecules, granzymes and perforin, may act in additional roles to modulate the immune response.
CD8 T cells have also been shown to restrict latent herpes simplex virus (HSV) reactivation from within infected neurons without destroying the cells (38, 39). Furthermore, it has previously been demonstrated that perforin-dependent neuropathology occurs in mice infected with Theiler's murine encephalomyelitis virus (TMEV) (40). This mechanism also occurs independently of detectable CD8 T cell killing (41, 42). Future work focused on defining how CD8 T cells regulate infection through alternative pathways will enhance our understanding of CD8 T cell biology. This knowledge could potentially translate to therapeutic intervention during cerebral malaria that would not block productive CD8 T cell functions.
Our use of gadolinium-enhanced T1-weighted MRI allowed us to track BBB disruption in live animals. We observed vascular leakage occurring during ECM that is localized to specific regions of the brain. The brainstems of P. berghei ANKA-infected mice exhibit extensive gadolinium leakage. Pathology in the brainstem during ECM correlates with the first symptoms of neurological deficit, including a lack of balance and strained respiration (43). Proper respiratory control is regulated through nerve centers innervating the medulla and pons of the brainstem (44). The cerebellum also relies upon signals originating within the brainstem, specifically the pontine nuclei, to effectively regulate motion and balance (44, 45). Perturbation of homeostasis in these vital regions disrupts the signals required for adequate brain function. This indicates that disruption of the BBB within the brainstem could induce the observed symptoms. These results complement recent work by Swanson et al. showing that ECM induces neuronal death (30). These results indicate that the extensive vascular leakage within the brainstem results in the death of critical neurons. These MRI results also raise the question of what makes vasculature in these discrete brain regions particularly susceptible to BBB disruption. It is clear that endothelial cells are highly heterogeneous in their functionality and surface protein expression (46). Identifying features unique to these distinct areas could present intriguing molecular targets for strengthening the BBB.
Our MRI analysis also revealed extensive gadolinium enhancement in the olfactory bulb. This region is populated with an intricate network of microvessels. Recently, Zhao et al. showed that microhemorrhages occur early during ECM in this region (47). Our data corroborate this finding and also show that functionally, these hemorrhages are associated with a leaky BBB. Further evidence depicts vascular leakage and edema initiating in the olfactory bulb and traveling down the rostral-migratory stream to the brainstem (48). We also see BBB disruption in these regions of the brain. This work provides mechanistic insight into the cause of pathology in ECM. We show that perforin-mediated tight-junction disruption leads to BBB disruption and edema during ECM. MRI analysis allows us to address questions that were previously inaccessible to researchers. Pairing this technique with a small-animal model in which we can attempt to manipulate the disease course pushes forward the possibility of better understanding the disease mechanism to discover targets for treatment. Future studies employing these methodologies have the potential to advance the understanding of human cerebral malaria.
Finally, this study provides insight into the deleterious effects that excessive vascular permeability has upon the brain. These data show that BBB breakdown (Fig. 2 and 3) results in edema (Fig. 4). More specifically, we show that perforin produced by CD8 T cells is required to initiate the fatal edema that occurs during ECM. The recent human MRI study from Seydel et al. implicated brain swelling from edema as the cause of fatal outcomes in HCM (7). Our findings indicate that the ECM model of P. berghei ANKA infection follows a similar pathogenesis. This study proposes a potential mechanism common to the human disease. This is an important discovery for a field that has been debating the similarities and differences between ECM and HCM pathogenesis (49). Importantly, the etiology of human brain swelling and edema is currently unknown. The data put forward in this study demonstrate that perforin has the capacity to induce edema, the leading indicator associated with fatal HCM. Further studies investigating the involvement of CD8 T cells in HCM are required to fully understand the relationship to the mouse model. These data show the benefits in using the ECM model for future preclinical work to test potential therapeutic interventions for preventing brain edema.
MATERIALS AND METHODS
Animals.
C57BL/6 and C57BL/6 perforin knockout (pfp−/−) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice were housed at the Mayo Clinic (Rochester, MN).
Plasmodium berghei ANKA infection.
