


Abstract
The Ensembl (http://www.ensembl.org/) project provides a comprehensive and integrated source of annotation of large genome sequences. Over the last year the number of genomes available from the Ensembl site has increased by 7 to 16, with the addition of the six vertebrate genomes of chimpanzee, dog, cow, chicken, tetraodon and frog and the insect genome of honeybee. The majority have been annotated automatically using the Ensembl gene build system, showing its flexibility to reliably annotate a wide variety of genomes. With the increased number of vertebrate genomes, the comparative analysis provided to users has been greatly improved, with new website interfaces allowing annotation of different genomes to be directly compared. The Ensembl software system is being increasingly widely reused in different projects showing the benefits of a completely open approach to software development and distribution.
INTRODUCTION
Genome sequences provide a natural framework about which to organize biological data. In the few years in which they have been available, genome databases have proved invaluable resources to researchers. Ensembl provides one of the most popular sources of automatic analysis and integration of large genome sequence data. It now contains 16 genomes, 7 of which have been added during the last year. These include 11 vertebrates: human, chimpanzee, mouse, rat, dog, cow, chicken, fugu, zebrafish, tetraodon and frog; two worms: Caenorhabditis briggsae and Caenorhabditis elegans; and three insects: fruitfly, mosquito and honeybee.
Ensembl provides access to these data in a variety of ways to suit different audiences and types of use. The largest number of researchers use the Ensembl website (http://www.ensembl.org/) and can rapidly locate individual items of interest either by entering keywords or from the built-in sequence similarity search interface. For cases where researchers are working with large groups of items, such as all of a particular class of genes, Ensembl provides a web-based data-mining interface called EnsMart as an alternative to browsing individual web pages. For bioinformaticians, Ensembl provides access to all the data behind the Ensembl website both as downloadable datasets and via direct access to databases containing that data which are hosted on the Ensembl site (access using mysql to ensembldb.ensembl.org, user anonymous). The complete software system for manipulating and storing genome information that has been created by the project is also freely available with source code for all to use.
This paper briefly outlines some of the main developments of the Ensembl project since the report last year (1) and their relevance to researchers. For information about new features and data contained in the monthly updates of Ensembl, researchers are recommended to read the ‘what's new’ pages accompanying every release. For more detailed information about Ensembl, researchers are referred to the series of papers published last year that describe both technical aspects of the software implementation and the scientific aspects of the genome annotation system (2–11).
GENE ANNOTATION
A core part of Ensembl is its automatic gene build system, which is used to consistently annotate genomes for which there is no curated geneset. The genome sequences of human, chimpanzee, mouse, rat, chicken, fugu, zebrafish, C.briggsae, mosquito and honeybee have been annotated in this way. Annotation of the newest genomes, dog, cow and frog, is in progress. The exceptions are fruitfly [annotation imported from flybase (12)], C.elegans [annotation imported from wormbase (13)] and tetraodon (annotation provided by Genoscope). Ensembl genesets have formed the basis for the initial analysis and publication of most vertebrate genomes. During the last year, the rat (14), C.briggsae (15) and chicken (16) genomes have been published.
Full descriptions of the gene build system (7) and the genewise family of algorithms that it uses (8) have recently been published. Briefly, the system is mainly based on building gene models from initial alignments of protein and cDNA sequences to a genome sequence. Where a genome has limited species-specific cDNA data, the number of genes in the geneset will reflect the number of homologous cDNAs from other related organisms that can be aligned. Where expressed sequence tag (EST) collections are thought to contain a significant number of artefact sequences, they are considered a less reliable source of evidence for gene structures and so separate gene builds are created from them (9). Where ESTs have been generated by a small number of groups and are thought to be of consistent quality they are used in the main gene build, but gene models are only built from them if there is no other evidence. This approach has been used in the chicken and honeybee gene builds.
Over the year, the flexibility of the gene build system has been exploited to create genesets for genomes such as zebrafish, honeybee and chicken, which have varied amounts of species-specific cDNA data. Of these three gene builds, the most difficult one has been for honeybee, which is evolutionarily very distant from other sequenced organisms. Zebrafish and chicken gene builds are much more complete, being evolutionarily closer to other sequenced vertebrates. Experimental validation of a randomly selected sample of gene models from the geneset made for the chicken genome (17) shows a low false positive rate of ∼4% (Eyras et al., submitted for publication). As cDNA resources for these genomes increase, it is fully expected that the number of genes in their genesets will increase until it is similar to those of comparable organisms.
