

Abstract
Chronic gastritis causes significant morbidity and mortality in captive cheetahs but is rare in wild cheetahs despite colonization by abundant spiral bacteria. This research aimed to identify the Helicobacter species that were associated with gastritis in captive cheetahs but are apparently commensal in wild cheetahs. Helicobacter species were characterized by PCR amplification and sequencing of the 16S rRNA, urease, and cagA genes and by transmission electron microscopy of frozen or formalin-fixed paraffin-embedded gastric samples from 33 cheetahs infected with Helicobacter organisms (10 wild without gastritis and 23 captive with gastritis). Samples were screened for mixed infections by denaturant gel gradient electrophoresis of the 16S rRNA gene and by transmission electron microscopy. There was no association between Helicobacter infection and the presence or severity of gastritis. Eight cheetahs had 16S rRNA sequences that were most similar (98 to 99%) to H. pylori. Twenty-five cheetahs had sequences that were most similar (97 to 99%) to “H. heilmannii” or H. felis. No cheetahs had mixed infections. The ultrastructural morphology of all bacteria was most consistent with “H. heilmannii,” even when 16S rRNA sequences were H. pylori-like. The urease gene from H. pylori-like bacteria could not be amplified with primers for either “H. heilmannii” or H. pylori urease, suggesting that this bacteria is neither H. pylori nor “H. heilmannii.” The cagA gene was not identified in any case. These findings question a direct role for Helicobacter infection in the pathogenesis of gastritis and support the premise that host factors account for the differences in disease between captive and wild cheetah populations.
Since the initial isolation of Helicobacter pylori from humans and its association with gastritis and peptic ulceration (23), Helicobacter spp. have been isolated from an ever-expanding range of host species (38). While Helicobacter pylori infection in humans has been associated with chronic gastritis, peptic ulceration, gastric adenocarcinoma, and lymphoma (23, 31, 32), Helicobacter pathogenicity in many species is less clear.
Worldwide, the majority of captive cheetahs (Acinonyx jubatus) have a progressive gastritis that causes vomiting, weight loss, and failure to thrive and is associated with Helicobacter infection (10, 25, 26). Moderate to severe gastritis was present in greater than 70% of cheetahs that have died since 1995 within the North American captive population. Many cheetahs develop systemic amyloidosis (type AA) secondary to gastritis that results in renal failure, a leading cause of death among captive cheetahs (30). Within the South African captive cheetah population, gastritis was a major cause of death or the reason for euthanasia in 69% of cheetahs (26).
In 1992, genetic and morphological analysis of spiral bacteria in cheetahs from a single captive facility identified a novel species, Helicobacter acinonychis (9). In these cheetahs, a second nonculturable spiral bacterium morphologically similar to “H. heilmannii” was identified by electron microscopy in some cases (11). However, there was no difference between the severity of gastritis in cheetahs colonized with H. acinonychis, “H. heilmannii,” or and coinfected animals.
Since the initial isolation of H. acinonychis, culture attempts have been unsuccessful from many cheetahs despite the presence of spiral bacteria histologically (K. Eaton, personal communication). This suggests that H. acinonychis may not be the most common Helicobacter sp. infecting captive cheetahs and that there may be additional unculturable species of Helicobacter important in the development of gastritis. Interestingly, gastritis is rare in wild cheetahs despite the presence of abundant spiral bacteria (L. Munson, unpublished data). Therefore, this study aimed to identify the Helicobacter spp. associated with gastritis in captive cheetahs and compare them with the apparent commensal organisms in wild cheetahs in order to understand the role of Helicobacter spp. in the pathogenesis of gastritis in this species. Furthermore, this study aimed to compare Helicobacter spp. within and among facilities to investigate whether geographic location determined bacterial type and could explain differences in gastritis severity within the captive cheetah population.
MATERIALS AND METHODS
Animals.
Gastric samples from 33 cheetahs, 10 wild and 23 captive, that were infected with Helicobacter organisms and housed in different facilities and had different severities of gastritis were selected for this study. Wild cheetahs were located in north central Namibia, and samples were obtained opportunistically at necropsy (5 of 10) or by endoscopy (5 of 10) under general anesthesia. None (0 of 10) of the wild cheetahs had any histological evidence of gastritis. Gastric samples were obtained from captive cheetahs at necropsy (3 of 23) or by endoscopic biopsy (20 of 23) under general anesthesia during routine annual examination as part of the health surveillance program of the American Zoo and Aquarium Association Cheetah Species Survival Plan. The 23 captive cheetahs had various degrees of gastritis, graded mild (6 of 23), moderate (7 of 23), or severe (10 of 23) based on previously published criteria (25). Cheetahs were housed at seven different North American facilities, with up to 10 animals housed at a single facility and between two and four animals housed at four facilities. Two facilities had only a single animal in this study.
Electron microscopy.