Mice were infected intravenously between 8 to 12 weeks of age with 106 parasitized red blood cells diluted in PBS. At 6 dpi, the mice were euthanized and their brains were harvested for experimentation.
Flow cytometry.
Flow cytometry for brain-infiltrating lymphocytes was performed as previously described (50). Briefly, brains were homogenized and digested in type IV collagenase. Following incubation, samples were spun through a Percoll gradient and the lymphocyte layer was extracted and stained.
FITC-albumin permeability assay.
Mice were injected intravenously with 10 mg FITC-albumin (Sigma-Aldrich, St. Louis, MO). One hour later, brains were harvested, flash frozen, and stored at −80°C. Fresh frozen brains were homogenized, and fluorescence readings were taken at 488-nm excitation and 525-nm emission wavelengths on a Synergy H1 plate reader (BioTek, Winooski, VT).
Confocal microscopy.
Samples were prepared as previously described (50). Mice were injected with 10 mg FITC-albumin 1 h before the brains were harvested. Samples were fresh frozen and embedded in O.C.T. compound (Tissue-Tek). Twenty-micrometer coronal sections of isocortex anterior to the hippocampal formation were taken. Samples were blocked with 5% normal goat serum for 1 h. Primary antibodies, mouse anti-claudin-5 (35-2500; Invitrogen) and rabbit anti-occludin (71-1500; Invitrogen), were applied and incubated overnight at room temperature (RT). Sections were stained with secondary antibodies, Alexa Fluor 532 goat anti-mouse IgG (H+L) (A-11002; Invitrogen) and Alexa Fluor 647 goat anti-rabbit IgG (H+L) (A-21244; Invitrogen), for 1 h at RT. During analysis, 15-μm z-stacks of brain sections were taken to ensure that vessels were within the visible plane. The images were acquired on a Leica DM 2500 confocal microscope and analyzed using LAS AF 6000 acquisition software (Leica Microsystems). Image-Pro Premier software (Media Cybernetics) was used to quantify the areas of positive fluorescence intensity for claudin-5 and occludin from tissue sections. Three randomly chosen fields were averaged for each sample.
Western blotting.
Brain tissue was homogenized in radioimmunoprecipitation assay (RIPA) buffer with complete ULTRA protease inhibitor supplement (Roche). Fifty micrograms of protein was added to reducing Laemmli buffer and separated using a 4-to-20% gradient gel by SDS-PAGE. Samples were transferred to a polyvinylidene difluoride (PVDF) membrane and blocked with 5% dry milk in PBS-T (PBS with Tween 20) for 1 h at RT. Primary antibodies against cleaved caspase 3 (9661; Cell Signaling Technology) or GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (5174; Cell Signaling Technology) were diluted 1:1,000 in 5% dried milk–PBS-T overnight at 4°C. Goat anti-rabbit secondary horseradish peroxidase (HRP)-conjugated antibodies were applied for 1 h at RT at a dilution of 1:5,000 in a 5% dried milk–PBS-T solution (sc-2004; Santa Cruz Biotechnology). Blots were developed with Western Lightning Plus-ECL chemiluminescence reagent (PerkinElmer).
Magnetic resonance imaging.
Magnetic resonance images were acquired using a Bruker Avance II 7 T vertical-bore small-animal MRI system (Bruker Biospin) as previously described (51). Mice were anesthetized under isoflurane for the duration of scanning. For T1-weighted scans, mice were given an intraperitoneal (i.p.) injection of gadolinium (100 mg/kg). After 15 min, a T1-weighted multislice multiecho (MSME) sequence with a repetition time (TR) of 300 ms, an echo time (TE) of 9.5 ms, a field of view (FOV) of 4.0 by 2.0 by 2.0 cm, and a matrix of 192 by 96 by 96 was used. For T2-weighted scans, a rapid acquisition with relaxation enhancement (RARE) pulse sequence was used with a TR of 1,500 ms, a TE of 70 ms, a RARE factor of 16, an FOV of 3.2 by 1.92 by 1.92 cm, and a matrix of 256 by 128 by 128. A T2-weighted MRI MSME sequence with a TR of 300 ms, a TE of 9.5 ms, an FOV of 4.0 by 2.0 by 2.0 cm, and a matrix of 192 by 96 by 96 was used. MRI scans were analyzed using Analyze 10.0 (Biomedical Imaging Resource, Mayo Clinic) to generate 3D volumes.
