

ABSTRACT
Staphylococcus aureus plays an important role in sepsis, pneumonia, wound infections, and cystic fibrosis (CF), which is caused by mutations of the cystic fibrosis transmembrane conductance regulator (Cftr). Pulmonary S. aureus infections in CF often occur very early and prior to colonization with other pathogens, in particular Pseudomonas aeruginosa. Here, we demonstrate that CF mice are highly susceptible to pulmonary infections with S. aureus and fail to clear the pathogen during infection. S. aureus is internalized by Cftr-deficient macrophages in the lung, but these macrophages are unable to kill intracellular bacteria. This failure might be caused by a defect in the fusion of phagosomes with lysosomes, while this process occurs rapidly in wild-type macrophages and serves to kill intracellular pathogens. Transplantation of infected Cftr-deficient alveolar macrophages into the lungs of noninfected CF mice is sufficient to induce pneumonia. This suggests that intracellular survival of S. aureus in macrophages may allow the pathogen to chronically infect CF lungs.
KEYWORDS: cystic fibrosis, Cftr deficiency, Staphylococcus aureus, lung, macrophages, intracellular survival, pneumonia
INTRODUCTION
Cystic fibrosis (CF) is the most common autosomal recessive disease, at least in Western countries. It is caused by mutations of the cystic fibrosis transmembrane conductance regulator (human gene symbol, CFTR; mouse gene symbol, Cftr) (1–3). The incidence is approximately one case per 2,500 births, affecting ∼80,000 individuals in the United States and Europe (4–6). Mutations of CFTR cause a variety of intestinal and pulmonary symptoms. While the intestinal problems are usually well controlled, pulmonary inflammation, chronic lung damage, and recurrent acute, as well as chronic bacterial infections of the lung lead to early destruction of the lung and mainly determine the reduced life expectancy of the patients. Thus, the lung disease is currently the primary cause of morbidity, responsible for ∼80% of mortality in cystic fibrosis patients (4, 7).
Many patients suffer from chronic pulmonary infections with Pseudomonas aeruginosa, but most of the initial and early pulmonary infections of CF patients are caused by Staphylococcus aureus (8–11). The overall prevalence of S. aureus infection in 6- to 10-year-old children with CF increased from 39.5% in 1995 to 63.0% in 2005 (12).
It has been speculated that S. aureus initiates changes in the lungs of CF patients that permit P. aeruginosa to finally infect these patients. It is therefore of great importance to define the molecular mechanisms that determine S. aureus infections in CF patients.
Macrophages are the most common immune cells present in the lung and play a critical role in its innate immunity (13–18). Macrophages eliminate pathogens directly by phagocytosis, which involves engulfment of the bacteria into the phagosome, fusion of the phagosome with late lysosomes, and the formation of a phagolysosome, which serves to finally degrade the bacteria (19). Macrophages from CF patients and mice have a dysfunction in inflammatory cytokine production, in particular the production of gamma interferon (IFN-γ), interleukin-1αβ (IL-1αβ), IL-6, IL-8/KC, and tumor necrosis factor alpha (TNF-α), and impaired killing of pathogens (20–27). Lysosomal functions of macrophages lacking Cftr might also be compromised, although conflicting data have been published on this issue. Thus, it was shown that the pH in lysosomes of Cftr-deficient macrophages is increased, which reduces the ability of these cells to kill pathogens (28, 29). The reduced influx of Cl− ions results in an impaired influx of H+, because Cl− ions serve as counterions for H+ (28, 29). However, other studies failed to show such a lack of acidification in Cftr-deficient macrophages (30). Our own studies demonstrated an increased pH in secretory lysosomes of Cftr-deficient cells, organelles that were not analyzed by the other studies (31). It might be possible that the different results are caused by the analysis of different lysosome populations expressing different sets of ion channels.
It is unknown whether S. aureus survives in CF alveolar macrophages in vivo and whether these cells then serve as Trojan horses to spread the infection and, thereby, may cause recurrent and chronic infections. Therefore, we investigated whether Cftr-deficient macrophages are able to internalize and kill intracellular S. aureus. We demonstrate that CF mice are highly susceptible to pulmonary infection with S. aureus and that the pathogen accumulates within alveolar macrophages of these mice in vivo. Cftr-deficient macrophages fail to fuse phagosomes and lysosomes, resulting in a lack of intracellular killing of S. aureus in these cells. In contrast, wild-type macrophages rapidly kill intracellular S. aureus. Likewise, macrophages in lung sections from human CF patients contain intracellular S. aureus cells. Most importantly, transfer of Cftr-deficient alveolar macrophages carrying engulfed S. aureus cells into the lungs of noninfected Cftr-deficient mice is sufficient to induce severe pneumonia, while wild-type macrophages are able to kill the pathogen and cannot transfer the infection into another host. Thus, the intracellular survival of S. aureus in CF alveolar macrophages in vivo might allow the pathogen to survive in and chronically infect CF lungs.
RESULTS
Cftr deficiency impairs S. aureus killing in mouse lungs in vivo.
S. aureus is considered to be a mostly extracellular pathogen (32, 33), and the pathophysiological significance of intracellular S. aureus remains to be determined. To investigate whether Cftr deficiency alters infections with S. aureus and permits intracellular survival of the pathogen, we intranasally infected wild-type and CF mice with S. aureus for 6 h. The lungs were then removed and homogenized without damaging the cells, and the homogenates were incubated with the cell-impermeable antibiotic gentamicin, which kills extracellular bacteria but leaves intracellular bacteria intact. The results showed that intracellular S. aureus cells were rapidly cleared in lungs of wild-type mice, while CF mice failed to kill intracellular bacteria in the lung and had significantly more bacteria in their lungs than wild-type mice (Fig. 1A).
FIG 1.