As the genome sequence of human has been finished, genesets for individual chromosomes have been manually curated and published [for a review see (18)]. Ensembl is part of a new international collaboration to refine the human geneset involving the Havana group (19) [which provides most of the curated annotation in the Vega database (20)], the NCBI groups [that curate RefSeq (21) and generate automatic gene builds], the UCSC browser group (22) and Uniprot (23). The aim is to resolve transcript sequence differences and generate and maintain a set of human genes with stable identifiers, where the CDS part of the gene structure can be agreed between all groups. The process of comparison of genesets has proved very fruitful and is leading to improved automatic gene building methods for both the Ensembl and NCBI. It is anticipated that this agreed geneset will progressively increase in size as the entire human genome is fully curated. The current Ensembl human gene build is already benefiting from this comparison, as where the CDS part of a Vega curated transcript or Ensembl transcript and a NCBI transcript agree perfectly and are complete (from ATG to stop codon), these propagate automatically into the next Ensembl geneset.
COMPARATIVE ANALYSIS
A major ongoing focus of the Ensembl project is to increase the integration between genomes through comparative analysis. Ensembl currently contains three types of similarity information: (i) Ensembl family entries (ENSF identifiers) cluster Ensembl peptides across all annotated genomes together with UniProt metazoan entries on the basis of protein similarity. (ii) Pairwise DNA similarity alignments are stored for each groups of genomes that can be aligned (i.e. vertebrates, insects and worms). These alignments are either generated locally using BLASTZ (24) and a variety of other algorithms or are imported BLASTZ alignments downloaded from the UCSC (22). These alignments are also grouped to generate large-scale synteny blocks. (3) Putative orthologues relationships are stored across all annotated genomes, generated using automatic algorithms that take into account transcript similarity (seeded using the widely used reciprocal best match approach) and synteny. During the year the amount of comparative data provided by Ensembl has grown considerably, both from the increase in the number of vertebrate genomes and the development of a computation pipeline to generate these data more rapidly. For example, alignment data for almost all possible pairwise DNA similarity comparisons is now available.
The most prominent addition to the website is multicontigview (Figure 1), which allows regions of genome sequence from multiple species to be viewed aligned to each other, something that was previously only possible using the external Apollo java browser (25). As well as making these alignments accessible to a wider audience, multicontigview allows the alignment of as many genomes as desired and is able to show in a single display both DNA similarity and putative orthologue relationships. Multicontigview is complementary to the display of regions of conservation in contigview. Whereas the latter is useful to identify important regions in a single genome, multicontigview allows researchers to compare annotation between genomes to look for places where annotation may be missing.
Figure 1.
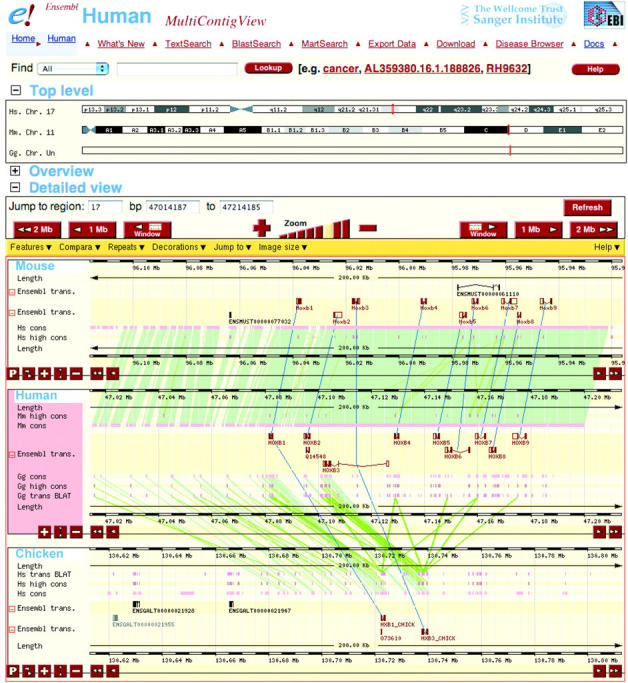
Screenshot of Ensembl multicontigview. The view shows genome sequence with annotation from human, mouse and chicken, aligned according to DNA–DNA similarity, shown in green. Pairwise similarity is shown between the ‘primary’ genome (human in this case) and each of the other genomes. Menus allow additional genomes to be added to the display. The ‘P’ button allows a different genome to be selected as the primary one. Genes automatically identified as putative orthologues are linked by blue lines. The region shown is centred around the HOXB3 gene in the HOX cluster on human chromosome 17 and is shown to be syntenic with a region on mouse chromosome 11. All Ensembl known gene structures are conserved and have been correctly identified as orthologues. Two novel Ensembl gene structures predicted in mouse are not