Adoptive transfer.
Donor mice were infected with 106 parasitized erythrocytes. At 6 dpi, splenocytes were isolated and CD8 T cells were purified using a CD8a+ T cell isolation kit (Miltenyi Biotec catalog no. 130-104-075). A total of 5 × 106 purified CD8 T cells were transferred intravenously into recipient pfp−/− animals. Recipient mice were infected with 106 parasitized red blood cells on the same day as the CD8 T cell transfer.
Ethics statement.
The Institutional Animal Care and Use Committee at the Mayo Clinic approved all experiments (protocol A57913). All experiments were conducted in accordance with the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals.
ACKNOWLEDGMENTS
M.H. designed experiments, performed the experiments, analyzed data, and wrote the manuscript. A.J.J. designed experiments and wrote the manuscript. H.L.J., F.J., A.N., L.M.H., and S.J.L. performed experiments and provided manuscript feedback. J.T.H. provided the plasmodium stock, experimental design, and manuscript feedback. N.S.B. provided technical assistance and manuscript feedback.
There are no competing interests to declare.
REFERENCES
- 1.WHO. 2015. World malaria report: 2015. WHO Global Malaria Program, Geneva, Switzerland. [Google Scholar]
- 2.WHO. 2000. Severe falciparum malaria. Trans R Soc Trop Med Hyg 94(Suppl 1):S1–S90. [PubMed] [Google Scholar]
- 3.Idro R, Jenkins NE, Newton CR. 2005. Pathogenesis, clinical features, and neurological outcome of cerebral malaria. Lancet Neurol 4:827–840. doi: 10.1016/S1474-4422(05)70247-7. [DOI] [PubMed] [Google Scholar]
- 4.Fernando SD, Rodrigo C, Rajapakse S. 2010. The ‘hidden’ burden of malaria: cognitive impairment following infection. Malar J 9:366. doi: 10.1186/1475-2875-9-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kihara M, Carter JA, Newton CR. 2006. The effect of Plasmodium falciparum on cognition: a systematic review. Trop Med Int Health 11:386–397. doi: 10.1111/j.1365-3156.2006.01579.x. [DOI] [PubMed] [Google Scholar]
- 6.Mishra SK, Newton CR. 2009. Diagnosis and management of the neurological complications of falciparum malaria. Nat Rev Neurol 5:189–198. doi: 10.1038/nrneurol.2009.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seydel KB, Kampondeni SD, Valim C, Potchen MJ, Milner DA, Muwalo FW, Birbeck GL, Bradley WG, Fox LL, Glover SJ, Hammond CA, Heyderman RS, Chilingulo CA, Molyneux ME, Taylor TE. 2015. Brain swelling and death in children with cerebral malaria. N Engl J Med 372:1126–1137. doi: 10.1056/NEJMoa1400116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rest JR. 1982. Cerebral malaria in inbred mice. I. A new model and its pathology. Trans R Soc Trop Med Hyg 76:410–415. doi: 10.1016/0035-9203(82)90203-6. [DOI] [PubMed] [Google Scholar]
- 9.Amani V, Boubou MI, Pied S, Marussig M, Walliker D, Mazier D, Renia L. 1998. Cloned lines of Plasmodium berghei ANKA differ in their abilities to induce experimental cerebral malaria. Infect Immun 66:4093–4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neill AL, Hunt NH. 1992. Pathology of fatal and resolving Plasmodium berghei cerebral malaria in mice. Parasitology 105:165–175. doi: 10.1017/S0031182000074072. [DOI] [PubMed] [Google Scholar]