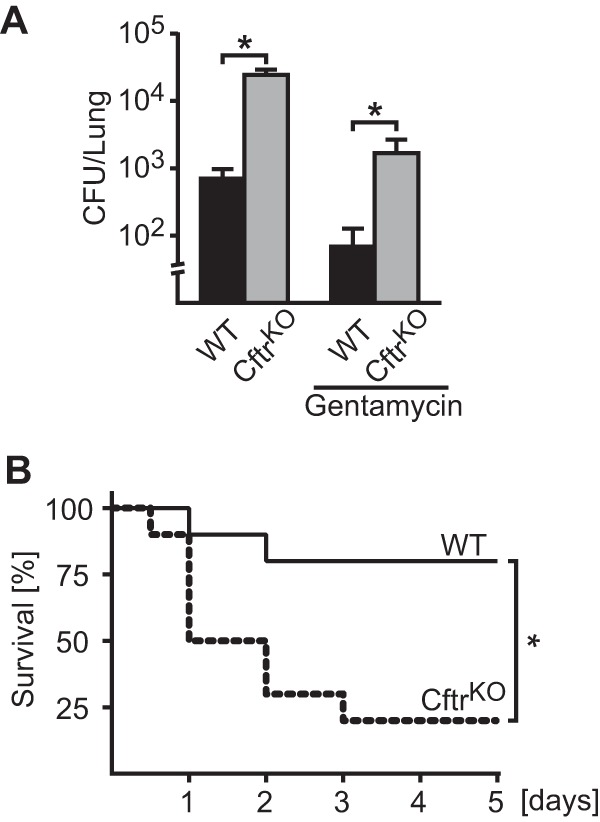
Cystic fibrosis mice fail to clear S. aureus after pulmonary infection. (A) Wild-type and CF mice were intranasally infected with 8 × 108 CFU S. aureus for 6 h. Mice were sacrificed, lungs were homogenized and incubated with gentamicin for 1 h to kill extracellular bacteria, and the numbers of intracellular bacteria were determined. The results reveal that intracellular S. aureus survive in Cftr-deficient lung cells, while wild-type lungs kill the pathogen. In addition, wild-type and CF mice were infected with S. aureus for 6 h, and 50 mg/kg gentamicin was administered by inhalation to kill extracellular bacteria in the lungs. Mice were sacrificed 24 h after infection (18 h after gentamicin application), and CFU were determined. Displayed are the mean results ± SD from 4 experiments. An asterisk indicates a significance difference (P < 0.05) between cystic fibrosis and wild-type mice, determined by t test. (B) Wild-type and Cftr-deficient mice were intranasally infected with 8 × 108 CFU S. aureus. Mice were observed for 5 days. The graph shows mortality for 10 mice per group. Comparisons of survival variables were performed by log-rank test, *, P < 0.05.
To confirm in vivo internalization of S. aureus cells, we infected wild-type and CF mice intranasally with S. aureus and administered gentamicin by inhalation after the inoculation. Gentamicin killed most bacteria in wild-type mice (Fig. 1A). Surprisingly, the bacterial counts in the lungs of CF mice were still high after inhalation of gentamicin, indicating that S. aureus is partially internalized and that Cftr-deficient cells fail to clear the bacteria (Fig. 1A).
The failure of Cftr-deficient cells to kill intracellular S. aureus correlated with high mortality of CF mice upon intranasal infection with S. aureus, while wild-type mice cured the infection (Fig. 1B).
Cftr-deficient macrophages fail to kill internalized S. aureus in vitro and in vivo.
To gain insight into the mechanisms allowing S. aureus to survive in CF lungs, we cultured macrophages from wild-type and CF mice and determined the uptake and killing of intracellular S. aureus by these macrophages in vitro. Cells were infected, incubated with gentamicin to kill extracellular bacteria, and washed, and intracellular CFU were determined. Both bone marrow-derived macrophages and freshly isolated alveolar macrophages from wild-type mice efficiently killed S. aureus between 2 and 8 h after internalization. In contrast, S. aureus survived and replicated in Cftr-deficient macrophages (Fig. 2A). The uptake of the pathogen was not affected by Cftr deficiency.
FIG 2.

Cftr deficiency impairs intracellular killing of S. aureus in macrophages in vivo. (A) Alveolar macrophages (AM) and bone marrow-derived macrophages (BMDM) from wild-type (WT) or CF (Cftr knockout [CftrKO]) mice were infected with S. aureus for the indicated times. After infection, gentamicin was added and cells were incubated for an additional 1 h to ensure killing of extracellular bacteria. Cells were then washed and lysed, and the numbers of viable S. aureus colonies derived from lysates were determined. Data are expressed as the mean results ± SD from 4 experiments. *, P < 0.05 by one-way ANOVA and Student-Newman-Keuls test. (B) Wild-type and CF mice were intranasally infected with S. aureus, and alveolar macrophages were isolated by bronchial lavage after 6 and 12 h. Alveolar macrophages were incubated with gentamicin for 1 h, and the numbers of internalized bacteria were determined. (C, D) The infection experiments were repeated with S. aureus strain Newman, with very similar results as for the clinical S. aureus strain. Values are the mean results ± SD from 4 independent experiments; *, P < 0.05 by one-way ANOVA and Student-Newman-Keuls test.
The killing of S. aureus in the lungs in vivo might be mediated by a variety of cells, including epithelial cells and alveolar macrophages. Here, we focused on alveolar macrophages and tested their capacity to kill S. aureus in vivo. To this end, wild-type and CF mice were infected with S. aureus for 6 and 12 h and then sacrificed. Alveolar macrophages were isolated from these mice by bronchoalveolar lavage (BAL), incubated with gentamicin, and washed, and the numbers of live intracellular bacteria were measured via a bacterial killing assay. These studies revealed that wild-type macrophages were able to kill intracellular S. aureus (Fig. 2B), whereas Cftr-deficient alveolar macrophages failed to kill intracellular bacteria in vivo (Fig. 2B). We repeated the bacterial killing experiments using the well-established S. aureus Newman strain. These studies had very similar results and demonstrated that Cftr deficiency impairs the killing of intracellular bacteria (Fig. 2C and D).
