

Summary
The innate immune system is the first line of defense against Neisseria gonorrhoeae (GC). Exposure of cells to GC lipooligosaccharides induces a strong immune response, leading to type I interferon (IFN) production via TLR4/MD-2. In addition to living freely in the extracellular space, GC can invade the cytoplasm to evade detection and elimination. Double-stranded DNA introduced into the cytosol binds and activates the enzyme cyclic-GMP-AMP synthase (cGAS), which produces 2′3′-cGAMP and triggers STING/TBK-1/IRF3 activation resulting in type I IFN expression. Here we revealed a cytosolic response to GC DNA that also contributes to type I IFN induction. We demonstrated that complete IFN-β induction by live GC depends on both cGAS and TLR4. Type I IFN is detrimental to the host and dysregulation of iron homeostasis genes may explain lower bacteria survival in cGAS−/− and TLR4−/− cells. Collectively, these observations reveal cooperation between TLRs and cGAS in immunity to GC infection.
Keywords: Type I interferon, cGAS, STING, TLR4, Neisseria gonorrhoeae
eTOC paragraph
Neisseria gonorrhoeae (GC) can invade the cytoplasm and evade detection and elimination by the innate immune system. Andrade et al. show that type I interferon induction by GC infection is dependent on both cGAS/STING and TLR4 signaling pathways, and that IFN-β is detrimental for macrophage bacterial killing.
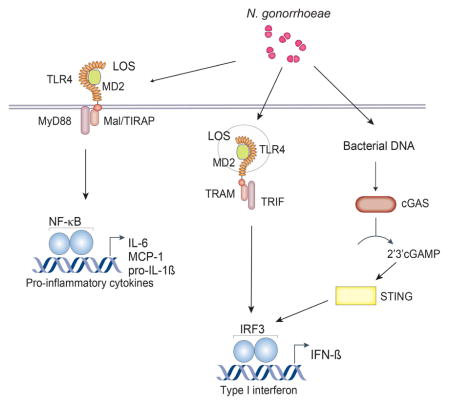
Introduction
The genus Neisseria contains commensal and pathogenic species that colonize human mucosal epithelia. The pathogenic species, Neisseria gonorrhoeae and Neisseria meningitidis, can induce inflammation and breach mucosal barriers. Each year N. gonorrhoeae and N. meningitidis cause an estimated 88 million cases of gonorrhea and 500,000 cases of meningococcal meningitis worldwide, respectively (Stephens, 2009). These pathogens are highly adapted to humans; rather than produce cytotoxins or secrete toxic products, they have evolved mechanisms to promote growth and persistence in the host. In addition to living freely in the extracellular space, N. gonorrhoeae can invade the cytoplasm of host phagocytes, thereby evading detection and elimination by the innate immune system (Criss and Seifert, 2012; Duncan et al., 2009).
The detection of pathogens occurs through the recognition of molecules from microorganisms known as pathogen-associated molecular patterns through several host pattern recognition receptors including Toll-like receptors (TLRs). For example, TLR4 mediates recognition of extracellular gonococcal lipooligosaccharide (LOS) (Pridmore et al., 2003). It has also been shown that lipopolysaccharide (LPS) and infection with different gram-negative and gram-positive bacteria induces IFN-β (Monroe et al., 2010).
The sensing of pathogen-derived nucleic acids (DNA and RNA) is a central strategy used by the innate immune system to initiate immune responses following microbial invasion (Barbalat et al., 2011). Accumulation of foreign or self-DNA in the cytosol can induce strong innate immune responses through a variety of sensors (Barber, 2011). An endoplasmic-reticulum-resident host protein called stimulator of IFN genes (STING) is required for a type I IFN response to cytosolic dsDNA (Ishikawa et al., 2009), and IFN responses to a family of unique bacterial nucleic acids that form cyclic dinucleotides (Sauer et al., 2011). STING has been shown to have critical roles in the innate immune responses to many bacterial pathogens (Manzanillo et al., 2012; Prantner et al., 2010).