- 11.Lou J, Lucas R, Grau GE. 2001. Pathogenesis of cerebral malaria: recent experimental data and possible applications for humans. Clin Microbiol Rev 14:810–820. doi: 10.1128/CMR.14.4.810-820.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nacer A, Movila A, Sohet F, Girgis NM, Gundra UM, Loke P, Daneman R, Frevert U. 2014. Experimental cerebral malaria pathogenesis–hemodynamics at the blood brain barrier. PLoS Pathog 10:e1004528. doi: 10.1371/journal.ppat.1004528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cabrales P, Zanini GM, Meays D, Frangos JA, Carvalho LJ. 2011. Nitric oxide protection against murine cerebral malaria is associated with improved cerebral microcirculatory physiology. J Infect Dis 203:1454–1463. doi: 10.1093/infdis/jir058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zanini GM, Martins YC, Cabrales P, Frangos JA, Carvalho LJ. 2012. S-nitrosoglutathione prevents experimental cerebral malaria. J Neuroimmune Pharmacol 7:477–487. doi: 10.1007/s11481-012-9343-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ong PK, Melchior B, Martins YC, Hofer A, Orjuela-Sanchez P, Cabrales P, Zanini GM, Frangos JA, Carvalho LJ. 2013. Nitric oxide synthase dysfunction contributes to impaired cerebroarteriolar reactivity in experimental cerebral malaria. PLoS Pathog 9:e1003444. doi: 10.1371/journal.ppat.1003444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yañez DM, Manning DD, Cooley AJ, Weidanz WP, van der Heyde HC. 1996. Participation of lymphocyte subpopulations in the pathogenesis of experimental murine cerebral malaria. J Immunol 157:1620–1624. [PubMed] [Google Scholar]
- 17.Hermsen C, van de Wiel T, Mommers E, Sauerwein R, Eling W. 1997. Depletion of CD4+ or CD8+ T-cells prevents Plasmodium berghei induced cerebral malaria in end-stage disease. Parasitology 114:7–12. doi: 10.1017/S0031182096008293. [DOI] [PubMed] [Google Scholar]
- 18.Boubou MI, Collette A, Voegtle D, Mazier D, Cazenave PA, Pied S. 1999. T cell response in malaria pathogenesis: selective increase in T cells carrying the TCR V(beta)8 during experimental cerebral malaria. Int Immunol 11:1553–1562. doi: 10.1093/intimm/11.9.1553. [DOI] [PubMed] [Google Scholar]
- 19.Finley RW, Mackey LJ, Lambert PH. 1982. Virulent P. berghei malaria: prolonged survival and decreased cerebral pathology in cell-dependent nude mice. J Immunol 129:2213–2218. [PubMed] [Google Scholar]
- 20.Nitcheu J, Bonduelle O, Combadiere C, Tefit M, Seilhean D, Mazier D, Combadiere B. 2003. Perforin-dependent brain-infiltrating cytotoxic CD8+ T lymphocytes mediate experimental cerebral malaria pathogenesis. J Immunol 170:2221–2228. doi: 10.4049/jimmunol.170.4.2221. [DOI] [PubMed] [Google Scholar]
- 21.Potter S, Chan-Ling T, Ball HJ, Mansour H, Mitchell A, Maluish L, Hunt NH. 2006. Perforin mediated apoptosis of cerebral microvascular endothelial cells during experimental cerebral malaria. Int J Parasitol 36:485–496. doi: 10.1016/j.ijpara.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 22.Haque A, Best SE, Unosson K, Amante FH, de Labastida F, Anstey NM, Karupiah G, Smyth MJ, Heath WR, Engwerda CR. 2011. Granzyme B expression by CD8+ T cells is required for the development of experimental cerebral malaria. J Immunol 186:6148–6156. doi: 10.4049/jimmunol.1003955. [DOI] [PubMed] [Google Scholar]
- 23.Lundie RJ, de Koning-Ward TF, Davey GM, Nie CQ, Hansen DS, Lau LS, Mintern JD, Belz GT, Schofield L, Carbone FR, Villadangos JA, Crabb BS, Heath WR. 2008. Blood-stage Plasmodium infection induces CD8+ T lymphocytes to parasite-expressed antigens, largely regulated by CD8alpha+ dendritic cells. Proc Natl Acad Sci U S A 105:14509–14514. doi: 10.1073/pnas.0806727105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miyakoda M, Kimura D, Yuda M, Chinzei Y, Shibata Y, Honma K, Yui K. 2008. Malaria-specific and nonspecific activation of CD8+ T cells during blood stage of Plasmodium berghei infection. J Immunol 181:1420–1428. doi: 10.4049/jimmunol.181.2.1420. [DOI] [PubMed] [Google Scholar]