Cftr-deficient alveolar macrophages infected with S. aureus form a reservoir to infect the host.
To study whether viable intracellular S. aureus cells might serve as a reservoir for the pathogen to chronically infect the lungs of CF mice, we infected wild-type and CF mice with S. aureus and isolated alveolar macrophages from these mice. We then reinjected these macrophages into wild-type or Cftr-deficient mice, respectively, and determined the CFU in the lungs of these animals (Fig. 3). The transplanted macrophages from wild-type mice did not infect either wild-type or CF mice (Fig. 3), indicating efficient killing of S. aureus in wild-type macrophages. Wild-type mice were also resistant to infected macrophages transferred from Cftr-deficient mice (Fig. 3). In contrast, injection of infected macrophages from CF mice into CF mice resulted in an infection, indicating that infected Cftr-deficient macrophages acted as a bacterial carrier (Fig. 3).
FIG 3.

Cftr-deficient alveolar macrophages are a reservoir for infection of S. aureus. Wild-type and CF mice were infected with S. aureus for 6 h. Mice were sacrificed, and alveolar macrophages were isolated. These infected macrophages were incubated with gentamicin for 1 h to kill extracellular bacteria, extensively washed, and reinjected into wild-type or CF mice. Mice were sacrificed 6 h after the macrophage transfer, and bacterial counts in the lungs were determined. The results show that only Cftr-deficient macrophages transplanted into CF mice caused a severe infection, while wild-type macrophages transplanted into wild-type mice or CF mice were resistant to infection with S. aureus. Data are presented as mean results ± SD from 4 independent experiments. *, P < 0.05 by one-way ANOVA and Student-Newman-Keuls test.
Human CF lungs contain intracellular S. aureus in macrophages.
To confirm these mouse data in humans, we stained human lung samples from CF patients and healthy individuals with antibodies against the macrophage marker CD11b and S. aureus. The immunofluorescence staining results revealed intracellular S. aureus within macrophages in CF lungs, while no bacteria were detected in healthy controls (Fig. 4). Note that we detected S. aureus in 4 of 9 CF patients but in none of the 3 samples from healthy donors. Among the infected CF samples, 19% ± 4% of macrophages were infected with S. aureus.
FIG 4.
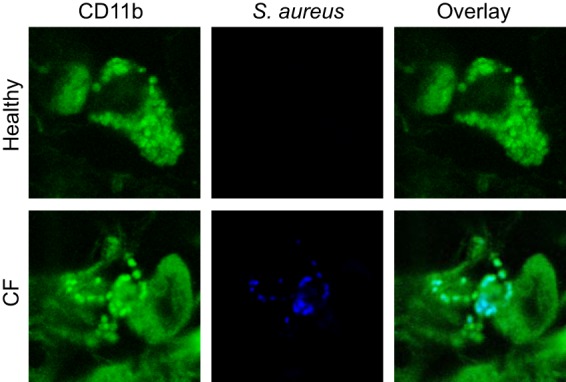
Macrophages in human CF lungs contain intracellular S. aureus cells. Sections of human lungs from CF patients or healthy donors were stained with FITC-coupled anti-CD11b and Cy5-labeled anti-S. aureus antibodies and analyzed by confocal immunofluorescence microscopy. The results show that S. aureus cells accumulate in macrophages in the lungs of CF patients. The staining results shown are representative of 3 (healthy donors) or 9 (CF patients) similar results.
Cftr-deficient macrophages fail to fuse lysosomes with phagosomes.
To gain insight into the molecular mechanisms of the impaired bacterial killing of S. aureus in CF mice and human CF lungs, we determined whether Cftr deficiency affects the fusion of phagosomes with lysosomes, one of the initial and central steps of intracellular killing of pathogens. To this end, we stained the macrophages with Cy5-labeled anti-S. aureus antibodies and LysoTracker red (Fig. 5A) or antibodies against cathepsin B (Fig. 5B). LysoTracker is a fluorescent dye for labeling and tracking acidic organelles in living cells. Most of the intracellular S. aureus cells colocalized with LysoTracker-labeled acidic-positive compartments in wild-type macrophages, whereas the fusion of S. aureus-positive phagosomes with LysoTracker-positive lysosomes was reduced in Cftr-deficient macrophages (Fig. 5A). In detail, over 55% of S. aureus cells colocalized with LysoTracker-positive lysosomes after infection of wild-type cells, while only 20% of bacteria colocalized with LysoTracker in Cftr-deficient cells (Fig. 5A, bottom). Similar results were obtained by staining cells with cathepsin D, a lysosome-specific marker. Cftr deficiency reduced the association of S. aureus with cathepsin D-labeled lysosomes upon infection (Fig. 5B): while approximately 60% of S. aureus cells colocalized with cathepsin D in wild-type cells, only 20% of all intracellular S. aureus cells localized to cathepsin D-positive vesicles in Cftr-deficient cells.
FIG 5.

Cftr deficiency affects phagosome-lysosome fusion in macrophages. Fusion of lysosomes with phagosomes is impaired in Cftr-deficient macrophages after infection with S. aureus. Wild-type or Cftr-deficient bone marrow-derived macrophages were stained with LysoTracker red (A) or Cy3-coupled anti-cathepsin D antibodies (B) and Cy5-labeled S. aureus and analyzed by confocal microscopy. Top, representative fluorescence images from four independent experiments are shown; bottom, calculated colocalization rates of LysoTracker or Cy3–anti-cathepsin D antibodies and Cy5-labeled S. aureus. All results are representative of or summarized from the results of 4 independent confocal studies per group. The quantitative results are the mean values ± SD for at least 100 cells. AOI, area of interest. *, P < 0.05 for wild-type versus CF by t test.
