

Abstract
The Vertebrate Genome Annotation (Vega) database (http://vega.sanger.ac.uk) has been designed to be a community resource for browsing manual annotation of finished sequences from a variety of vertebrate genomes. Its core database is based on an Ensembl-style schema, extended to incorporate curation-specific metadata. In collaboration with the genome sequencing centres, Vega attempts to present consistent high-quality annotation of the published human chromosome sequences. In addition, it is also possible to view various finished regions from other vertebrates, including mouse and zebrafish. Vega displays only manually annotated gene structures built using transcriptional evidence, which can be examined in the browser. Attempts have been made to standardize the annotation procedure across each vertebrate genome, which should aid comparative analysis of orthologues across the different finished regions.
INTRODUCTION
In 1999 the DNA sequence of chromosome 22, the first human chromosome to be fully sequenced, was published (1). It provided a snapshot of the complexity of genes within a chromosomal landscape and set the standard for manual annotation, which the rest of the community was to follow. Yet as sequencing methods improved and researchers wanted to analyse unfinished, as well as finished, sequence data, new automated annotation methods were established and genome browsers such as Ensembl (2) and the UCSC Genome Browser (3) provided automatic genome annotation for the draft human genome assembly finished in 2001 (4). After the announcement of the finishing of the human genome in 2003, attention turned to producing a gold standard manually curated view of the human gene set.
The Vertebrate Genome Annotation (Vega) database is specifically dedicated to the browsing and maintenance of manually annotated data. Initially designed to view the manual annotation produced by the Havana group at the Sanger Institute (http://www.sanger.ac.uk/HGP/havana/), the project has expanded to include the manual annotation from the major centres (including RIKEN, the Joint Genome Institute, Genoscope and Washington University Genome Sequencing Center) involved in the sequencing and annotation of the human genome. Currently, it contains the annotation for 10 human chromosomes (6, 7, 9, 10, 13, 14, 20, 22, X and Y), but as the public consortium aims to complete the publication of its analysis by the end of 2004, it is planned that Vega will contain the complete manual annotation of the human genome by the beginning of 2005. Manual annotation is currently more accurate at identifying splice variants, pseudogenes, polyadenylation [poly(A)] features, non-coding genes and complex gene arrangements and clusters than automated methods. At the time of writing, the Vega human database contains over 15 000 gene loci and approximately 29 500 transcripts. In addition, Vega contains manual annotation of other vertebrate species and it is possible to view small chromosomal regions, e.g. mouse Del36H (5) and non-contiguous finished clone annotation of zebrafish. Figure 1 represents an overview of the processes and software involved in producing the data shown in Vega.
Figure 1.
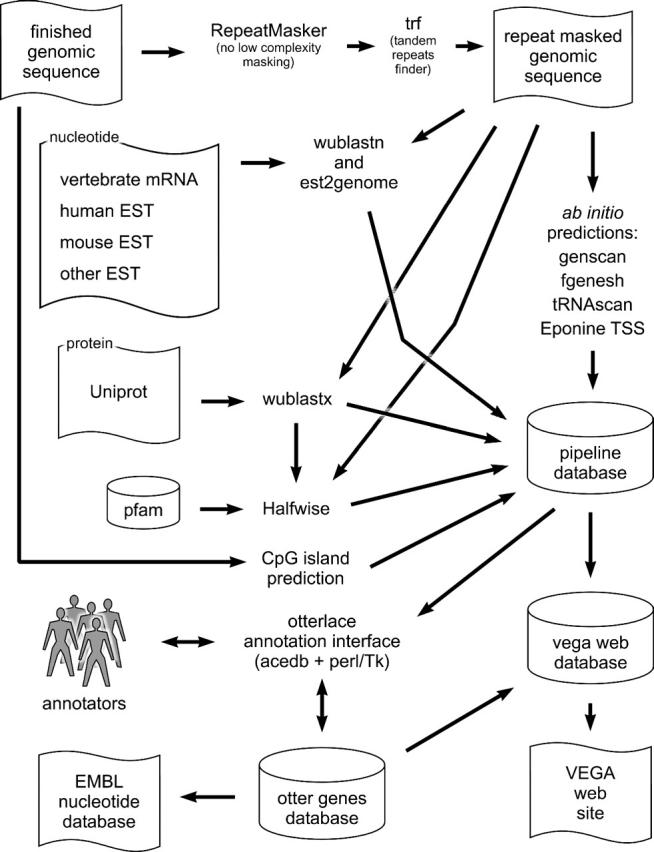