Gastric samples were available for electron microscopy from a subset of the cases (. = 25, 18 captive and 7 wild cheetahs). Samples were fixed and stored in either 4% glutaraldehyde or 10% neutral buffered formalin and processed based on previously described methods (14). Briefly, samples were postfixed in reduced 2% osmium tetroxide in 2.5% potassium ferrocyanide in the microwave for 2.5 min with a 37°C temperature restriction, dehydrated in a graded acetone series, and embedded in a 1:1 Epon-Spurr's resin mixture. Thin sections were cut with a diamond knife, mounted on 300-mesh thin-bar copper grids, stained with 2% aqueous uranyl acetate, and examined on a Zeiss 10C transmission electron microscope (Carl Zeiss, Thornwood, N.Y.). For each cheetah, the morphological characteristics of at least 50 different bacteria in 10 different fields were measured from print images of known magnification. Morphological characteristics between bacteria with different 16S rRNA sequences were compared with the Mann-Whitney . test.
DNA extraction.
Because of the widespread geographic distribution of the study animals, including wild cheetahs in Africa, bacterial culture and isolation were not feasible. Furthermore, culture of biopsies had previously yielded negative results despite histological evidence of bacteria. Therefore, molecular and ultrastructural methods were utilized to characterize bacteria. DNA was extracted from frozen (−70°C) or formalin-fixed paraffin-embedded tissues with modifications of previously described methods (17, 36). For formalin-fixed paraffin-embedded samples, three 15-μm sections were deparaffinized in xylene and rehydrated in graded alcohols. Samples were minced under sterile conditions and placed in 300 μl of digestion buffer containing 100 mM Tris (pH 8.5) and 5 mM EDTA plus 1% sodium dodecyl sulfate (Fisher Scientific, Pittsburgh, Pa.) and 500 μg of proteinase K (Sigma, St. Louis, Mo.) per ml. Samples were incubated at 55°C for 12 h and then at 94°C for 10 min to denature the proteinase K.
The remaining cellular debris was sedimented, the supernatant was withdrawn, and the DNA was extracted with phenol-chloroform-isoamyl alcohol and then precipitated in 100% ethanol at −20°C for 12 h. DNA was pelleted by centrifugation, reconstituted in 100 μl of sterile distilled H2O, and stored at −20°C pending analysis. For fresh-frozen samples, a single, frozen (−70°C) endoscopic biopsy sample was placed directly into 200 μl of digestion buffer of 50 mM Tris (pH 9)-1 mM EDTA containing 1% Laureth 12 (PPG/Mazer Chemicals, Gurnee, Ill.) and 500 μg of proteinase K per ml to thaw. Samples were minced under sterile conditions and incubated at 37°C for 12 h and then at 94°C for 10 min to denature the proteinase K. The remaining cellular debris was sedimented, and the supernatant was withdrawn and frozen at −20°C.
Amplification and sequencing of 16S rRNA, urease, and cagA genes.
Universal and genus-specific primers (Table 1) were synthesized and designed as needed based on homologous regions of the 16S rRNA gene in other known species of Helicobacter and preliminary sequence data from the cheetah isolates. Amplification for the urease gene was performed with primer sets specific for known strains of “H. heilmannii” and H. pylori (Table 2). Samples were screened for the presence of the cag pathogenicity island with primers D008 F (ATAATGCTAAATTAGACAACTTCAGCGA) and R008 R (TTAGAATAATCAACAAACATCACGCCAT), which amplified a 270-bp fragment corresponding to positions 1232 to 1502 of the cagA gene.
TABLE 1.
Oligonucleotide primers used for amplification and sequencing of Helicobacter 16S rRNA from cheetahs
| Primer. | E. coli 16S rRNA positions | Sequence (5′ to 3′) |
|---|---|---|
| 8 F | 8-27 | AGAGTTTGATCCTGGCTCAG |
| 133Hh F | 110-133 | CGGGTCAGTAACGCATAGATGACA |
| 133Hp F | 110-133 | CGGGTCAGTAACGCATAGGTCATG |
| 244 F | 234-254 | CTATGTCCTATCAGCTTGTTG |
| 274 R | 300-274 | TCTCAGGCCGGATACCCGTCATAGC |
| 455 F | 445-489 | GAGAAGATAATGACGGTATC |
| 515 F | 515-533 | GTGCCAGCAGCCGCGGTAA |
| 528 R | 537-518 | CGTATTACCGCGGCTGCTGG |
| 613 F | 613-630 | TATGGCTTAACCATAGAA |
| 621 R | 621-604 | TAAGCCATAGGATTTCAC |
| 714 F | 704-725 | GAGATCAAGAGGAATACTCATTG |
| 795 R | 799-783 | CCAGGGTATCTAATCCTG |
| 840 F | 840-860 | GCTTTGTCTTTCCAGTAATGCA |
| 941 F | 935-954 | ACAAGCGGTGGA T/G G/C ATGTGG |
| 985 R | 994-972 | CAAGCCTAGGTAAGGTTCTTCG |
| 1062 F | 1062-1079 | TCGTCAGCTCGTGTCGT |
| 1098 R | 1115-1098 | AGGGTTGCGCTCGTTGCG |
| 1174 F | 1156-1175 | AGACTGCCTGCGTAAGCAGG |
| 1174C F | 1156-1175 | ATACTGCCTCCGTAAGGAGG |
| 1216 R | 1233-1216 | CACGTGTGTAGCCCTAGG |
| 1328 F | 1328-1347 | CATGAAGTCGGAATCGCTAG |
| 1383 R | 1383-1366 | GGAACGTATTCACCGCAA |
| 1492 R | 1512-1492 | CCGGGTTACCTTGTTACGACTT |
F and R designate forward and reverse primers, respectively.