DISCUSSION
Here, we demonstrate that S. aureus is internalized by alveolar and bone marrow-derived macrophages. We used, wherever possible, two different types of macrophages to ensure that all findings can be generalized and are not restricted to a certain macrophage cell type. Wild-type macrophages rapidly killed the pathogen within the cell, whereas Cftr-deficient macrophages failed to kill the intracellular pathogen. S. aureus was long thought to be an extracellular pathogen, and our studies certainly do not exclude that many of the symptoms of S. aureus pneumonia are caused by extracellular S. aureus and toxins. However, our results indicate that after internalization into macrophages, Cftr-deficient cells fail to kill the intracellular pathogen, which is most likely caused by a defect of the fusion of lysosomes and phagosomes containing the pathogen. In addition, the lysosomes in S. aureus-infected Cftr-deficient macrophages seem to be less acidic than those in wild-type cells, although it is unknown whether the acidification defect is secondary to the lack of phagosome-lysosome fusion in these macrophages.
In the present study, we demonstrate reduced fusion of phagosomes containing S. aureus cells with lysosomes in Cftr-deficient macrophages and suggest that this defect results in intracellular survival of S. aureus in these macrophages. Most importantly, we present evidence for the first time that those ingested S. aureus cells are viable and are able to serve as a novel infection focus. Thus, Cftr-deficient alveolar macrophages can be seen as Trojan horses, spreading the pathogen in CF lungs even after a limited, initially local infection. They might also serve the pathogen as a carrier to chronically infect the host. The intracellular survival of S. aureus in CF lungs might finally protect the pathogen from the hostile extracellular environment in an inflamed area, the specific immune system, and antibiotics applied to treat the infection (T- and B-lymphocytes). The human studies that were performed with CF end-stage lungs suggest that S. aureus survives for a long time within Cftr-deficient macrophages. If this can be confirmed in mice, our data may provide evidence for the following pathophysiological scenario: the initial infection of Cftr-deficient alveolar macrophages with S. aureus results in intracellular survival and even proliferation of S. aureus, which in turn triggers a continuous activation of macrophages and incoming neutrophils that results in chronic inflammation of cystic fibrosis lungs, one of the hallmarks of lung disease in cystic fibrosis. The chronic infection with S. aureus and the chronic inflammation of the lung then finally alter the physiology of the lung. This allows further, even more serious pathogens like the most common pathogen, Pseudomonas aeruginosa, to infect the lung and to induce a severe chronic pneumonia in CF patients.
Several previous studies suggested a defect of vesicular pH regulation in cystic fibrosis. Thus, Barasch et al. demonstrated a regulation of the pH in trans-Golgi vesicles by CFTR and an alkalinization of these vesicles in cells lacking CFTR (28). Several more recent studies revealed that Cftr also contributes to acidification of phagolysosomes in macrophages (20, 29, 34–36). These studies showed that Cftr determines the influx of chloride ions into lysosomes (29). The negatively charged chloride ions serve as counterions for protons pumped into phagolysosomes by the lysosomal V-type H+-ATPase (37, 38). A lack of Cftr thereby prevents the accumulation of protons in phagolysosomes and therefore impairs acidification of these vesicles, resulting in alkalinization of phagolysosomes (28, 29, 34–36). Since the acidification of phagolysosomes is required for killing pathogens and the full activity of lysosomal enzymes, these studies concluded that the defect of Cftr-deficient alveolar macrophages to kill pathogens is caused by the increased pH in lysosomes in Cftr-deficient cells (29, 36). We showed that Cftr-deficient cells have an increased pH in secretory lysosomes (20, 31). Such a specific role of Cftr in the control of the pH of a distinct subset of vesicles might also explain controversial findings of other groups (30, 39) reporting that Cftr deficiency does not result in changes of the lysosomal pH. However, in contrast to the above-mentioned and our studies, these studies investigated the pH in vesicles employing zymosan conjugates containing fluorescein and tetramethylrhodamine-dextran. Thus, these studies report the pH in vesicles that were generated upon endocytosis of these dyes (30, 39), and it might be possible that specific phagosomes or phagolysosomes differ from these vesicles. In summary, this suggests that Cftr only determines the pH in distinct vesicle populations, most likely prelysosomes, secretory lysosomes, and phagolysosomes.
In the present study, we did not focus on an exact measurement of the pH in lysosomes but instead, investigated the fusion of phagosomes containing S. aureus cells with lysosomes in wild-type and Cftr-deficient macrophages.
At present, the molecular mechanisms by which Cftr deficiency prevents fusion of phagosomes with lysosomes are unknown. We have previously shown that ceramide accumulates in Cftr-deficient airway epithelial cells and macrophages, while sphingosine expression decreases in the epithelial cells of the upper respiratory tract of CF mice (40). The subcellular distribution of ceramide and sphingosine in Cftr-deficient macrophages cells was not investigated in these studies, and it is unknown whether the change in these two lipids in Cftr-deficient cells has an impact on vesicular trafficking and fusion.
Previous data have demonstrated that alveolar macrophages from CF patients internalize Pseudomonas aeruginosa and S. aureus at similar rates and amounts as wild-type macrophages (41). This was also observed in a study comparing phagocytosis by CF and wild-type macrophages and polymorphonuclear leukocytes, which concluded that the latter have a higher killing capacity than macrophages. Phagocytosis did not differ between CF and wild-type cells (42). These studies are consistent with the present findings. However, our findings extend these studies and investigate events that occur after internalization. We show that intracellular killing of S. aureus is reduced in CF alveolar macrophages in vitro and in vivo compared to that in wild-type macrophages.
In summary, our data provide a novel model for the initiation of a chronic infection in CF lungs. We demonstrate that S. aureus survives in alveolar macrophages and that these cells are able to initiate a new infection after transfer into previously uninfected mice. These data suggest a novel mechanism to explain how CF lungs are chronically infected with S. aureus, which may be a prerequisite for further infections with Pseudomonas aeruginosa and other pathogens frequently detected in end-stage CF lungs.
MATERIALS AND METHODS
Human samples.