seen in human. It would be interesting to investigate the corresponding region in human to understand why they were not predicted there. Features such as alignments to cDNAs and proteins can be turned on using the menus to facilitate such a comparison. Putative orthologue prediction and DNA similarity show a much weaker and incomplete link to a region in the chicken genome; however this is on chromosome Un, which is a fake chromosome composed of fragments that could not be mapped onto chromosomes in the current assembly. Whereas the chicken fragment contains HOXB3 and HOXB1, HOXB4 and others are absent. The putative chicken orthologue for human HOXB4 is found in another chicken fragment in the fake Un chromosome (data not shown), suggesting that the chicken equivalent of the human chromosome 17 HOX cluster is fragmented in the current chicken assembly.
With a comprehensive set of comparative data now available, links to comparative data views have been added throughout the Ensembl website. For example, in geneview, putative orthologues are listed in all other organisms and links are provided to a multicontigview display showing putative orthologues, aligned across genome sequence and to alignview showing alignments of homologous transcripts.
ENSEMBL WEBSITE
As well as the major new website view multicontigview discussed above, the core web code has also been substantially rewritten to make it a more modular set of software components and thereby improve its flexibility (4). This has allowed new data views to be constructed quickly in response to user requests, such as genesnpview (Figure 2), which shows information about single nucleotide polymorphisms (SNPs) for a single gene at genome, transcript and translation level in a single page. All of the data on genesnpview is available on other web pages, but researchers working in detail on individual genes have found it very convenient to have a single web page containing everything relating to the genome variation around a single gene structure. The Ensembl website has a large number of different data ‘views’ and to make it easier for users to discover and explore them, the revamped sitemap page (http://www.ensembl.org/sitemap/) provides links to each of them with example entry points that are auto generated to ensure they continue to work when the underlying genome data is updated. The sitemap pages for each species only show the ‘views’ that are available for a particular genome, for example there is no mapview for fugu as the genome sequence is currently available only as a set of unmapped fragments that have not been positioned on chromosomes.
Figure 2.

Screenshot of Ensembl genesnpview. This new gene-centric view shows in a single display the genomic context of a gene and its surrounding SNPs. The figure shows the region of the human genome around the HOXB3 gene in the HOX cluster on chromosome 17. The display shows three different resolutions: the genes over a 270 kb region are shown around HOXB3; the HOXB3 gene itself (gene id ENSG00000120093) and the HOXB3 transcript (transcript id ENST00000311626) with intragenic sequence and introns truncated so that it is mainly CDS and untranslated region (UTR) sequence that is shown. By default the flanking regions are truncated to 50 bp. This can be changed with the ‘Context’ menu and in this case has been set to 200 bp, revealing six intronic SNPs. It can be seen that the CDS of the transcript includes one known protein domain (the Pfam PF00046 homeobox domain). There are only two SNPs that fall within the CDS and only one of these is non-synonyomous leading to a proline (P) to threonine (T) amino acid change in the second exon as a result of an A to C base change. There is a further flanking SNP (C to T change) in the 3′-UTR. A table immediately below the figure provides more information about the SNPs that intersect the transcript being viewed. The three menu bars ‘SNP class’, ‘Validation’ and ‘SNP type’ allow the SNPs being displayed to be filtered. If there were multiple transcripts for the gene selected, they would each be displayed. The view thus combines data in a single view that can partly be found in contigview, transview, protview and snpview.