TABLE 2.
Oligonucleotide primers used for amplification of the urease gene in Helicobacter isolates from cheetahs
| Species specificity | Primer. | Sequence (5′ to 3′) | Size of expected product (bp) |
|---|---|---|---|
| “H. heilmannii” | HhU3351F | CTATCAACTGCGGTTGCGAC | 140 |
| HhU3481R | TCGCCATAAGTGGTGCAGTC | ||
| HhU3351F | CTATCAACTGCGGTTGCGAC | 403 | |
| HhU3716R | TTCTGTAGCAGGTCCTACGC | ||
| H. pylori | HpU1286F | ACGCAACCACTATCACTCC | 115 |
| HpU1400R | CTTGCATCGTTAGAAGCGTTA | ||
| HpU1204F | TCCCCCCAACAAATCCCTAC | 297 | |
| HpU1501R | TGTCCGCAACATCTAACGC |
F and R designate forward and reverse primers, respectively.
DNA extracts were thawed on ice, and 2.5 μl was added to a 25-μl reaction volume containing standard amounts of GeneAmp reagents (Perkin-Elmer, Foster City, Calif.), 25 pmol of each primer, and 1.5 mM MgCl2. Amplification (94°C for 2 min; 35 cycles of 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min; and 72°C for 2 min) was performed in a thermocycler (Perkin-Elmer model 9700). Negative (sterile water substituted for DNA extract) and positive (H. felis ATCC 49179 and a cagA-positive strain of H. pylori [clinical isolate 87A300; California State Health Department]) controls were included in each set of reactions. The cloned urease gene from “H. heilmannii” (complete ureAB in pBSII; Stratagene, La Jolla, Calif.) (37) was also included in the urease reactions to confirm the specificity of the primers. PCR products were visualized by agarose gel electrophoresis and purified with a Centricon 30 concentrator according to the manufacturer's instructions (Amicon, Beverly, Mass.). For all cases, the nucleotide sequences of both strands of the 16S rRNA gene were determined by dideoxy nucleotide methodology with an automated sequencer (ABI Prism 377, Applied Biosystems, Foster City, Calif.). In addition, a subset (. = 10) of the PCR products from the urease gene reactions was sequenced to confirm their identity. The 16S rRNA and urease gene sequences were compared to sequences in the GenBank database and to each other with the Internet-based program SeqWeb (Genetics Computer Group) with Pileup and Growtree. Similarity matrices for the 16S rRNA gene were constructed from aligned sequences with only the 1,188-bp region available from the shortest sequence.
Denaturant gradient gel electrophoresis.
To determine if more than one type of Helicobacter organism was present within the stomachs of cheetahs, a fragment of the 16S rRNA gene was amplified from gastric samples with universal primers (244f and 528r from Table 1). A GC clamp was attached to the 5′ end of the forward primer (CGCCCGCCGC GCCCCGCGCC CGGCCCGCCG CCGCCGCTAT GTCCTATCAG CTTGTTG) to improve separation of closely related sequences (1). PCR conditions were similar to those previously described, and amplified products were separated in a 12% polyacrylamide gel with a 20 to 60% gradient of urea-formamide (DCode electrophoresis reagent kit, Bio-Rad, Richmond, Calif.) run in TAE buffer at 60°C and 300 V for 4 h in a Bio-Rad D-Gene apparatus (Bio-Rad, Richmond, Calif.). Positive controls were amplified along with cheetah samples and included DNA isolated from pure cultures of H. pylori (clinical isolate 87A300, California State Health Department) and H. felis (ATCC 49179), DNA extracted from a cheetah isolate most similar to “H. heilmannii,” as well as a mixture of DNA from these three isolates. DNA was stained with a fluorescent nucleic acid stain (GelStar nucleic acid gel stain, Cambrex, Rockland, Maine) and examined under UV light.
RESULTS
Ultrastructural morphology.
Long, tightly coiled bacteria were present within the gastric glands and parietal cell canaliculi in all cases examined. The ultrastructural morphology of bacteria in all cheetahs was similar irrespective of the severity of gastritis (Fig. 1). Bacterial size and shape were more consistent with “H. heilmannii” than H. pylori. The bacteria with the 16S rRNA sequences most similar to H. pylori were helical, 4 to 6.4 by 0.35 to 0.54 μm with three to four polar flagella. Bacteria with 16S rRNA sequences most similar to “H. heilmannii” were helical, 3.4 to 7.9 by 0.4 to 0.64 μm with three to six polar flagella. There was no difference in the overall measurements between bacteria with different 16S rRNA sequences (. = 0.344). Mixed infections with bacteria exhibiting different morphological characteristics were not detected. In most cases, three to five polar flagella could be visualized along at least one pole. Periplasmic fibrils were not identified in any of the cases.