Human samples were obtained from nine patients with CF in end stage and three healthy donors aged between 19 and 46. Specimens were provided by the West German Heart and Vascular Center Essen, Essen, Germany. The study was approved by the local ethics committee of the Medical Faculty of the University of Duisburg-Essen, and patients gave informed consent. Lung tissue specimens were fixed in 10% formaldehyde and embedded in paraffin. All CF patient samples were end-stage lung disease, and the patients were known to suffer from chronic infection with pathogenic bacteria, including P. aeruginosa and S. aureus. The donor samples were from healthy donors whose death was caused by accidents but who were otherwise healthy and had no lung disease.
Mice.
Cftrtm1Unc-Tg(FABPCFTR) were obtained from Jackson Laboratories (Bar Harbor, ME, USA). These mice lack Cftr but express human CFTR in the gut under the control of a fatty acid binding protein promoter, allowing a normal diet. These mice therefore do not develop malnutrition and are usually of the same weight as wild-type controls. The mice were backcrossed for more than 10 generations onto a C57BL/6 background. The controls were wild-type littermates. The genotype was verified by PCR. Mice were bred in the animal facility of the University Duisburg-Essen under specific-pathogen-free (SPF) conditions according to the criteria of the Federation of Laboratory Animal Science. All procedures performed on mice were approved by the Bezirksregierung Düsseldorf, Düsseldorf, Germany, or the local ethics committee.
Staining of human lung samples.
Lung paraffin sections were dewaxed and rehydrated. For fluorescence staining, lung sections were incubated in pepsin (Invitrogen) for 30 min at 37°C, washed, and treated for 15 min in phosphate-buffered saline (PBS) supplemented with 5% fetal calf serum (FCS). Samples were incubated with anti-CD11b antibody (diluted 1:100; BioLegend) and rabbit polyclonal IgG anti-S. aureus antibody (diluted 1:200; Abcam) for 1 h at room temperature. Samples were then washed 3 times for 5 min each with 0.05% Tween 20–PBS and incubated with fluorescein isothiocyanate (FITC)-conjugated F(ab′)2 fragments of goat anti-rat IgG and Alexa Fluor 647-conjugated F(ab′)2 fragments of donkey anti-rabbit IgG (final concentration of each, 1.5 μg/ml; Jackson ImmunoResearch) for 45 min at room temperature. The samples were washed again 3 times with PBS–0.05% Tween 20 and once in PBS, and the cells were mounted in Mowiol. Samples were analyzed with a Leica true confocal scanner (TCS) 5-channel spectrophotometry (SP5) confocal microscope employing a 100× oil lens, and images were analyzed with Leica LCS software (Leica Microsystems). At least 25 macrophages per specimen were quantified for calculation.
Staphylococcus aureus.
The S. aureus strain used in the present study has been previously described and extensively characterized (43). The strain was isolated from a patient with sepsis and expresses alpha-toxin and enterotoxin D but not the Panton-Valentine leukocidin or toxic shock syndrome toxin (43). To exclude strain-specific results, we repeated the principal experiments with the well-characterized S. aureus strain Newman (ATCC 25904) (43). Bacteria were grown overnight on 5% sheep blood Trypticase soy agar plates (BD), removed from the plates, resuspended in 40 ml tryptic soy broth (BD) at an optical density of 0.2 to 0.25, and incubated at 37°C with shaking at 125 rpm for 75 min. The bacteria were then pelleted at 2,800 rpm for 10 min, washed twice in RPMI 1640 (Gibco) supplemented with 10 mM HEPES, and finally resuspended in HEPES-saline (H-S) buffer consisting of 132 mM NaCl, 20 mM HEPES, pH 7.4, 5 mM KCl, 1 mM CaCl2, 0.7 mM MgCl2, and 0.8 mM MgSO4. Cells or mice were then infected within the next 10 min.
In vivo infections.
Mice were intranasally infected with 8 × 108 CFU S. aureus and sacrificed 6 h after infection or observed over 5 days to determine survival. This dose was lethal for CF mice. The lung tissue was collected, homogenized into very small pieces, and either incubated with 100 μg/ml gentamicin (Sigma) in PBS for 60 min to kill extracellular bacteria or left untreated. The samples were extensively washed to remove any traces of gentamicin. Mammalian cells were then lysed in 5 mg/ml saponin-PBS to release intracellular S. aureus. This buffer does not lyse S. aureus. The samples were washed again, and the numbers of intracellular or total (intra- and extracellular) bacteria, respectively, were determined by plating aliquots and quantifying colonies after overnight growth.
Alternatively, gentamicin was dissolved in PBS at a concentration of 50 mg/kg of body weight and administered via inhalation, employing a PARI Boy SX nebulizer (PARI GmbH, Starnberg, Germany). The nebulizer generates a fine aerosol by pumping the fluid with an air jet. Mice were administered the aerosol via a mask that was adapted to the size of their noses.
Bronchoalveolar lavage and alveolar macrophage isolation.
Alveolar macrophages were isolated by performing BAL (44). To this end, mice were sacrificed, the trachea was exposed, a small catheter was inserted and fixed, and the lung was washed out 10 times with 1 ml of PBS each time. Cells were centrifuged for 5 min at 300 × g at 4°C and resuspended in minimal essential medium (MEM) (Gibco)-HEPES supplemented with 2 mM l-glutamine, 1 mM sodium pyruvate, 100 μM nonessential amino acids. Cells were aliquoted into well plates and used for experiments within the next 60 min.
Isolation of bone marrow-derived macrophages.