Recent studies have reported that the enzyme cyclic GMP-AMP synthase (cGAS) acts as an intracellular DNA sensor for cytosolic DNA, generating the second messenger cGAMP. cGAMP binds to STING and leads to IRF3 phosphorylation and type I IFN production (Sun et al., 2013; Wu et al., 2013; Zhang et al., 2013). Induction of type I IFN by some bacterial infections is partially STING dependent, but in most cases the DNA sensor upstream of STING was unknown (Barker et al., 2013; Gratz et al., 2011). Type I IFN induction by several bacteria including Chlamydia trachomatis, Mycobacterium tuberculosis and Listeria monocytogenes is dependent on cGAS (Watson et al., 2015; Zhang et al., 2014).
In this study, we report that N. gonorrhoeae was able to induce IFN-β in both human and murine cells. As previously reported for other bacteria, type I IFN induction by GC was partially dependent on STING. Using cells deficient in cGAS, IFI16 and TLR4, in combination with the TLR4/MD-2 inhibitor E5564, or cGAS/TLR4 double knockout cells, we found that IFN-β induction by N. gonorrhoeae was dependent on both cGAS and TLR4, but independent of IFI16. We also demonstrated by mass spectrometry that 2′3′-cGAMP is produced both by GC DNA delivered to the cytosol by transfection and by GC infection. Our data strongly indicates that gonococcus induces type I IFNs by two distinct and, most likely, coordinated mechanisms involving TLR4/MD2 and cGAS, and that IFN-β is detrimental to bacterial killing.
Results
Induction of type I interferon by GC is both STING and TLR4-dependent
Recent studies have revealed that type I IFNs might be beneficial or harmful to the host, depending on the bacteria. STING has a critical role in the innate immune response to many bacterial pathogens (Monroe et al., 2010). To evaluate whether type I IFNs play a role in N. gonorrhoeae infection, we infected WT and STING knockout (KO) bone marrow-derived macrophages (BMDMs) with GC strains FA1090, a widely used experimental strain (Jordan et al., 2005). The induction of IFN-β (Figure 1A and 1B), but not TNF-α (Figure 1C and 1D), IL-6, and IL-12 (data not shown) was strongly, but not entirely, dependent on STING.
Figure 1. Induction of type I IFN by GC is both STING- and TLR4-dependent.

(A-D) WT and STING−/− BMDMs were infected with GC strain FA1090, stimulated with LOS (100ng/ml) or infected with Sendai virus (SeV, 200HAU/ml) for 6 hr; in (B) polydA:dT (pdAdT, 5μg/ml) was used as an additional stimulant. (E and F) WT, TLR4−/− and STING−/− BMDMs were infected with GC strain FA1090, stimulated with LOS (100ng/ml), or transfected with GC DNA (3μg/ml)(F). Levels of IFN-β mRNA (A and E) and TNF-α mRNA (C) were determined by qPCR and normalized to GAPDH levels. Levels of IFN-β protein (B and F) and TNF-α protein (D) were measured by ELISA after 18 hr. Data are represented as mean ± SD of three experiments that yielded similar results. Asterisks indicate that differences are statistically significant (*p < 0.05. **p < 0.01, and ***p < 0.001). ND: not detected. See also Figure S1.
In general, TLR4/MD-2 stimulation by LPS (or LOS) results in the activation of two signaling pathways, Mal/MyD88 and TRAM/TRIF, leading to the production of different subsets of proinflammatory cytokines and interferons. GC LOS induces several cytokines (TNF-α, IL-8, IFN-β) through TLR4/MD-2/MyD88/TRIF activation (Liu et al., 2010). Our results showed that induction of IFN-β by LOS or Sendai Virus (SeV) was independent of STING (Figure 1A and 1B). Our data is consistent with that of others who have also shown that IFN-β induction by LOS is dependent on TLR4 (Liu et al., 2010) and that IFN-β induction by SeV is dependent on RIG-I and independent of STING (Strahle et al., 2007).