The VEGA annotation pipeline. The pipeline shown here is for human. The automated analysis for other species has slight differences. The searches are run on our computer farm and stored in an Ensembl MySQL database using the Ensembl analysis pipeline system (20). Nearly all searches and prediction algorithms are run on repeat masked sequence, the exception being CpG island prediction [see cpgreport in the EMBOSS (21) application suite]. RepeatMasker (http://ftp.genome.washington.edu/RM/RepeatMasker.html) is used to mask interspersed repeats, followed by TRF (22) to mask tandem repeats. Nucleotide sequence databases are searched with wuBLASTN (http://blast.wustl.edu), and significant hits are re-aligned to the unmasked genomic sequence using est2genome (23). The Uniprot protein database (http://www.uniprot.org) is searched with wuBLASTX, and the accession numbers of significant hits are looked up in the Pfam database (24). The hidden Markov models for Pfam protein domains are aligned against the genomic sequence using Genewise (25) to provide annotation of protein domains (Halfwise in the figure). We also run a number of ab initio prediction algorithms: genscan (26) and fgenesh (27) for genes, tRNAscan (28) to find tRNA genes and Eponine TSS (29), which predicts transcription start sites. The annotators use the Otterlace interface to create and edit genes, which are stored in the Otter database (13). Where predicted transcript structures from Ensembl are available these can be viewed from within the Otterlace interface and may be used as starting templates for gene curation. Annotation in the Otter database is submitted to the EMBL/GenBank/DDBJ nucleotide database. The database for the VEGA website is periodically created by a publishing process that involves the copying and reformatting of data from the Otter genes and automated pipeline databases.
GENE CLASSIFICATION AND STANDARDIZATION OF ANNOTATION
Since different research groups are performing high-quality manual annotation of different chromosomes, it has been essential to standardize a set of definitions to describe the annotation of different gene features. A common factor is that all annotated gene structures must be supported by transcriptional evidence, either from cDNA, expressed sequence tag (EST) or protein sequences. The following are the gene indices used in human chromosome 20 annotation (6) and adopted by the Vega database as standard:
Known genes: identical to human cDNA or protein sequences identified by LocusLink ID in the LocusLink database (http://www.ncbi.nlm.nih.gov/LocusLink/).
Novel genes: have an open reading frame (ORF) and are identical or homologous to known cDNAs (vertebrates) and/or proteins (all species).
Novel transcripts: similar to novel genes but no ORF can be unambiguously assigned.
Putative genes: homologous to spliced ESTs (vertebrates) but devoid of significant ORF/CDS.
Pseudogenes: sequences homologous to proteins (over ≥50% of the subject length) with a disrupted CDS and for which an active gene can generally be found at another locus.
These definitions have also been used in the recent annotation of chromosome 14 together with an additional classification ‘predicted genes’. Genoscope used this new classification to describe a gene based on ab initio predictions for which at least one exon is covered by biological or similarity data (unspliced ESTs, mouse or Tetraodon genomes or expression data from Rosetta) (7). These predicted genes as well as putative genes provide targets for experimental validation (8). Immunoglobulin segments and pseudogenes found on chromosomes 22 (1) and 14 (7) have also been given unique tags. These classifications have been extended across all the species in Vega with the only exception being that the specific model organism databases, e.g. the Mouse Genome Database (MGD) (http://www.informatics.jax.org/) (9) and the Zebrafish Information Network (ZFIN) nomenclature database (http://zfin.org/zf_info/nomen.html) (10), are used as the point of reference for known genes in place of LocusLink (11).