Mice were sacrificed, and bone marrow cells were flushed though femurs and tibias with MEM supplemented with 10% fetal bovine serum (FBS; Gibco), 10 mM HEPES, pH 7.4, 2 mM l-glutamine, 1 mM sodium pyruvate, 100 μM nonessential amino acids, 100 U/ml penicillin, and 100 μg/ml streptomycin (all from Thermo Fisher Scientific). Single cells were isolated by passing the samples through a 23-gauge needle and were cultured for 24 h in small tissue culture flasks. The nonadherent cells were removed and counted, and 3 × 104 or 1.5 × 105 cells were cultured in 6- or 24-well plates containing MEM supplemented as described above plus 20% supernatant from L cells, which served as a source of macrophage colony-stimulating factor. After 4 days in culture, macrophages were supplied with fresh MEM–10% FCS–20% L cell supernatant medium, and mature macrophages were used on day 10 of culture for experiments.
Infection of cells.
Macrophages were cultured or seeded in a 6-well plate for measuring intracellular S. aureus and/or glass coverslips in 24-well plates for all immunofluorescence studies. Bone marrow-derived macrophages were infected with S. aureus for the indicated times at a multiplicity of infection (MOI) of 100 bacteria/1 macrophage; alveolar macrophages were infected at an MOI of 50:1. Infections were stopped by washing the samples in cold PBS, followed by either fixation in 4% paraformaldehyde (PFA) buffered to pH 7.4 for all immunofluorescence studies or incubation for 60 min with 100 μg/ml gentamicin for all internalization/killing studies.
Bacterial killing assays in vitro.
For in vitro assays, cells were infected as described above for the indicated times, gently washed with cold sterile PBS, and incubated for 1 h at 37°C in MEM-HEPES with or without 100 μg/ml gentamicin to determine the numbers of internalized bacterial or the total numbers of internalized and attached bacteria, respectively. The cells were then extensively washed to remove all gentamicin and lysed in 5 mg/ml saponin-PBS for 10 min to release intracellular bacteria. The samples were transferred into 15-ml tubes, and bacteria were pelleted by 10 min of centrifugation at 3,200 rpm and resuspended in PBS. Then, 50-μl aliquots were plated on LB-agar plates, and the CFU were quantified after overnight growth at 37°C.
As a control, we tested the killing of S. aureus by gentamicin. Gentamicin at the concentration used kills 99.97% ± 0.01% of the clinical S. aureus strain used here, and 99.9998% ± 0.0004% of the Newman strain. For this purpose, we added 107 CFU S. aureus to bone marrow-derived macrophages (BMDMs) and alveolar macrophages. Since approximately 99.9% of the bacteria were washed off before adding gentamicin, the maximum numbers of extracellular bacteria that would not have been killed were less than 10 CFU (theoretically 3) for the clinical strain and even lower for the Newman strain.
Alveolar macrophage transplantation.
Briefly, wild-type and Cftr-deficient mice were infected with S. aureus and sacrificed after 6 h. Alveolar macrophages were isolated by BAL as described above. Wild-type and Cftr-deficient alveolar macrophages were incubated with 100 μg/ml gentamicin for 60 min to kill extracellular bacteria. Cells were then washed once by centrifugation at 300 × g and resuspended in 20 μl PBS. Cell suspensions were noninvasively administered intranasally to wild-type and Cftr-deficient mice anesthetized by ether.
Phagosome-lysosome detection.
Macrophages were preincubated with 60 nM LysoTracker red DND-99 (ThermoFisher) for 60 min or left untreated for cathepsin D staining. Macrophages were then infected with S. aureus for the indicated times. Infection was terminated by fixation in 4% PFA–PBS for 10 min. Cells were washed 3 times with H-S buffer, permeabilized with 0.1% Triton X-100–PBS for 5 min at room temperature, washed again with PBS, and incubated for 1 h with goat anti-Armenian hamster IgG(H+L) antibodies (dilution of 1:100; Jackson ImmunoResearch) to block nonspecific binding. Samples were washed and incubated for 1 h at room temperature with a rabbit polyclonal IgG anti-S. aureus antibody (diluted 1:200; Abcam) for costaining with LysoTracker or for 1 h with anti-cathepsin D and anti-S. aureus antibodies (diluted 1:200; Abcam). Samples were washed 3 times for 5 min each time with 0.05% Tween 20–PBS and incubated with Cy3-conjugated F(ab′)2 fragments of donkey anti-rabbit IgG (final concentration, 1.5 μg/ml; Jackson ImmunoResearch) or Cy5-conjugated F(ab′)2 fragments of donkey anti-mouse IgG for 45 min at room temperature. The samples were washed again 3 times with PBS–0.05% Tween 20 and once in PBS, and the cells were mounted in Mowiol. Samples were analyzed with a Leica TCS SP5 confocal microscope employing a 100× oil lens, and images were analyzed using Leica LCS software (Leica Microsystems). The colocalization rate of S. aureus with LysoTracker or cathepsin D was calculated by counting the number of bacteria per cell that colocalized with the lysosomal markers compared to the total number of bacteria within the cell.
Statistics.
Data are expressed as arithmetic means ± standard deviations (SD) unless otherwise indicated. One-way analysis of variance (ANOVA) followed by the Student-Newman-Keuls test was used to test between-group and within-group differences. Pairwise comparisons were made with Student's t test. Comparisons of survival variables were performed by log-rank test. Statistical significance was set at a P value of <0.05. All data were obtained from independent measurements. The GraphPad Prism statistical software program (GraphPad Software) was used for the analyses.
ACKNOWLEDGMENTS
The study was supported by DFG grant GR 1697/2-1 to H.G. and DFG grant GU 335/33-1 to E.G.