FIG. 1.

Gastric histopathology, demonstrating the absence of gastritis in wild cheetahs (A and C) and severe lymphoplasmacytic gastritis with glandular destruction in captive cheetahs (B and D) infected with similar bacteria. Insets in each panel demonstrate the typical ultrastructural characteristics of the bacteria infecting that cheetah. The cheetahs in panels A and B were infected with H. pylori-like (based on 16S rRNA sequence) bacteria, while those in panels C and D were infected with “H. heilmannii”.like bacteria. In the large panels (hematoxylin and eosin stain), the bar equals 20 μm. In the insets (transmission electron microscopy), the bar equals 0.5 μm.
16S rRNA, urease, and cagA gene analyses.
The 16S rRNA sequences were amplified by PCR from the stomachs of all 33 cheetahs. Sequences represented nearly the entire 16S rRNA gene with 1,188 to 1,427 bp (77 to 92% of the 16S rRNA gene) of readable sequence determined from bacterial DNA isolated from cheetahs. On the basis of these sequences, all 33 cheetahs were infected with bacteria that were consistent with the genus Helicobacter. The Helicobacter spp. infecting the cheetahs clustered into three groups irrespective of severity of gastritis (Fig. 2). Bacteria with identical sequences were present in cheetahs with and without gastritis. Bacterial types varied within and among captive facilities. Two facilities had multiple types of bacteria present within their population. Three facilities housing more than one cheetah had only a single type of bacteria. At one of these facilities, samples from a mother and her three cubs had similar (99.62 to 99.91%) but not identical sequences.
FIG. 2.

Phylogenetic tree based on 16S rRNA sequences demonstrating the relationship between Helicobacter spp. from 33 cheetahs and previously validated or provisional species of helicobacters. The first number in brackets is the severity of gastritis; none of the wild cheetahs had gastritis (grade 0), while all of the captive cheetahs had some degree of gastritis (grades 1 to 3). The number in parentheses is the GenBank accession number.
Although all cheetahs were infected with Helicobacter spp. that were morphologically indistinguishable, the 16S rRNA sequences resembled H. pylori in some cases and “H. heilmannii” in others. The 16S rRNA sequences in eight cheetahs (five wild and three captive) were most similar (98 to 99%) to H. pylori. Six (three captive and three wild) of these H. pylori-like sequences were similar (>98%) to each other, while sequences from the other two wild cheetahs were 94 to 95% similar to the other H. pylori-like sequences. These sequences were only approximately 95% similar to H. acinonychis, the species of Helicobacter previously isolated from cheetahs (9). Twenty-five cheetahs (5 wild and 20 captive) had sequences that were 97 to 99% similar to “H. heilmannii” isolated from a domestic cat (AF058768) or H. felis (U51870) isolated from a domestic dog. These cheetah sequences had >98% similarity to each other.
PCR products of the expected sizes (140 and 403 bp) were amplified with primers for “H. heilmannii” urease from 25 of 25 cheetahs with bacterial 16S rRNA sequences most similar to “H. heilmannii” or H. felis. Of the 10 cases in which the urease gene was sequenced, nine were most similar to “H. heilmannii” (95 to 98% similarity) and one was most similar (94%) to H. felis. The urease gene could not be amplified with either primer set from any of the cases in which Helicobacter 16S rRNA sequences were similar to H. pylori. The cagA gene could not be amplified from any of the 33 cheetah isolates, including those with 16S rRNA sequences most similar to H. pylori.
Denaturant gradient gel electrophoresis.
No mixed Helicobacter infections were identified by denaturant gradient gel electrophoresis analysis of a 284-bp fragment of the 16S rRNA gene. In all cases, the denaturation patterns of bands matched the results of the 16S rRNA sequences (Fig. 3).
FIG. 3.
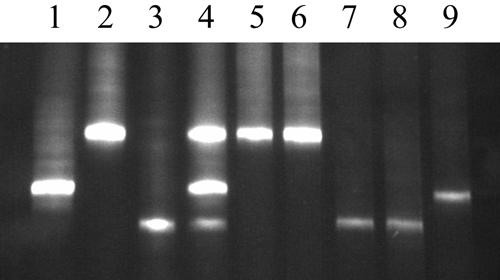
Results of denaturant gradient gel electrophoresis for previously characterized and cheetah strains of Helicobacter. Lane 1, H. felis; lane 2, H. pylori; lane 3, “H. heilmannii”.like bacteria from a cheetah; lane 4, mixture of DNA extracts from the isolates in lanes 1 to 3; lane 5, wild cheetah; lane 6, captive cheetah; lane 7, wild cheetah; lane 8, captive cheetah; lane 9, captive cheetah.