Using correct gene nomenclature is an important method for maintaining consistency in an annotation database, especially when comparing haplotypes or syntenic regions. The annotation staff involved in the Vega project, therefore, interact closely with the nomenclature committees from the Human Genome Organisation (HUGO, HGNC) (12), ZFIN and MGD. If an approved symbol is not available for a gene locus, an interim internal identifier is used, which is usually in the format clonename.number, e.g. RP11-694B14.5.
The locus and its associated transcripts and exons are also attributed stable, versioned database IDs (e.g. OTTHUMG00000017411, OTTHUMT00000046000), generated and tracked within the Otter database (see Figure 2). Whenever a gene locus is edited the version number will increase and the date of the change will be saved, allowing the user to find out when the annotation was last updated. Otter is an extended Ensembl database with an associated client/server system that is able to support interactive updating of annotation (13). The annotation stored in the Otter backend for the Vega database is either curated directly using Otterlace (a Perl/TK curation interface wrapped around Acedb) or via Otter XML uploads, such as from external groups. Multiple versions of any genome assembly can be stored in the system, with tools to migrate annotation to the latest assembly. Although the finished sequence is highly accurate (better than 1 base error in 10 000) over megabase regions, assemblies are frequently revised as chromosomal regions are finished, particularly in regions of genome duplication and frequently in conjunction with feedback from manual annotation. For reference sequences we can also expect assemblies to be revised as re-sequencing reveals a more common haplotype.
Figure 2.

Curated Locus Report giving information about the PAX2 locus on chromosome 10.
In an attempt to define a common standard for manual annotation across the human genome, collaborators involved in submitting annotation data to Vega have held a series of human annotation workshops (HAWK) (http://www.sanger.ac.uk/HGP/havana/hawk.shtml; see http://www.sanger.ac.uk/HGP/havana/docs/guidelines.pdf). Currently, there are many different transcript structures available in different browsers for various loci. With the aim of producing a single gold standard gene set, NCBI, UCSC, Ensembl and the Sanger Institute have started a collaboration to analyse the human gene sets produced by RefSeq, Ensembl and Vega and to define a non-redundant set of protein coding transcripts (HCDSs) that all collaborators can agree on.
ADDITIONAL FEATURES IN Vega
Unlike most of the browsers currently available containing automated gene builds, the manually annotated data shown in Vega does not have a particular emphasis on displaying only coding transcripts. Of transcripts annotated within Vega, ∼50% have no ORF associated with them. There are many possible properties of these transcripts, such as their being non-coding RNAs, transcripts involved in nonsense mediated mRNA decay (possibly regulating coding genes) (14) or partial transcripts where the ORF has not yet been experimentally determined. Each transcript constructed has spliced evidence associated with it, which can be viewed in Vega, so the user can assess the validity of each transcript. In addition to coding genes, ∼30% of gene structures are pseudogenes. These have been subdivided into unprocessed and processed categories in the recently finished chromosomes, so the user can identify whether the pseudogene has arisen from a duplication event or retrotransposition.
Polyadenylation sites and signals, identified manually by examining 3′ EST and cDNA data, are visible within the ContigView webpage of Vega (see Figure 3). The features are not associated with a particular transcript as it is difficult using 3′ EST data to associate a poly(A) feature with a particular alternative variant when they share the same 3′-untranslated region. Single nucleotide polymorphisms (SNPs) can also be viewed in ContigView and are mapped from the Glovar database (http://www.glovar.org/Homo_sapiens/) onto the clones within Vega. Glovar contains all the human dbSNP data in addition to SNPs derived from comparing public human reads from the trace repository (http://trace.ensembl.org/) with the current genome build. The functional classification of the SNPs (coding, untranslated region, Intronic, Other) is derived from mapping onto the Vega annotation. Currently SNPs are available only for the human chromosomes but they will eventually be available for all genomic sequences within Vega.
Figure 3.

ContigView webpage from human chromosome 6 Vega displaying poly(A) signals/sites and SNPs associated with SLC29A1 and HSPCB loci.