REFERENCES
- 1.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, Drumm ML, Iannuzzi MC, Collins FS, Tsui LC. 1989. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 2.Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, Zsiga M, Buchwald M, Riordan JR, Tsui LC, Collins FS. 1989. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 245:1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- 3.Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. 1989. Identification of the cystic fibrosis gene: genetic analysis. Science 245:1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- 4.Cutting GR. 2015. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet 16:45–56. doi: 10.1038/nrg3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies JC, Alton EW, Bush A. 2007. Cystic fibrosis. BMJ 335:1255–1259. doi: 10.1136/bmj.39391.713229.AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Sullivan BP, Freedman SD. 2009. Cystic fibrosis. Lancet 373:1891–1904. doi: 10.1016/S0140-6736(09)60327-5. [DOI] [PubMed] [Google Scholar]
- 7.Stoltz DA, Meyerholz DK, Welsh MJ. 2015. Origins of cystic fibrosis lung disease. N Engl J Med 372:351–362. doi: 10.1056/NEJMra1300109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stone A, Saiman L. 2007. Update on the epidemiology and management of Staphylococcus aureus, including methicillin-resistant Staphylococcus aureus, in patients with cystic fibrosis. Curr Opin Pulm Med 13:515–521. doi: 10.1097/MCP.0b013e3282efbbac. [DOI] [PubMed] [Google Scholar]
- 9.Goss CH, Muhlebach MS. 2011. Review: Staphylococcus aureus and MRSA in cystic fibrosis. J Cyst Fibros 10:298–306. doi: 10.1016/j.jcf.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Kahl BC. 2010. Impact of Staphylococcus aureus on the pathogenesis of chronic cystic fibrosis lung disease. Int J Med Microbiol 300:514–519. doi: 10.1016/j.ijmm.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Wong JK, Ranganathan SC, Hart E, Australian Respiratory Early Surveillance Team for Cystic Fibrosis. 2013. Staphylococcus aureus in early cystic fibrosis lung disease. Pediatr Pulmonol 48:1151–1159. doi: 10.1002/ppul.22863. [DOI] [PubMed] [Google Scholar]
- 12.Razvi S, Quittell L, Sewall A, Quinton H, Marshall B, Saiman L. 2009. Respiratory microbiology of patients with cystic fibrosis in the United States, 1995 to 2005. Chest 136:1554–1560. doi: 10.1378/chest.09-0132. [DOI] [PubMed] [Google Scholar]
- 13.Pozzi C, Lofano G, Mancini F, Soldaini E, Speziale P, De Gregorio E, Rappuoli R, Bertholet S, Grandi G, Bagnoli F. 2015. Phagocyte subsets and lymphocyte clonal deletion behind ineffective immune response to Staphylococcus aureus. FEMS Microbiol Rev 39:750–763. doi: 10.1093/femsre/fuv024. [DOI] [PubMed] [Google Scholar]
- 14.Murray PJ, Wynn TA. 2011. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fournier B, Philpott DJ. 2005. Recognition of Staphylococcus aureus by the innate immune system. Clin Microbiol Rev 18:521–540. doi: 10.1128/CMR.18.3.521-540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foster TJ. 2005. Immune evasion by staphylococci. Nat Rev Microbiol 3:948–958. doi: 10.1038/nrmicro1289. [DOI] [PubMed] [Google Scholar]
- 17.Varol C, Mildner A, Jung S. 2015. Macrophages: development and tissue specialization. Annu Rev Immunol 33:643–675. doi: 10.1146/annurev-immunol-032414-112220. [DOI] [PubMed] [Google Scholar]
- 18.Wynn TA, Chawla A, Pollard JW. 2013. Macrophage biology in development, homeostasis and disease. Nature 496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tabary O, Escotte S, Couetil JP, Hubert D, Dusser D, Puchelle E, Jacquot J. 2001. Relationship between IkappaBalpha deficiency, NFkappaB activity and interleukin-8 production in CF human airway epithelial cells. Pflugers 443(Suppl 1):S40–S44. doi: 10.1007/s004240100642. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Li X, Grassme H, Doring G, Gulbins E. 2010. Alterations in ceramide concentration and pH determine the release of reactive oxygen species by Cftr-deficient macrophages on infection. J Immunol 184:5104–5111. doi: 10.4049/jimmunol.0902851. [DOI] [PubMed] [Google Scholar]
- 21.Inoue H, Massion PP, Ueki IF, Grattan KM, Hara M, Dohrman AF, Chan B, Lausier JA, Golden JA, Nadel JA. 1994. Pseudomonas stimulates interleukin-8 mRNA expression selectively in airway epithelium, in gland ducts, and in recruited neutrophils. Am J Respir Cell Mol Biol 11:651–663. doi: 10.1165/ajrcmb.11.6.7946394. [DOI] [PubMed] [Google Scholar]
- 22.Oceandy D, McMorran BJ, Smith SN, Schreiber R, Kunzelmann K, Alton EW, Hume DA, Wainwright BJ. 2002. Gene complementation of airway epithelium in the cystic fibrosis mouse is necessary and sufficient to correct the pathogen clearance and inflammatory abnormalities. Hum Mol Genet 11:1059–1067. doi: 10.1093/hmg/11.9.1059. [DOI] [PubMed] [Google Scholar]
- 23.Tang A, Sharma A, Jen R, Hirschfeld AF, Chilvers MA, Lavoie PM, Turvey SE. 2012. Inflammasome-mediated IL-1beta production in humans with cystic fibrosis. PLoS One 7:e37689. doi: 10.1371/journal.pone.0037689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schultz MJ, Rijneveld AW, Florquin S, Edwards CK, Dinarello CA, van der Poll T. 2002. Role of interleukin-1 in the pulmonary immune response during Pseudomonas aeruginosa pneumonia. Am J Physiol Lung Cell Mol Physiol 282:L285–290. doi: 10.1152/ajplung.00461.2000. [DOI] [PubMed] [Google Scholar]