DISCUSSION
No single strain of Helicobacter was associated with gastritis in cheetahs, and in some cases apparently identical strains were present in cheetahs with and without disease. Among the cheetahs with gastritis, the severity of gastritis was also not associated with any one type of Helicobacter, and no specific types were present at any one facility. Sequences from captive cheetahs were most homologous with H. pylori, H. felis, or “H. heilmannii,” while sequences from wild cheetahs were most homologous to H. pylori and “H. heilmannii.” A greater percentage of the wild cheetahs in this study were infected with H.pylori-like Helicobacter spp. Irrespective of this difference, there were striking differences in disease severity between cheetahs infected with seemingly similar bacteria. Additionally, the samples from wild cheetahs came from one geographic region within Namibia, which may have biased the types of Helicobacter identified.
Despite the previous identification of small numbers of coinfections in cheetahs (11), no coinfections were identified in this study either by electron microscopy or by denaturant gradient gel electrophoresis analysis of a 16S rRNA gene fragment. In addition to the strains identified in the current study, H. acinonychis was previously isolated from captive cheetahs at one facility (9, 10). Taken together, these data suggest that at least four different helicobacters are present in the stomachs of captive cheetahs with gastritis. Similar strains are apparently commensal organisms in wild cheetahs, bringing into question the role of specific Helicobacter spp. in the pathogenesis of gastritis in cheetahs.
Despite the previous isolation of H. acinonychis and naming of this organism after the cheetah, none of the cheetahs sampled in this study were infected by this Helicobacter spp. This observation is consistent with the negative results of more recent culture attempts from other cheetahs (K. Eaton, personal communication). It is possible that H. acinonychis was not representative of the Helicobacter spp. infecting the cheetah population as a whole. H. acinonychis has been isolated from other species of exotic felids and may have been transmitted to the cheetahs in the previously studied collection (4, 35). Alternatively, these bacteria may have historically been more prevalent in cheetahs but become less so due to selective breeding efforts in captive institutions and maternal transmission of other Helicobacter spp. (15).
It is unlikely that the organisms with 16S rRNA sequences most similar to H. pylori truly represent H. pylori, as urease could not be detected with primers specific for conserved regions of the H. pylori urease gene. It is presumed that these organisms have urease genes, given their colonization of the gastric microenvironment. However, the urease sequence of these bacteria is likely different from that of either H. pylori or “H. heilmannii.” Discrepancies between genetic and morphological characteristics further complicate the appropriate classification of these organisms in cheetahs. While H. pylori has been shown to assume the morphology of “H. heilmannii” under certain culture conditions (12), this phenomenon has not been reported in vivo.
Organisms have been characterized in this study as H. pylori-like solely on the basis of the 16S rRNA sequences, which may not be the most accurate method of classification (19, 41). Despite proposals to utilize the 23S rRNA subunit (16, 18) or the urease gene (5), the 16S rRNA gene sequence currently remains the standard gene for classification of Helicobacter spp. (7). Because most of the bacteria in cheetahs are currently unculturable, information on the biochemical characteristics was unavailable. These results suggest that these H. pylori-like organisms may represent a distinct species. In some of the cheetahs with Helicobacter most similar to “H. heilmannii” or H. felis, the 16S rRNA sequences were phylogenetically equidistant from both of these organisms. Because the ultrastructural morphology of these organisms can be indistinguishable (8), many of these bacteria might be better classified as belonging to the H. felis-like clade of gastrospirilla.
In other species, the pathogenesis of Helicobacter gastritis is dependent on bacterial as well as host factors. It has been suggested that disease develops when either the host gastric microenvironment is altered or the bacteria acquire characteristics, such as the cag pathogenicity island, that may be evolutionarily beneficial to the bacteria (3). The cagA gene, a marker for virulence factors important in the induction of neutrophilic inflammation (33), could not be identified in any of the cheetah samples analyzed. This result was not surprising because neutrophils are an uncommon feature of gastritis in cheetahs (10, 25). Although it is possible that other, not yet identified, pathogenicity factors are present in the Helicobacter spp. associated with gastritis in cheetahs, it is more likely that host factors are responsible for the disparity in disease occurrence between captive and wild cheetahs.
Differences in occurrence and intensity of inflammation may be due to host genotypic differences (22, 24, 42). Cheetahs are homogenic for major histocompatibility complex (MHC) genes, a characteristic that has been proposed as an explanation for their unique susceptibility to some infectious diseases (28, 29). However, homogeneity is a feature of both captive and wild cheetah populations (28), yet only captive cheetahs commonly develop gastritis. Additionally, the founders of the captive population originated from the same region of Africa as the wild population in this study. The contributions of both MHC and non-MHC genes appear to influence the degree of inflammation in MHC-congenic mice infected with H. felis, as do polymorphisms in genes encoding inflammatory mediators in humans (24, 40). Therefore, genotypic differences in MHC are not likely the basis for the occurrence of inflammatory reactions only in captive cheetahs. Investigation of polymorphisms in other genes potentially important in the development of gastritis is warranted.