There is far more biological data that can be displayed as features on genomic coordinates than it is practical to import into core Ensembl databases. For example, laboratories around the world are generating large amounts of sequence data that can be aligned to a reference genome and both experimentalists and bioinformaticians are generating annotation of features in promoters. Rather than centralize the integration of these data, we have enabled Ensembl with the DAS (distributed annotation system) (26) to allow researchers to view data of their choice in the context of the annotation provided by Ensembl. This might be data they have generated themselves, but it can also be third-party data. DAS enabled viewers such as Ensembl contigview can be configured to display data from any DAS server as an extra track on the display. Users can configure this using the DAS menu of contigview. In cases where researchers have data that they wish to display, but do not want to setup their own DAS server, they can also upload their data in a simple flat file format into the Ensembl DAS server using the same menu.
During the year DAS support has been extended. DAS sources can now also be attached to protview, allowing external annotation on protein sequences to be displayed just as it can be on genomic coordinates in contigview. Ensembl has also recently extended the DAS specification to add a new DAS data type based on identifiers (such as gene names, Uniprot identifiers, etc.) rather than coordinates, referred to as geneDAS. This allows textual annotation such as descriptions from UniProt (23) and InterPro (27) and references from pubmed to be added via DAS to geneview and protview pages.
ENSEMBL INFRASTRUCTURE
The Ensembl software system provides an efficient way of representing genome data in a relational database and providing access to it via an object-oriented application programming interface (API) (3). This API is used by computational pipelines (5) to generate and store genome annotation. The API is also used by the website (4) and EnsMart data-mining system (11) to provide researchers with access to the database. Bioinformaticans can use the API to access ensembl databases remotely (from ensembldb.ensembl.org) or local databases containing their own data.
The database representation and API are being continuously developed to address bottlenecks affecting website and pipeline performance and increase flexibility. For example, during the year there was a substantial change to the database schema to make it easier to support multiple haplotype sequences and different genome assemblies. These features have been used to handle the pseudoautosomal regions shared between the human X and Y chromosome and the alternative MHC haplotype sequence on human chromosome 6. The API was also extended to provide support for generic mapping operations between genome assemblies that were previously carried out in external software. This should make it easier to transfer and compare gene annotation across genome assemblies, which should in turn lead to better stability of gene stable identifiers for users. Efforts have also been made to increase the degree of automation of processes such as gene building and comparative analysis by extending the pipeline system. This automation is essential to allow Ensembl to scale to the increasing number of vertebrate genomes, but also makes it easier for others to adopt Ensembl technology.
As part of the development of the software infrastructure to support comparative analysis, a rationalization of the database structure has been carried out, so that all inter-genome data is contained in a single database, ensembl-compara. The availability of the this database for download as well as external access (ensembldb.ensembl.org), alongside the core annotation databases for each organism, provides a rich source of structured and pre-calculated data that can be used as a starting point for many comparative genome bioformatics projects. APIs to ensembl-compara are provided in Perl and Java from Ensembl and as well as through BioJava (28).
REUSING ENSEMBL SOFTWARE
Ensembl has developed a strong network of bioinformatician users in both academia and industry and Ensembl software is being installed both to mirror Ensembl generated data and used as a software foundation for user projects. For example, NASC, the European Arabidopsis stock centre, has set up an Ensembl browser for the Arabidopsis genome (http://atensembl.arabidopsis.info/); the Temasek Life Sciences Laboratory in Singapore is using Ensembl to both generate and display annotation for two Ciona genomes savingyi and intestinalis (http://www2.bioinformatics.tll.org.sg/) and a researcher at the Swiss Institute of Bioinformatics has setup an Ensembl browser for the cotton pathogen Ashbya gossypii genome (http://agd.unibas.ch/).
Ensembl technology is also being extensively reused at the Wellcome Trust Genome Campus to support other projects. At the European Bioinformatics Institute (EBI) the EnsMart data-mining system has been spun out into a separate project (BioMart; http://www.ebi.ac.uk/biomart/) to enable it to provide data-mining front ends to a variety of databases [e.g. Uniprot and the Macromolecular Structure Database (MSD)] and allow chained data-mining queries between them. At the Sanger Institute, both the Vega (Vertebrate Genome Annotation Database, http://vega.sanger.ac.uk/) (10,20) and Glovar (Global Variation Database, http://www.glovar.org/) projects are reusing parts of the Ensembl system for databases and web front ends. The Epigenomics Consortium (29) (http://www.epigenome.org/) has built a viewer based on Ensembl web drawing code and DAS sources (http://www.sanger.ac.uk/PostGenomics/epigenome/). DECIPHER (DatabasE of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources) and other projects are starting to embed images of karyotypes, gene regions etc. into their web pages, which are generated dynamically from Ensembl databases or DAS sources using Ensembl drawing code.