In contrast to the response to LOS, the IFN-β response to GC infection was partially STING-dependent. To better characterize IFN-β induction by GC infection, we infected TLR4−/− and STING−/− BMDMs, as well as MyD88/TRIF double Kos macrophages to evaluate the role of STING and TLR4/MD-2/MyD88/TRIF pathways in type I IFN induction during infection. Induction of IFN-β was partially dependent on TLR4, STING (Figure 1E and 1F) and MyD88/TRIF (Figure S1A, available online), but TNF-α and IL-6 inductions were STING independent and partially TLR4 dependent (Figure S1B and S1C). IFN-β was also induced when GC DNA was introduced into the cytosol by transfection, however the IFN response to transfected GC DNA occurred in the absence of TLR4, but not STING (Figure 1F). As expected, TLR4 was required for IFN-β induction by LOS (Figure 1E and 1F). When we pretreated TLR4 and MyD88/TRIF deficient cells with cytochalasin D (which blocks phagocytosis) or bafilomycin A (which inhibits phagosome acidification), IFN-β production was significantly decreased after GC infection (Figure S1A), indicating that phagocytosis and phagosome acidification are important for the type I IFN response to GC, similar to other bacteria (Charrel-Dennis et al., 2008). Our results indicate that IFN-β induction by GC infection is both TLR4- and STING-dependent, but TNF-α and IL-6 are partially TLR4-dependent and STING-independent, suggesting predominantly IRF3 activation, but not NF-κB through STING (Collins et al., 2015).
Type I interferon induction by GC infection is dependent on cGAS, but not IFI16
Our results demonstrated that IFN-β induction by GC infection is partially dependent on TLRs, but is also partially dependent on STING, indicating a role for cytosolic DNA sensors. Recent studies reported that cGAS acts as an intracellular DNA sensor and generates the second messenger cGAMP (Sun et al., 2013; Wu et al., 2013), and that cGAS is involved in type I IFN induction by Streptococcus pyogenes and Francisella novicida (Collins et al., 2015; Storek et al., 2015). To investigate the role of intracellular DNA sensors in GC infection of human macrophages, we used two different THP-1 KO cell lines, a cGAS-deficient cell line (Figure S2A) and an IFI16-deficient line (Figure S2B), both engineered using the CRISPR/Cas9 system (Wassermann et al., 2015). We found that THP-1 cells in which cGAS was knocked out had impaired IFN-β (Figure 2A) and IP-10 (Figure S2C) production after GC infection or transfection with GC DNA. The PYHIN family member, IFI16 has been shown to be an important DNA sensor and inducer of type I IFNs by macrophages in M. tuberculosis (Manzanillo et al., 2012) and L. monocytogenes infections (Hansen et al., 2014). IFI16 KO THP-1 cells infected with GC induced normal levels of IFN-β (Figure 2B), suggesting that, in contrast to cGAS, the DNA sensor IFI16 does not play a role in the type I IFN response to GC infection.
Figure 2. Induction of IFN-β by GC infection is dependent on cGAS but independent of IFI16.

(A and C) Undifferentiated cGAS+/+ and cGAS KO THP-1 cells were infected with GC strains FA1090 and MS11, transfected with GC DNA (3μg/ml), or stimulated with LOS (100ng/ml) for 6 hr. Cells were pretreated with cytochalasin D (Cyto D) for 1 hr. (B and D) Undifferentiated WT and IFI16 KO THP-1 cells (clones 1 and 2) were infected with GC or stimulated with LOS (100ng/ml) for 6 hr. Levels of IFN-β (A and B) and TNF-α (C and D) were determined by qPCR and normalized to GAPDH levels. Data are represented as mean ± SD of one experiment of four experiments (each performed in triplicate) (A–D) that yielded similar results. Asterisks indicate that differences are statistically significant (**p < 0.01). See also Figure S2.