In addition to displaying the latest working assembly for each chromosome from individual public human sequencing consortia, Vega contains the annotated haplotype sequence available for the major histocompatability complex (MHC) region on chromosome 6 (15) (see Figure 4). The two common HLA haplotypes, PG and CX, are strongly associated with auto-immune diseases including type 1 diabetes and multiple sclerosis. The two haplotypes differ in their complement component C4 genes and MHC class II HLA-DRB genes, and this comparison can be easily made within ContigView. In a future Vega release we are planning to use the Ensembl Compara genome comparison framework to allow us to support MultiContigView pages so that haplotypes can be examined in parallel and to facilitate browsing the four more human MHC haplotype regions that will be available at the end of the year.
Figure 4.

Different chromosomes and regions annotated from the three different vertebrates currently available in Vega.
The mouse annotation browser within Vega, besides displaying finished regions from the reference mouse strain (C57BL/6), also displays three regions from the non-obese diabetic (Nod) strain (16) (see Figure 4). Since several insulin dependent diabetes (IDD) susceptibility loci have been mapped onto mouse chromosomes 1 and 3, comparison of the genomic sequence and genes between the two strains could be used to highlight functionally important SNPs (17). The zebrafish genome sequence will be finished and manually annotated solely by the Sanger Institute. Vega will be the main site for browsing the annotated data and at present there are 1164 loci, mostly from individual BAC and PAC clones. The genome is currently displayed in chromosomes/linkages groups 1–25 (see Figure 4). In addition, there are two ‘artificial’ chromosomes, U containing all the clones that could not be mapped onto the chromosomes yet, and AB containing clones from the AB strain. Clones that have not yet been annotated are displayed with all their features derived from automated computational analysis (repeat masking, ab initio gene predictions, BLAST searches, etc.) but are shaded in grey to avoid confusion with the annotated ones.
ACCESSING AND QUERYING DATA
The Vega browser, which is based on the Ensembl web code and infrastructure, provides a number of standard entry points such as sequence similarity (BLAST and SSAHA search) and keyword search. Data can be downloaded using ExportView, which can dump data in a variety of formats including FASTA, Gene Feature Format (GFF) and as flat files. Annotation can also be accessed directly via distributed annotation server (DAS) data sources. At present, we do not directly provide data mining via BioMart (http://www.ebi.ac.uk/biomart/) since Vega is designed to be updated weekly, which currently makes rebuilding BioMart impractical. It is possible to use EnsMart (18), available at Ensembl (http://www.ensembl.org/Multi/martview), to query a recent version of the gene structures from the Vega human database (which are displayed on the Ensembl website in ContigView). However, the Vega annotation shown in Ensembl would have been mapped from the latest chromosome assembly, upon which the annotation was curated and which is displayed in Vega, onto the current international genome assembly, which inevitably lags behind. If the assembly in Ensembl differs from that in Vega, only the annotation that can be cleanly transferred is present. For the informatician, a more comprehensive search of the Vega data can be performed using the Ensembl API (http://www.ensembl.org/Docs).
FEEDBACK AND SUBMITTING DATA
Vega is a community annotation database and feedback from researchers is essential to produce a gold standard annotation of the genomes available. Therefore, a webform is provided on the website (http://vega.sanger.ac.uk/helpdesk/index.html) to enable the user to contact the Vega team directly and improve the annotation if additional evidence is available. Since the browser is not restricted to annotation of whole genomes, we encourage users to contact vega@sanger.ac.uk to submit manual annotation of vertebrate finished regions they have sequenced, provided it has been peer reviewed and/or meets the HAWK standard for annotation.
FUTURE PLANS
We aim to have a fully manually annotated human genome available by the beginning of 2005. With community support, we hope to maintain the annotation and update on a weekly basis. We will also display the latest manual annotation of the regions as part of the ENCODE project (http://www.genome.gov/10005107) (19). In addition, annotated sequences from the mouse and zebrafish genomes, finished at the Sanger Institute or by the public sequencing centres, will be released on a chromosome basis in Vega. In collaboration with the MHC consortium we are also planning to release additional human MHC haplotypes, as well as MHC regions from dog, cat, pig and rat. Using the comparative analysis pipeline designed by the Ensembl team we are looking into producing comparative views for these data to enable the user to browse easily among different species.