Another theory to explain inflammatory reactions to similar Helicobacter types is modulation of the host inflammatory response to Helicobacter spp. by enteric helminth infections (13). It is possible that enteric parasite infections in the wild cheetahs reduced the inflammatory response, whereas captive cats that receive regular antihelminthic medication as part of their routine health care lack this suppressive effect. However, of the five wild cheetahs from which samples were obtained at necropsy, only two animals had documented enteric cestode or nematode infections. Additionally, other species of captive and domesticated felids that receive antihelminthic treatment commonly have minimal to no inflammation associated with Helicobacter infections (20, 21, 27). These findings suggest that the gastric inflammatory reaction that occurs solely in captive cheetahs is likely due to aspects of captivity other than the absence of helminth infections.
Environmental differences between captive and wild cheetahs are almost certainly important in the development of gastritis. Diet alone is not likely the cause of gastritis, as captive cheetahs in South African facilities are fed a diet closely resembling that of wild cheetahs and yet gastritis is prevalent within this population (26). Cheetahs in the wild are generally spared many of the diseases afflicting captive cheetahs worldwide (L. Munson, unpublished data), suggesting that cheetahs are maladapted to some as yet unknown aspect of the captive environment. This maladaptation is evidenced by increased adrenocortical function in captive but not wild cheetahs (39). Because of the immunomodulatory affects of the glucocorticoids, it is possible that captive cheetahs have an altered systemic or local immune response that accounts for their reaction to otherwise commensal bacteria (2, 6, 34). This hypothesis would also explain the presence of gastritis in captive cheetahs infected with apparently different organisms. Ongoing research characterizing gastric cytokine profiles in captive and wild cheetahs aims to determine if elevated corticosteroids are affecting the local gastric immune response.
In summary, based on 16S rRNA sequences, urease sequences, and ultrastructural characteristics, multiple types of Helicobacter were identified in captive cheetahs with gastritis. Similar organisms were present in cheetahs with and without gastritis, suggesting that host factors are more important than bacteria in the pathogenesis of gastritis in cheetahs. The distinct differences in the occurrence of gastritis in captive and wild cheetahs, despite infection with similar Helicobacter organisms, provides an interesting natural disease model for analysis of host factors important in the development of gastritis.
Acknowledgments
We thank the following institutions and their veterinarians for contributing tissues for this study: Cleveland Metroparks Zoo, Fossil Rim Wildlife Center, The Living Desert, Rio Grande Zoo, Sacramento Zoo, White Oak Conservation Center, Wildlife Safari, and a special thanks to the Cheetah Conservation Fund in Namibia, particularly Bonnie Schumann for collecting tissues from wild cheetahs. We also thank Lori Hanson, Sima Torabian, Julie Fong, Jennifer Ingram, and Angela Hughes for technical assistance with the DNA sequencing and Robert Nordhausen for assistance with electron microscopy.
This study was funded by the Morris Animal Foundation (D00Z00-19). Work in the laboratory of J. V. Solnick was supported by grants A142081and RR14298 from the National Institutes of Health.
REFERENCES
- 1.Aldridge, B. M., S. M. McGuirk, R. J. Clark, L. A. Knapp, D. I. Watkins, and D. P. Lunn. 1998. Denaturing gradient gel electrophoresis: a rapid method for differentiating BoLA-DRB3 alleles. Anim. Genet. 29:389-394. [DOI] [PubMed] [Google Scholar]
- 2.Almawi, W. Y., M. M. Abou Jaode, and X. C. Li. 2002. Transcriptional and post-translational mechanisms of glucocorticoid antiproliferative effects. Hematol. Oncol. 20:17-32. [DOI] [PubMed] [Google Scholar]
- 3.Blaser, M. J. 1997. Ecology of Helicobacter pylori in the human stomach. J. Clin. Investig. 100:759-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cattoli, G., A. Bart, P. S. J. Klaver, R. J. Robijin, H. J. Beumer, R. van Vugt, R. G. J. Pot, I. van der Gaag, C. M. J. E. Vandenbroucke-Grauls, E. J. Kuipers, and J. G. Kusters. 2000. Helicobacter acinonychis eradication leading to the resolution of gastric lesions in tigers. Vet. Rec. 147:164-165. [DOI] [PubMed] [Google Scholar]