In contrast, TNF-α (Figure 2C), IL-6, and IL-12 (Figure S2D and S2E) responses were unaffected in cGAS-deficient cells, and TNF-α was also unaffected in IFI16 deficient cells (Figure 2D), indicating an important role for cGAS in IFN-β and IP-10, but not TNF-α, IL-6, and IL-12 induction by GC infection.
To determine the role of bacterial uptake in IFN-β induction, we pretreated cells with cytochalasin D to block phagocytosis. Levels of IFN-β were strongly reduced in WT cells treated with cytochalasin D (Figure 2A), indicating a role for phagocytosis in IFN-β induction. These results also suggested a cGAS-independent means of activating IFN-α/β production that could be observed in WT cells.
Complete activation of IFN-β by GC infection is an additive effect of both cGAS/STING and TLR4 pathways in human and murine cells
To further characterize the role of STING and cGAS in GC infection, we infected STING- and cGAS-deficient mouse BMDMs and evaluated IFN-β induction. After GC infection, IFN-β induction was reduced in both cGAS−/− and STING−/− BMDMs (Figure 3A). The active component of LPS/LOS is lipid A, a partially conserved glycolipid that anchors LPS into the outer membrane of gram-negative bacteria, including GC. E5564 is a well-characterized synthetic lipid A analog that interferes with TLR4 signaling by acting as a competitive inhibitor of LPS/LOS to block the binding of lipid A to the TLR4 co-receptor, MD-2 (Kim et al., 2007). When WT, cGAS- and STING-deficient cells were infected with GC in the presence of E5564, IFN-β production was reduced in WT cells and abrogated in cGAS- and STING-deficient cells (Figure 3A). These data indicate that complete induction of IFN-β is dependent on both the cGAS/STING and TLR4/MD-2 signaling pathways.
Figure 3. Induction of IFN-β by GC infection is completely dependent on both cGAS/STING and TLR4 pathways.

(A) WT, cGAS−/−, and STING−/− BMDMs were infected with GC strain FA1090 in the absence or presence of E5564 (10 μg/ml) for 6 hr. (B–D) WT, TLR4−/−, cGAS−/−, and TLR4/cGAS−/− BMDMs were infected with GC strain FA1090, transfected with GC DNA (3μg/ml) or stimulated with LOS (100ng/ml). Levels of IFN-β mRNA (A) were determined by qPCR and normalized to GAPDH levels. Levels of IFN-β (B), TNF-α (C) and IL-6 (D) were measured by ELISA after 18 hr. Data are represented as mean ± SD of one experiment of three experiments (each performed in triplicate) (A–D) that yielded similar results. Asterisks indicate that differences are statistically significant (*p < 0.05, **p < 0.01, and ***p < 0.001). ND: not detected. See also Figure S3.
To validate our findings on the role of LOS in GC-induced IFN-β production in human cells, WT and cGAS KO THP-1 cells were infected in the absence and presence of E5564. WT cells infected in the presence of the drug, had reduced IFN-β levels. Strikingly, the IFN-β response in cGAS KO cells infected in the presence of E5564 was completely abrogated (Figure S3), indicating that both cGAS and TLR4 are responsible for IFN-β induction after GC infection both in human and murine cells. Consistent with its known role in inhibiting LPS-induced innate immune activation, E5564 abrogated the effects of GC LOS in the activation of type I IFN (Figure S3). To confirm our data, we used a genetic approach and generated a TLR4/cGAS double-deficient mouse line and evaluated type I IFN induction by GC infection in BMDMs. Confirming our previous data, type I IFN induction by GC infection was partially reduced in TLR4- and cGAS-deficient BMDMs, and more importantly, we did not observe IFN-β induction in TLR4/cGAS double KO BMDMs (Figure 3B), showing an additive effect of the two different pathways (TLR4 and cGAS) in type I IFN induction by GC. As observed in THP-1 cells, TNF-α and IL-6 production were normal in cGAS−/− BMDMs, and partially decreased in TLR4−/− and TLR4/cGAS double KO BMDMs (Figure 3C and 3D).