In a world with a plethora of databases, these examples of reuse should be a positive trend both for researchers and bioinformaticians. Not only can bioinformaticians develop interfaces to new data faster through software reuse, but researchers using websites built from common components are more likely to already be familiar with the interface from other sites. Similarly, using DAS servers to reuse existing data when presenting it in new ways should improve data consistency for researchers, since the data remain in a single location and when it is updated the view in all interfaces built upon it changes simultaneously.
FUTURE DIRECTIONS
Ensembl remains focused on providing an integrated dataset and website covering all vertebrate genomes and a genome information infrastructure of use to many researchers. It continues to be a challenge to evolve the system to handle and represent the ever increasing number of genomes. In particular over the next year, a number of low coverage whole genome shotgun vertebrate genomes are expected. Development of comparative genome analysis will continue to be a major focus. For example, the present pairwise storage of genome comparisons will not scale well to the expected number of new genomes, so new ways to calculate and represent the data are being developed. Finally, with the anticipated completion of the human HapMap project a further rich source of variation data will become available, which Ensembl will integrate.
CONTACTING ENSEMBL
Ensembl is a joint project of the EBI and the Wellcome Trust Sanger Institute (WTSI), both of which are located on the Wellcome Trust Genome Campus, Cambridge, UK. To receive announcements about updates, subscribe to the ‘announce’ mailing list: majordomo@ebi.ac.uk ‘subscribe ensembl-announce’. To follow the day-to-day development of Ensembl, subscribe to the ‘development’ mailing list: majordomo@ebi.ac.uk ‘subscribe ensembl-dev’. Requests for information and support can be sent to helpdesk@ensembl.org, which is a fully supported helpdesk. Extensive additional documentation can be found on the Ensembl website, including installation guides and tutorials, both about using the software system and the web interface.
Acknowledgments
ACKNOWLEDGEMENTS
The Ensembl project is principally funded by the Wellcome Trust with additional funding from EMBL, NIH-NIAID and BBSRC. We are grateful to users of our website and the developers on our mailing lists for much useful feedback and discussion.
REFERENCES
- 1.Birney E., Andrews,D., Bevan,P., Caccamo,M., Cameron,G., Chen,Y., Clarke,L., Coates,G., Cox,T., Cuff,J. et al. (2004) Ensembl 2004. Nucleic Acids Res., 32, D468–D470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Birney E., Andrews,T.D., Bevan,P., Caccamo,M., Chen,Y., Clarke,L., Coates,G., Cuff,J., Curwen,V., Cutts,T. et al. (2004) An overview of Ensembl. Genome Res., 14, 925–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stabenau A., McVicker,G., Melsopp,C., Proctor,G., Clamp,M. and Birney,E. (2004) The Ensembl core software libraries. Genome Res., 14, 929–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stalker J., Gibbins,B., Meidl,P., Smith,J., Spooner,W., Hotz,H.R. and Cox,A.V. (2004) The Ensembl Web site: mechanics of a genome browser. Genome Res., 14, 951–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Potter S.C., Clarke,L., Curwen,V., Keenan,S., Mongin,E., Searle,S.M., Stabenau,A., Storey,R. and Clamp,M. (2004) The Ensembl analysis pipeline. Genome Res., 14, 934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cuff J.A., Coates,G.M., Cutts,T.J. and Rae,M. (2004) The Ensembl computing architecture. Genome Res., 14, 971–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curwen V., Eyras,E., Andrews,T.D., Clarke,L., Mongin,E., Searle,S.M. and Clamp,M. (2004) The Ensembl automatic gene annotation system. Genome Res., 14, 942–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birney E., Clamp,M. and Durbin,R. (2004) GeneWise and Genomewise. Genome Res., 14, 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eyras E., Caccamo,M., Curwen,V. and Clamp,M. (2004) ESTGenes: alternative splicing from