Acknowledgments
ACKNOWLEDGEMENTS
We thank the Havana, Chromosome 22, Genoscope and WashU genome annotation groups for providing the annotation data that have been currently incorporated into Vega. We also thank the MHC haplotype consortium for providing sequence and analysis of the MHC haplotypes. We thank the Ensembl Project for the software that is the basis of the Vega website and the Otter client/server system and the Glovar project for providing support for SNPs on Vega.
REFERENCES
- 1.Dunham I., Shimizu,N., Roe,B.A., Chissoe,S., Hunt,A.R., Collins,J.E., Bruskiewich,R., Beare,D.M., Clamp,M., Smink,L.J. et al. (1999) The DNA sequence of human chromosome 22. Nature, 402, 489–495. [DOI] [PubMed] [Google Scholar]
- 2.Birney E., Andrews,T.D., Bevan,P., Caccamo,M., Chen,Y., Clarke,L., Coates,G., Cuff,J., Curwen,V., Cutts,T. et al. (2004) An overview of Ensembl. Genome Res., 14, 925–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kent W.J., Sugnet,C.W., Furey,T.S., Roskin,K.M., Pringle,T.H., Zahler,A.M. and Haussler,D. (2002) The human genome browser at UCSC. Genome Res., 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lander E.S., Linton,L.M., Birren,B., Nusbaum,C., Zody,M.C., Baldwin,J., Devon,K., Dewar,K., Doyle,M., FitzHugh,W. et al. (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 5.Mallon A.-M., Wilming,L., Weekes,J., Gilbert,J.G.R., Ashurst,J., Peyrefitte,S., Matthews,L., Cadman,M., McKeone,R., Sellick,C.A. et al. (2004) Organization and evolution of a gene-rich region of the mouse genome: A 12.7-Mb region deleted in the Del(13)Svea36H mouse. Genome Res., 14, 1888–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deloukas P., Matthews,L.H., Ashurst,J., Burton,J., Gilbert,J.G., Jones,M., Stavrides,G., Almeida,J.P., Babbage,A.K., Bagguley,C.L. et al. (2001) The DNA sequence and comparative analysis of human chromosome 20. Nature, 414, 865–871. [DOI] [PubMed] [Google Scholar]
- 7.Heilig R., Eckenberg,R., Petit,J.L., Fonknechten,N., Da Silva,C., Cattolico,L., Levy,M., Barbe,V., De Berardinis,V., Ureta-Vidal,A. et al. (2003) The DNA sequence and analysis of human chromosome 14. Nature, 421, 601–607. [DOI] [PubMed] [Google Scholar]
- 8.Ashurst J.L. and Collins,J.E. (2003) Gene annotation: prediction and testing. Annu. Rev. Genomics Hum. Genet., 4, 69–88. [DOI] [PubMed] [Google Scholar]
- 9.Blake J.A., Richardson,J.E., Bult,C.J., Kadin,J.A. and Eppig,J.T. (2003) MGD: the Mouse Genome Database. Nucleic Acids Res., 31, 193–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sprague J., Clements,D., Conlin,T., Edwards,P., Frazer,K., Schaper,K., Segerdell,E., Song,P., Sprunger,B. and Westerfield,M. (2003) The Zebrafish Information Network (ZFIN): the zebrafish model organism database. Nucleic Acids Res., 31, 241–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wheeler D.L., Church,D.M., Edgar,R., Federhen,S., Helmberg,W., Madden,T.L., Pontius,J.U., Schuler,G.D., Schriml,L.M., Sequeira,E. et al. (2004) Database resources of the National Center for Biotechnology Information: update. Nucleic Acids Res., 32, D35–D40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wain H.M., Lush,M.J., Ducluzeau,F., Khodiyar,V.K. and Povey,S. (2004) Genew: the Human Gene Nomenclature Database, 2004 updates. Nucleic Acids Res., 32, D255–D257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Searle S.M., Gilbert,J., Iyer,V. and Clamp,M. (2004) The otter annotation system. Genome Res., 14, 963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winter J., Lehmann,T., Krauss,S., Trockenbacher,A., Kijas,Z., Foerster,J., Suckow,V., Yaspo,M.L., Kulozik,A., Kalscheuer,V. et al. (2004) Regulation of the MID1 protein function is fine-tuned by a complex pattern of alternative splicing. Hum. Genet., 114, 541–552. [DOI] [PubMed] [Google Scholar]