cGAMP is induced after GC infection and GC DNA transfection
When double-stranded DNA is delivered inside the cytosol of eukaryotic cells, DNA is sensed by cGAS, which synthesizes the second messenger 2′3′-cGAMP. Our data demonstrated that cGAS is involved in type I IFN induction by GC infection. To further characterize this response, we transfected THP-1 cells with GC DNA, or infected the cells with living gonococci and characterized cGAMP production. We began by heat-treating cytosolic extracts of transfected or infected cells; as cGAMP is heat stable, heat-treated lysates can be used in a bioassay for cGAS activity. These lysates were used to stimulate type I IFN production from THP-1 cells permeabilized with digitonin, as described before (Zhang et al., 2014). IFN-β was induced in THP-1 cells stimulated for four hours with transferred cytosol from either transfected or infected cells (Figure 4A), consistent with the conclusion that cGAMP was induced after both GC DNA transfection and N. gonorrhoeae infection. To further confirm the induction of cGAMP by GC, we analyzed the same heat-resistant lysates by liquid chromatography combined with tandem mass spectrometry (MS). In both GC DNA transfected and FA1090 infected THP-1 cells (Figure 4B), 2′3′-cGAMP was produced (the cGAMP peak is represented by one asterisk, left panel). As a control, the samples were spiked with synthetic 3′3′-cGAMP (represented by two asterisks, left panel), which has a distinct mass spectra (Zhang et al., 2013). MS/MS of 2′3′-cGAMP corresponding peak revealed several fragmented ions with the expected m/z values for product ions of cGAMP, and were observed in similar ratios (Figure 4B, right panel) (Wu et al., 2013), confirming the induction of 2′3′-cGAMP by GC infection. To measure the levels of cGAMP induced by GC infection, we infected WT and cGAS KO THP-1 cells and quantified cGAMP by MS. WT THP-1 cells produced around 12500 cGAMP molecules per cell after GC infection, but no cGAMP was detected in GC infected cGAS KO THP-1 cells (Figure 4C).
Figure 4. Neisseria gonorrhoeae infection induces 2′3′-cGAMP.

(A) Undifferentiated THP-1 cells were transfected with GC DNA (6h), infected with GC (8h), or left untreated, and cytosolic extracts from those cells were incubated with digitonin-permeabilized THP-1 cells for 4 hr. Levels of IFN-β mRNA were measured by qPCR and normalized to GAPDH levels. Data are represented as the mean ± SD of one experiment of three experiments (each performed in triplicate) that yielded similar results. (B) LC-MS/MS profile of THP-1 isolates. Left panels: reconstructed ion chromatograms (XIC) of the cGAMP fragment ion at m/z 312.0492 (10 ppm tolerance) following LC-MS/MS fragmentation of the cGAMP protonated ion (m/z 675.1). Peaks marked with (*) are from 2′3′-cGAMP; (**) is from 3′3′-cGAMP, spiked at 500 pg to each isolate. Right panels: tandem mass spectra (MS/MS) of the peak observed at 9.3 min (left panels). Peaks marked with (*) are 2′3′-cGAMP fragment ions and are observed in similar ratios. (C) cGAS+/+ and cGAS KO THP-1 cells were infected with GC (8h) or left untreated and cGAMP levels were determined by quantitative mass spectrometry.
Type I interferon induction by GC is independent of the type IV secretion system
Bacteria can stimulate cytosolic signaling pathways in different ways. One common mechanism is the delivery of bacterial ligands to the host cytosol via specialized secretion systems (Burdette et al., 2011). The type IV secretion system (T4SS) is present in a number of bacterial pathogens (e.g., Bordetella pertussis and Legionella pneumophila) and is involved in the transport of proteins and DNA that are required for pathogenesis (Backert and Meyer, 2006). To evaluate the role of T4SS in IFN-β induction we used two different strains of GC, MS11, which naturally expresses T4SS, and FA1090, which does not express a T4SS. We also used genetically modified strains of MS11 (HH549-traH and HH557-traH/+traH), where traH present in the Gonococcal Genetic Island (GGI) and essential for T4SS function was deleted and/or complemented (Dominguez et al., 2011; Hamilton et al., 2005). All of the commonly used laboratory strains (FA1090, MS11), genetically modified strains (HH549-traH, HH557-traH/+traH, ND500) and clinical strains (#39, #61) were able to induce IFN-β (Figure 5A) and TNF-α (Figure 5B) to similar levels. Our results indicate that T4SS is not involved in IFN-β induction by GC infection in vitro, since the presence or absence of T4SS did not affect type I IFN production.