- 25.Howland SW, Poh CM, Gun SY, Claser C, Malleret B, Shastri N, Ginhoux F, Grotenbreg GM, Renia L. 2013. Brain microvessel cross-presentation is a hallmark of experimental cerebral malaria. EMBO Mol Med 5:984–999. doi: 10.1002/emmm.201202273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolburg H, Lippoldt A. 2002. Tight junctions of the blood-brain barrier: development, composition and regulation. Vascul Pharmacol 38:323–337. doi: 10.1016/S1537-1891(02)00200-8. [DOI] [PubMed] [Google Scholar]
- 27.Abbott NJ, Ronnback L, Hansson E. 2006. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 28.Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S. 1993. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol 123:1777–1788. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morita K, Sasaki H, Furuse M, Tsukita S. 1999. Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol 147:185–194. doi: 10.1083/jcb.147.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swanson PA II, Hart GT, Russo MV, Nayak D, Yazew T, Pena M, Khan SM, Janse CJ, Pierce SK, McGavern DB. 2016. CD8+ T cells induce fatal brainstem pathology during cerebral malaria via luminal antigen-specific engagement of brain vasculature. PLoS Pathog 12:e1006022. doi: 10.1371/journal.ppat.1006022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Howland SW, Poh CM, Renia L. 2015. Activated brain endothelial cells cross-present malaria antigen. PLoS Pathog 11:e1004963. doi: 10.1371/journal.ppat.1004963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaw TN, Stewart-Hutchinson PJ, Strangward P, Dandamudi DB, Coles JA, Villegas-Mendez A, Gallego-Delgado J, van Rooijen N, Zindy E, Rodriguez A, Brewer JM, Couper KN, Dustin ML. 2015. Perivascular arrest of CD8+ T cells is a signature of experimental cerebral malaria. PLoS Pathog 11:e1005210. doi: 10.1371/journal.ppat.1005210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nacer A, Movila A, Baer K, Mikolajczak SA, Kappe SH, Frevert U. 2012. Neuroimmunological blood brain barrier opening in experimental cerebral malaria. PLoS Pathog 8:e1002982. doi: 10.1371/journal.ppat.1002982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wassmer SC, de Souza JB, Frere C, Candal FJ, Juhan-Vague I, Grau GE. 2006. TGF-beta1 released from activated platelets can induce TNF-stimulated human brain endothelium apoptosis: a new mechanism for microvascular lesion during cerebral malaria. J Immunol 176:1180–1184. doi: 10.4049/jimmunol.176.2.1180. [DOI] [PubMed] [Google Scholar]
- 35.Lau LS, Fernandez Ruiz D, Davey GM, de Koning-Ward TF, Papenfuss AT, Carbone FR, Brooks AG, Crabb BS, Heath WR. 2011. Blood-stage Plasmodium berghei infection generates a potent, specific CD8+ T-cell response despite residence largely in cells lacking MHC I processing machinery. J Infect Dis 204:1989–1996. doi: 10.1093/infdis/jir656. [DOI] [PubMed] [Google Scholar]
- 36.Poh CM, Howland SW, Grotenbreg GM, Renia L. 2014. Damage to the blood-brain barrier during experimental cerebral malaria results from synergistic effects of CD8+ T cells with different specificities. Infect Immun 82:4854–4864. doi: 10.1128/IAI.02180-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prakash MD, Munoz MA, Jain R, Tong PL, Koskinen A, Regner M, Kleifeld O, Ho B, Olson M, Turner SJ, Mrass P, Weninger W, Bird PI. 2014. Granzyme B promotes cytotoxic lymphocyte transmigration via basement membrane remodeling. Immunity 41:960–972. doi: 10.1016/j.immuni.2014.11.012. [DOI] [PubMed] [Google Scholar]