ESTs in Ensembl. Genome Res., 14, 976–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Searle S.M., Gilbert,J., Iyer,V. and Clamp,M. (2004) The otter annotation system. Genome Res., 14, 963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kasprzyk A., Keefe,D., Smedley,D., London,D., Spooner,W., Melsopp,C., Hammond,M., Rocca-Serra,P., Cox,T. and Birney,E. (2004) EnsMart: a generic system for fast and flexible access to biological data. Genome Res., 14, 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flybase Consortium (2003) The FlyBase database of the Drosophila genome projects and community literature. Nucleic Acids Res., 31, 172–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris T.W., Chen,N., Cunningham,F., Tello-Ruiz,M., Antoshechkin,I., Bastiani,C., Bieri,T., Blasiar,D., Bradnam,K., Chan,J.et al. (2004) WormBase: a multi-species resource for nematode biology and genomics. Nucleic Acids Res., 32, D411–D417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rat Genome Sequencing Consortium (2004) Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature, 428, 493–521. [DOI] [PubMed] [Google Scholar]
- 15.Stein L.D., Bao,Z., Blasiar,D., Blumenthal,T., Brent,M.R., Chen,N., Chinwalla,A., Clarke,L., Clee,C., Coghlan,A. et al. (2003) The Genome Sequence of Caenorhabditis briggsae: a Platform for Comparative Genomics. PLoS Biol., 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.International Human Genome Sequencing Consortium (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 17.International Chicken Genome Sequencing Consortium (2004) Sequencing and comparative analysis of the chicken genome. Nature, in press. [Google Scholar]
- 18.International Human Genome Sequencing Consortium (2004) Finishing the euchromatic sequence of the human genome. Nature, 431, 931–945. [DOI] [PubMed] [Google Scholar]
- 19.Ashurst J.L. and Collins,J.E. (2003) Gene annotation: prediction and testing. Annu. Rev. Genomics Hum. Genet., 4, 69–88. [DOI] [PubMed] [Google Scholar]
- 20.Ashurst J., Chen,C.-K., Gilbert,J., Jekosch,K., Keenan,S., Meidl,P., Searle,S., Stalker,J., Storey,R., Trevanion,S. et al. (2004) The Vertebrate Genome Annotation (Vega) database. Nucleic Acids Res., 33, D459–D465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pruitt K.D., Tatusova,T. and Maglott,D.R. (2003) NCBI Reference Sequence project: update and current status. Nucleic Acids Res., 31, 34–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karolchik D., Baertsch,R., Diekhans,M., Furey,T.S., Hinrichs,A., Lu,Y.T., Roskin,K.M., Schwartz,M., Sugnet,C.W., Thomas,D.J. et al. (2003) The UCSC Genome Browser Database. Nucleic Acids Res., 31, 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leinonen R., Diez,F.G., Binns,D., Fleischmann,W., Lopez,R. and Apweiler,R. (2004) UniProt Archive. Bioinformatics, in press. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz S., Kent,W.J., Smit,A., Zhang,Z., Baertsch,R., Hardison,R.C., Haussler,D. and Miller,W. (2003) Human–mouse alignments with BLASTZ. Genome Res., 13, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lewis S.E., Searle,S.M., Harris,N., Gibson,M., Lyer,V., Richter,J., Wiel,C., Bayraktaroglir,L., Birney,E., Crosby,M.A. et al. (2002) Apollo: a sequence annotation editor. Genome Biol., 3, RESEARCH0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dowell R.D., Jokerst,R.M., Day,A., Eddy,S.R. and Stein,L. (2001) The Distributed Annotation System. BMC Bioinformatics, 2, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mulder N.J., Apweiler,R., Attwood,T.K., Bairoch,A., Barrell,D., Bateman,A., Binns,D., Biswas,M., Bradley,P., Bork,P. et al. (2003) The InterPro Database, 2003 brings increased coverage and new features. Nucleic Acids Res., 31, 315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pocock M.R., Down,T. and Hubbard,T.J.P. (2000) BioJava: Open Source Components for Bioinformatics. Sigbio Newslett., 20, 10–12. [Google Scholar]
- 29.Novik K.L., Nimmrich,I., Genc,B., Maier,S., Piepenbrock,C., Olek,A. and Beck,S. (2002) Epigenomics: genome-wide study of methylation phenomena. Curr. Issues Mol. Biol., 4, 111–128. [PubMed] [Google Scholar]