- 25.Del Porto P, Cifani N, Guarnieri S, Di Domenico EG, Mariggio MA, Spadaro F, Guglietta S, Anile M, Venuta F, Quattrucci S, Ascenzioni F. 2011. Dysfunctional CFTR alters the bactericidal activity of human macrophages against Pseudomonas aeruginosa. PLoS One 6:e19970. doi: 10.1371/journal.pone.0019970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murphy BS, Bush HM, Sundareshan V, Davis C, Hagadone J, Cory TJ, Hoy H, Hayes D Jr, Anstead MI, Feola DJ. 2010. Characterization of macrophage activation states in patients with cystic fibrosis. J Cyst Fibros 9:314–322. doi: 10.1016/j.jcf.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 27.Tirouvanziam R, de Bentzmann S, Hubeau C, Hinnrasky J, Jacquot J, Peault B, Puchelle E. 2000. Inflammation and infection in naive human cystic fibrosis airway grafts. Am J Respir Cell Mol Biol 23:121–127. doi: 10.1165/ajrcmb.23.2.4214. [DOI] [PubMed] [Google Scholar]
- 28.Barasch J, Kiss B, Prince A, Saiman L, Gruenert D, al-Awqati Q. 1991. Defective acidification of intracellular organelles in cystic fibrosis. Nature 352:70–73. doi: 10.1038/352070a0. [DOI] [PubMed] [Google Scholar]
- 29.Di A, Brown ME, Deriy LV, Li C, Szeto FL, Chen Y, Huang P, Tong J, Naren AP, Bindokas V, Palfrey HC, Nelson DJ. 2006. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat Cell Biol 8:933–944. doi: 10.1038/ncb1456. [DOI] [PubMed] [Google Scholar]
- 30.Haggie PM, Verkman AS. 2009. Unimpaired lysosomal acidification in respiratory epithelial cells in cystic fibrosis. J Biol Chem 284:7681–7686. doi: 10.1074/jbc.M809161200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teichgraber V, Ulrich M, Endlich N, Riethmuller J, Wilker B, De Oliveira-Munding CC, van Heeckeren AM, Barr ML, von Kurthy G, Schmid KW, Weller M, Tummler B, Lang F, Grassme H, Doring G, Gulbins E. 2008. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med 14:382–391. doi: 10.1038/nm1748. [DOI] [PubMed] [Google Scholar]
- 32.Lowy FD. 2000. Is Staphylococcus aureus an intracellular pathogen? Trends Microbiol 8:341–343. doi: 10.1016/S0966-842X(00)01803-5. [DOI] [PubMed] [Google Scholar]
- 33.Sinha B, Fraunholz M. 2010. Staphylococcus aureus host cell invasion and post-invasion events. Int J Med Microbiol 300:170–175. doi: 10.1016/j.ijmm.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 34.Barriere H, Bagdany M, Bossard F, Okiyoneda T, Wojewodka G, Gruenert D, Radzioch D, Lukacs GL. 2009. Revisiting the role of cystic fibrosis transmembrane conductance regulator and counterion permeability in the pH regulation of endocytic organelles. Mol Biol Cell 20:3125–3141. doi: 10.1091/mbc.E09-01-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J, Lu W, Guha S, Baltazar GC, Coffey EE, Laties AM, Rubenstein RC, Reenstra WW, Mitchell CH. 2012. Cystic fibrosis transmembrane conductance regulator contributes to reacidification of alkalinized lysosomes in RPE cells. Am J Physiol Cell Physiol 303:C160–C169. doi: 10.1152/ajpcell.00278.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deriy LV, Gomez EA, Zhang G, Beacham DW, Hopson JA, Gallan AJ, Shevchenko PD, Bindokas VP, Nelson DJ. 2009. Disease-causing mutations in the cystic fibrosis transmembrane conductance regulator determine the functional responses of alveolar macrophages. J Biol Chem 284:35926–35938. doi: 10.1074/jbc.M109.057372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohkuma S, Moriyama Y, Takano T. 1982. Identification and characterization of a proton pump on lysosomes by fluorescein-isothiocyanate-dextran fluorescence. Proc Natl Acad Sci U S A 79:2758–2762. doi: 10.1073/pnas.79.9.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mindell JA. 2012. Lysosomal acidification mechanisms. Annu Rev Physiol 74:69–86. doi: 10.1146/annurev-physiol-012110-142317. [DOI] [PubMed] [Google Scholar]
- 39.Haggie PM, Verkman AS. 2007. Cystic fibrosis transmembrane conductance regulator-independent phagosomal acidification in macrophages. J Biol Chem 282:31422–31428. doi: 10.1074/jbc.M705296200. [DOI] [PubMed] [Google Scholar]
- 40.Pewzner-Jung Y, Tavakoli Tabazavareh S, Grassme H, Becker KA, Japtok L, Steinmann J, Joseph T, Lang S, Tuemmler B, Schuchman EH, Lentsch AB, Kleuser B, Edwards MJ, Futerman AH, Gulbins E. 2014. Sphingoid long chain bases prevent lung infection by Pseudomonas aeruginosa. EMBO Mol Med 6:1205–1214. doi: 10.15252/emmm.201404075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomassen MJ, Demko CA, Wood RE, Tandler B, Dearborn DG, Boxerbaum B, Kuchenbrod PJ. 1980. Ultrastructure and function of alveolar macrophages from cystic fibrosis patients. Pediatr Res 14:715–721. doi: 10.1203/00006450-198005000-00003. [DOI] [PubMed] [Google Scholar]
- 42.Lee DA, Hoidal JR, Garlich DJ, Clawson CC, Quie PG, Peterson PK. 1984. Opsonin-independent phagocytosis of surface-adherent bacteria by human alveolar macrophages. J Leukoc Biol 36:689–701. [DOI] [PubMed] [Google Scholar]
- 43.Peng H, Li C, Kadow S, Henry BD, Steinmann J, Becker KA, Riehle A, Beckmann N, Wilker B, Li PL, Pritts T, Edwards MJ, Zhang Y, Gulbins E, Grassme H. 2015. Acid sphingomyelinase inhibition protects mice from lung edema and lethal Staphylococcus aureus sepsis. J Mol Med (Berl) 93:675–689. doi: 10.1007/s00109-014-1246-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Li X, Carpinteiro A, Goettel JA, Soddemann M, Gulbins E. 2011. Kinase suppressor of Ras-1 protects against pulmonary Pseudomonas aeruginosa infections. Nat Med 17:341–346. doi: 10.1038/nm.2296. [DOI] [PubMed] [Google Scholar]