- 5.Cattoli, G., R. van Vugt, V. Sanguinetti, C. M. J. E. Vandenbroucke-Grauls, and J. G. Kusters. 1999. Differentiation of Gastrospirillum-like organisms by a UREAB based PCR. Gastroenterology 116:A133. [Google Scholar]
- 6.Chiapelli, F., E. Manfrini, C. Franceschi, A. Cossarizza, and K. L. Black. 1994. Steroid regulation of cytokines: relevance for Th1 to Th2 shift? Ann. N. Y. Acad. Sci. 746:204-215. [DOI] [PubMed] [Google Scholar]
- 7.Dewhirst, F. E., J. G. Fox, and S. L. W. On. 2000. Proposal on minimal standards for describing new species of the genus Helicobacter. Int. J. Syst. Evol. Microbiol. 50:2231-2237. [DOI] [PubMed] [Google Scholar]
- 8.Eaton, K. A., F. E. Dewhirst, B. J. Paster, N. Tzellas, B. Coleman, J. Paola, and A. Sherding. 1996. Prevalence and varieties of Helicobacter species in dogs from random sources and pet dogs: animal and public health implications. J. Clin. Microbiol. 34:3165-3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eaton, K. A., F. E. Dewhirst, M. J. Radin, J. G. Fox, B. J. Paster, S. Krakowka, and D. R. Morgan. 1993. Helicobacter acinonyx sp. nov., isolated from cheetahs with gastritis. Int. J. Syst. Bacteriol. 43:99-106. [DOI] [PubMed] [Google Scholar]
- 10.Eaton, K. A., M. J. Radin, L. W. Kramer, R. F. Wack, R. Sherding, S. Krakowka, J. G. Fox, and D. R. Morgan. 1993. Epizootic gastritis associated with gastric spiral bacilli in cheetahs (Acinonyx jubatus). Vet. Pathol. 30:55-63. [DOI] [PubMed] [Google Scholar]
- 11.Eaton, K. A., M. J. Radin, L. W. Kramer, R. F. Wack, R. Sherding, S. Krakowka, and D. R. Morgan. 1991. Gastric spiral bacilli in captive cheetahs. Scand. J. Gastroenterol. 26:38-42. [DOI] [PubMed] [Google Scholar]
- 12.Fawcett, P. T., K. M. Gibney, and K. M. B. Vinette. 1999. Helicobacter pylori can be induced to assume the morphology of Helicobacter heilmannii. J. Clin. Microbiol. 37:1045-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fox, J. G., P. Beck, C. A. Dangler, M. T. Whary, T. C. Wang, H. Ning Shi, and C. Nagler-Anderson. 2000. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces helicobacter-induced gastric atrophy. Nat. Med. 6:536-542. [DOI] [PubMed] [Google Scholar]
- 14.Giberson, R. T., R. S. Jr. Demaree, and R. W. Nordhausen. 1997. Four-hour processing of clinical/diagnostic specimens for electron microscopy using microwave technique. J. Vet. Diagn. Investig. 9:61-67. [DOI] [PubMed] [Google Scholar]
- 15.Hanninen, M.-L., I. Happonen, and K. Jalava. 1998. Transmission of canine gastric Helicobacter salmonis infection from dam to offspring and between puppies. Vet. Microbiol. 62:47-58. [DOI] [PubMed] [Google Scholar]
- 16.Hurtado, A., and R. J. Owen. 1997. A rapid identification scheme for Helicobacter pylori and other species of Helicobacter based on 23S rRNA gene polymorphisms. Syst. Appl. Microbiol. 20:222-231. [Google Scholar]
- 17.Innis, M. A. 1990. PCR protocols: a guide to methods and applications. Academic Press, San Diego, Calif.
- 18.Jalava, K., S. Hielm, U. Hirvi, and M.-L. Hanninen. 1999. Evaluation of a molecular identification scheme based on 23S rRNA polymorphisms for differentiating canine and feline gastric Helicobacter spp. Lett. Appl. Microbiol. 28:269-274. [DOI] [PubMed] [Google Scholar]
- 19.Jalava, K., M. Kaartinen, M. Utriainen, I. Happonen, and M.-L. Hanninen. 1997. Helicobacter salomonis sp. nov., a canine gastric Helicobacter sp. related to Helicobacter felis and Helicobacter bizzozeronii. Int. J. Syst. Bacteriol. 47:975-982. [DOI] [PubMed] [Google Scholar]
- 20.Kinsel, M. J., M. B. Briggs, K. Venzke, O. Forge, and R. D. Murnane. 1998. Gastric spiral bacteria and intramuscular sarcocysts in African lions from Namibia. J. Wildl. Dis. 34:317-324. [DOI] [PubMed] [Google Scholar]
- 21.Kinsel, M. J., P. Kovarik, and R. D. Murnane. 1998. Gastric spiral bacteria in small felids. J. Zoo Wildl. Med. 29:214-220. [PubMed] [Google Scholar]
- 22.Mähler, M., C. Janke, S. Wagner, and H. J. Hedrich. 2002. Differential susceptibility of inbred mouse strains to Helicobacter pylori infection. Scand. J. Gastroenterol. 37:267-278. [DOI] [PubMed] [Google Scholar]
- 23.Marshall, B. J., and J. R. Warren. 1984. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet i:1311-1315. [DOI] [PubMed] [Google Scholar]