- 15.Stewart C.A., Horton,R., Allcock,R.J., Ashurst,J.L., Atrazhev,A.M., Coggill,P., Dunham,I., Forbes,S., Halls,K., Howson,J.M. et al. (2004) Complete MHC haplotype sequencing for common disease gene mapping. Genome Res., 14, 1176–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hill N.J., Lyons,P.A., Armitage,N., Todd,J.A., Wicker,L.S. and Peterson,L.B. (2000) NOD Idd5 locus controls insulitis and diabetes and overlaps the orthologous CTLA4/IDDM12 and NRAMP1 loci in humans. Diabetes, 49, 1744–1747. [DOI] [PubMed] [Google Scholar]
- 17.Wicker L.S., Chamberlain,G., Hunter,K., Rainbow,D., Howlett,S., Tiffen,P., Clark,J., Gonzalez-Munoz,A., Cumiskey,A.M., Rosa,R.L. et al. (2004) Fine mapping, gene content, comparative sequencing, and expression analyses support Ctla4 and Nramp1 as candidates for Idd5.1 and Idd5.2 in the nonobese diabetic mouse. J. Immunol., 173, 164–173. [DOI] [PubMed] [Google Scholar]
- 18.Kasprzyk A., Keefe,D., Smedley,D., London,D., Spooner,W., Melsopp,C., Hammond,M., Rocca-Serra,P., Cox,T. and Birney,E. (2004) EnsMart: a generic system for fast and flexible access to biological data. Genome Res., 14, 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The ENCODE Project Consortium (2004) The ENCODE (ENCyclopedia Of DNA Elements) Project. Science, 306, 636–640. [DOI] [PubMed] [Google Scholar]
- 20.Potter S.C., Clarke,L., Curwen,V., Keenan,S., Mongin,E., Searle,S.M.J., Stabenau,A., Storey,R. and Clamp,M. (2004) The Ensembl Analysis Pipeline. Genome Res., 14, 934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rice P., Longden,I. and Bleasby,A. (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet., 16, 276–277. [DOI] [PubMed] [Google Scholar]
- 22.Benson G. (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res., 27, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mott R. (1997) EST_GENOME: a program to align spliced DNA sequences to unspliced genomic DNA. Comput. Appl., 13, 477–478. [DOI] [PubMed] [Google Scholar]
- 24.Bateman A., Coin,L., Durbin,R., Finn,R.D., Hollich,V., Griffiths-Jones,S., Khanna,A., Marshall,M., Moxon,S., Sonnhammer,E.L.L. et al. (2004) The Pfam Protein Families Database. Nucleic Acids Res., 32, D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Birney E., Clamp,M. and Durbin,R. (2004) GeneWise and Genomewise. Genome Res., 14, 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burge C. and Karlin,S. (1997) Prediction of complete gene structures in human genomic DNA. J. Mol. Biol., 268, 78–94. [DOI] [PubMed] [Google Scholar]
- 27.Salamov A.A. and Solovyev,V.V. (2000) Ab initio gene finding in Drosophila genomic DNA. Genome Res., 10, 516–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowe T.M. and Eddy,S.R. (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res., 25, 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Down T.A. and Hubbard,T.J.P. (2002) Computational detection and location of transcription start sites in mammalian genomic DNA. Genome Res., 12, 458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]