Figure 5. IFN-β induction by GC is independent of T4SS.

Undifferentiated THP-1 cells were infected for 6 hr with different strains of GC, which were either capable or incapable of expressing T4SS. The presence of T4SS is indicated by (+) and (−) signs. Levels of IFN-β (A) and TNF-α (B) mRNA were measured by qPCR and normalized to GAPDH levels. Data are represented as mean ± SD of one experiment of two experiments (each performed in triplicate) that yielded similar results.
Type I interferon induced by GC infection impairs bacterial clearance
Our results demonstrated that GC induced type I interferon in human monocytes and murine macrophages. Although the role of type I IFNs in viral infection has been well described, the exact effect of type I IFNs in bacterial infection is variable and controversial (Monroe et al., 2010). To test whether type I IFNs are important for GC killing in macrophages, we infected unprimed and IFN-β-primed WT BMDMs and evaluated in vitro killing. When WT BMDMs were primed with IFN-β, we observed increased GC survival when compared to unprimed cells (Figure 6A) suggesting that IFN-β plays an important role in GC survival in macrophages.
Figure 6. Type I IFN-induced by GC infection impairs bacterial clearance.

(A) Survival of gonococci in WT BMDMs unprimed or IFN-β-primed, were evaluated at 1, 6, 12, and 18 hr post infection. (B) Survival of gonococci in human neutrophils unprimed or primed with type I IFN were evaluated at 1, 2, 6 and 12 hr post infection; n=3 donors. (C–D) Survival of gonococci in WT, TLR4−/−, and cGAS−/− BMDMs unprimed (C), or IFN-β-primed (D), were evaluated at 1, 6, 12, 18, 24, and 36 hr post infection. (A–D) Cells were infected with GC FA1090 (MOI=5) and live gonococci were retrieved and counted. Data are represented as pool ± SD of two independent experiments (each performed in triplicate) that yielded similar results. Asterisks indicate that differences are statistically significant using a non-parametric Mann-Whitney (A–B) or non-parametric ANOVA test (C–D) (*p < 0.05, **p < 0.01, and ***p < 0.001).
Polymorphonuclear leukocytes (PMNs) are one of the first inflammatory cell types to respond and migrate towards inflammation sites, especially in bacterial infections. Human infection with GC triggers a potent local inflammatory response driven by PMNs. GC is phagocytosed and destroyed by neutrophils, but it was already described that GC can survive inside these cells, which can also serve as a replicative niche, allowing dissemination (Criss and Seifert, 2012). To evaluate if type I IFN plays a similar role in human neutrophils as in murine BMDMs, and increases GC survival, we purified PMNs from healthy donors and infected IFN-β primed or unprimed neutrophils with GC strain FA1090 and evaluated bacterial survival. As demonstrated for mouse BMDMs, IFN-β primed neutrophils were less efficient in GC killing (Figure 6B), supporting our data that type I IFN is detrimental to GC infection.
Our results demonstrated that both TLR4 and cGAS are important to type I IFN induction in GC infection, and that type I IFN increases GC survival. To test the role of TLR4 and cGAS in the control of GC infection, we infected WT, TLR4−/−, and cGAS−/− BMDMs and evaluated in vitro killing. TLR4- and cGAS-deficiency enhanced bacterial in vitro killing compared to WT cells after 6 h of infection (Figure 6C). The lack of TLR4 or cGAS might have other consequences besides decreased IFN-β levels, therefore we evaluated whether IFN-β played a role in GC killing by performing the same assay in IFN-β-primed BMDMs. When TLR4−/− and cGAS−/− BMDMs were primed with IFN-β, the enhanced killing efficiency was abrogated (Figure 6D), thus promoting bacterial survival.