- 24.Mohammadi, M., R. W. Redline, J. G. Nedrud, and S. J. Czinn. 1996. Role of the host in pathogenesis of Helicobacter-associated gastritis: H. felis infection of inbred and congenic mouse strains. Infect. Immun. 64:238-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munson, L. 1993. Diseases of captive cheetahs (Acinonyx jubatus): results of the Cheetah Research Council Pathology Survey. Zoo Biol. 12:105-124. [Google Scholar]
- 26.Munson, L., J. W. Nesbit, D. G. A. Meltzer, L. P. Colly, L. Bolton, and N. P. J. Kriek. 1999. Diseases of captive cheetahs (Acinonyx jubatus jubatus) in South Africa: a 20 year retrospective survey. J. Zoo Wildl. Med. 30:342-347. [PubMed] [Google Scholar]
- 27.Norris, C. R., S. L. Marks, K. A. Eaton, S. Z. Torabian, R. J. Munn, and J. V. Solnick. 1999. Healthy cats are commonly colonized with “Helicobacter heilmannii” that is associated with minimal gastritis. J. Clin. Microbiol. 37:189-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Brien, S. J., M. E. Roelke, L. Marker, A. Newman, C. A. Winkler, D. Meltzer, L. Colly, J. F. Evermann, M. Bush, and D. E. Wildt. 1985. Genetic basis for species vulnerability in the cheetah. Science 227:1428-1434. [DOI] [PubMed] [Google Scholar]
- 29.O'Brien, S. J., D. E. Wildt, D. Goldman, C. R. Merril, and M. Bush. 1983. The cheetah is depauperate in genetic variation. Science 221:459-462. [DOI] [PubMed] [Google Scholar]
- 30.Papendick, R. E., L. Munson, T. D. O'Brien, and K. H. Johnson. 1997. Systemic AA amyloidosis in captive cheetahs (Acinonyx jubatus). Vet. Pathol. 34:549-556. [DOI] [PubMed] [Google Scholar]
- 31.Parsonnet, J., G. D. Friedman, D. P. Vandersteen, Y. Chang, J. H. Vogelman, N. Orentreich, and R. K. Sibley. 1991. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 325:1127-1136. [DOI] [PubMed] [Google Scholar]
- 32.Parsonnet, J., S. Hansen, L. Rodriguez, A. B. Gelb, R. A. Warnke, E. Jellum, N. Orentreich, J. H. Vogelman, and G. D. Friedman. 1994. Helicobacter pylori infection and gastric lymphoma. N. Engl. J. Med. 330:1267-1271. [DOI] [PubMed] [Google Scholar]
- 33.Peek, R. M., and M. J. Blaser. 1996. Pathophysiology of Helicobacter pylori-induced gastritis and peptic ulcer disease. Am. J. Med. 102:200-207. [DOI] [PubMed] [Google Scholar]
- 34.Riccardi, C., S. Bruscoli, and G. Migliorati. 2002. Molecular mechanisms of immunomodulatory activity of glucocorticoids. Pharmacol. Res. 45:361-368. [DOI] [PubMed] [Google Scholar]
- 35.Schroder, H.-D., C. Ludwig, W. Jakob, U. Reischl, M. Stolte, and N. Lehn. 1998. Chronic gastritis in tigers associated with Helicobacter acinonyx. J. Comp. Pathol. 119:67-73. [DOI] [PubMed] [Google Scholar]
- 36.Solnick, J. V., J. O'Rourke, A. Lee, B. J. Paster, F. E. Dewhirst, and L. S. Tompkins. 1993. An uncultured gastric spiral organism is a newly identified Helicobacter in humans. J. Infect. Dis. 168:379-385. [DOI] [PubMed] [Google Scholar]
- 37.Solnick, J. V., J. O'Rourke, A. Lee, and L. S. Tompkins. 1994. Molecular analysis of urease genes from a newly identified uncultured species of Helicobacter. Infect. Immun. 62:1631-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Solnick, J. V., and D. B. Schauer. 2001. Emergence of diverse Helicobacter species in the pathogenesis of gastric and enterohepatic diseases. Clin. Microbiol. Rev. 14:59-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Terio, K. A., L. Marker, and L. Munson. 2004. Evidence for chronic stress in captive but not free-ranging cheetahs (Acinonyx jubatus) based on adrenal morphology and function. J. Wildl. Dis. 40:259-266. [DOI] [PubMed] [Google Scholar]
- 40.Thye, T., G. D. Burchard, M. Nilius, B. Müller-Myhsok, and R. D. Horstmann. 2003. Genomewide linkage analysis identifies polymorphism in the human interferon-γ receptor affecting Helicobacter pylori infection. Am. J. Hum. Genet. 72:448-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vandamme, P., C. S. Harrington, K. Jalava, and S. L. W. On. 2000. Misidentifying helicobacters: the Helicobacter cinaedi example. J. Clin. Microbiol. 38:2261-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Doorn, N. E. M., F. Namavar, M. Sparrius, J. Stoof, E. P. Van Rees, L.-J. van Doorn, and C. M. J. E. Vandenbroucke-Grauls. 1999. Helicobacter pylori-associated gastritis in mice is host and strain specific. Infect. Immun. 67:3040-3046. [DOI] [PMC free article] [PubMed] [Google Scholar]