- 38.Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, Hendricks RL. 2008. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 322:268–271. doi: 10.1126/science.1164164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu T, Khanna KM, Chen X, Fink DJ, Hendricks RL. 2000. CD8(+) T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J Exp Med 191:1459–1466. doi: 10.1084/jem.191.9.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suidan GL, McDole JR, Chen Y, Pirko I, Johnson AJ. 2008. Induction of blood brain barrier tight junction protein alterations by CD8 T cells. PLoS One 3:e3037. doi: 10.1371/journal.pone.0003037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson HL, Chen Y, Suidan GL, McDole JR, Lohrey AK, Hanson LM, Jin F, Pirko I, Johnson AJ. 2012. A hematopoietic contribution to microhemorrhage formation during antiviral CD8 T cell-initiated blood-brain barrier disruption. J Neuroinflammation 9:60. doi: 10.1186/1742-2094-9-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson HL, Jin F, Pirko I, Johnson AJ. 2013. Theiler's murine encephalomyelitis virus as an experimental model system to study the mechanism of blood-brain barrier disruption. J Neurovirol 20:107–112. doi: 10.1007/s13365-013-0187-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hearn J, Rayment N, Landon DN, Katz DR, de Souza JB. 2000. Immunopathology of cerebral malaria: morphological evidence of parasite sequestration in murine brain microvasculature. Infect Immun 68:5364–5376. doi: 10.1128/IAI.68.9.5364-5376.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Purves D, Augustine GJ, Fitzpatrick D, Hall WC, LaMantia A-S, White LE. 2012. Neuroscience, 5th ed Sinauer Associates, Sunderland, Mass. [Google Scholar]
- 45.Allen GI, Tsukahara N. 1974. Cerebrocerebellar communication systems. Physiol Rev 54:957–1006. [DOI] [PubMed] [Google Scholar]
- 46.Aird WC. 2007. Phenotypic heterogeneity of the endothelium. I. Structure, function, and mechanisms. Circ Res 100:158–173. [DOI] [PubMed] [Google Scholar]
- 47.Zhao H, Aoshi T, Kawai S, Mori Y, Konishi A, Ozkan M, Fujita Y, Haseda Y, Shimizu M, Kohyama M, Kobiyama K, Eto K, Nabekura J, Horii T, Ishino T, Yuda M, Hemmi H, Kaisho T, Akira S, Kinoshita M, Tohyama K, Yoshioka Y, Ishii KJ, Coban C. 2014. Olfactory plays a key role in spatiotemporal pathogenesis of cerebral malaria. Cell Host Microbe 15:551–563. doi: 10.1016/j.chom.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 48.Hoffmann A, Pfeil J, Alfonso J, Kurz FT, Sahm F, Heiland S, Monyer H, Bendszus M, Mueller AK, Helluy X, Pham M. 2016. Experimental cerebral malaria spreads along the rostral migratory stream. PLoS Pathog 12:e1005470. doi: 10.1371/journal.ppat.1005470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Craig AG, Grau GE, Janse C, Kazura JW, Milner D, Barnwell JW, Turner G, Langhorne J, participants of the Hinxton Retreat meeting on Animal Models for Research on Severe Malaria. 2012. The role of animal models for research on severe malaria. PLoS Pathog 8:e1002401. doi: 10.1371/journal.ppat.1002401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson HL, Chen Y, Jin F, Hanson LM, Gamez JD, Pirko I, Johnson AJ. 2012. CD8 T cell-initiated blood-brain barrier disruption is independent of neutrophil support. J Immunol 189:1937–1945. doi: 10.4049/jimmunol.1200658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Renner DN, Jin F, Litterman AJ, Balgeman AJ, Hanson LM, Gamez JD, Chae M, Carlson BL, Sarkaria JN, Parney IF, Ohlfest JR, Pirko I, Pavelko KD, Johnson AJ. 2015. Effective treatment of established GL261 murine gliomas through picornavirus vaccination-enhanced tumor antigen-specific CD8+ T cell responses. PLoS One 10:e0125565. doi: 10.1371/journal.pone.0125565. [DOI] [PMC free article] [PubMed] [Google Scholar]