Type I interferon controls iron intracellular pool affecting GC survival
Bacteria are limited in their capacity to multiply in vivo due to several defense mechanisms present in the host such iron deprivation (Payne, 1993). Like many other human pathogens, GC adapts to the environment encountered during infection through regulation of several genes, including iron-responsive genes (Agarwal et al., 2005). It was demonstrated both in vitro and in vivo that iron regulated genes (i.e. tbpA, tbpB, fbpA) are upregulated in GC, suggesting a low iron environment during infection (Agarwal et al., 2008; McClure et al., 2015).
To evaluate the transcriptional response to GC during in vitro infection, we infected WT, TLR4−/−, and cGAS−/− BMDMs with GC FA1090 and measured the expression of GC iron-responsive genes (mpeR, tbpB, and fetA) induced in low iron conditions, and a non-iron-responsive gene serC (Zughaier et al., 2014). As previously reported, mpeR, tbpB, and fetA were induced in GC after infection in WT BMDMs (Figure 7A), suggesting an iron-limiting environment. The same genes were even more upregulated in both TLR4−/− and cGAS−/− BMDMs (Figure 7A), suggesting a lower intracellular iron pool in TLR4−/− and cGAS−/− BMDMs compared to WT BMDMs, which may account for the decreased bacterial survival. We found that these iron-responsive genes were significantly upregulated in monocyte-associated gonococci at 6 hr, compared to 1 hr after infection. In contrast, expression of serC was largely unchanged. Taken together, the data suggest that N. gonorrhoeae survives in association with macrophages and responds to the iron-limiting environment by upregulating iron-responsive genes to facilitate iron acquisition and survival, and that TLR4 and cGAS would affects this response.
Figure 7. cGAS and TLR4 pathways control the iron intracellular pool and GC survival.

(A) WT, TLR4−/−, and cGAS−/− BMDMs were infected with GC FA1090 (MOI=5) and iron-responsive and unresponsive genes in GC FA1090 at 6 hr post infection compared to 1 hr post infection were evaluated by qPCR and normalized to RNA16S levels. (B–D) WT, TLR4−/− and cGAS−/− BMDMs unprimed (B and D), or IFN-β-primed (C) were infected with GC FA1090 (MOI=5) for 6, 12, and 18 hr. Levels of Hamp1 (B and C) and Ferroportin (D) were determined by qPCR and normalized to GAPDH levels. Data are represented as mean ± SD of two (A), and three (B–D) experiments (each performed in triplicate) that yielded similar results. Asterisks indicate that differences are statistically significant (*p < 0.05, **p < 0.01, and *** p < 0.001).
Macrophages play a role in innate immunity and iron homeostasis, and a key cellular iron regulator involved in iron-limiting host defense is hepcidin (Ganz, 2002). It was demonstrated that GC infection induced the expression of murine Hamp1 (Hepcidin homologue), thus enhancing the intracellular iron pool, which is beneficial to bacterial growth (Zughaier et al., 2014). When we infected TLR4−/− and cGAS−/− BMDMs with GC FA1090, Hamp1 induction was substantially reduced in both TLR4−/− and cGAS−/− cells compared to WT cells (Figure 7B), but when primed with IFN-β, cGAS−/− BMDMs fully recovered, and TLR4−/− BMDMs partially recovered Hamp1 induction (Figure 7C). These results suggest a role for TLR4 and cGAS induced type I IFN and regulation of the intracellular iron pool that may affect GC survival.
Another gene involved in the control of intracellular iron is Ferroportin, the only iron export gene identified, whose expression is regulated by Hamp1 (Liu et al., 2005). Hamp1 binds to ferroportin and both are internalized and degraded, contributing to an increase in the intracellular iron pool (Nemeth et al., 2004). Once iron acquisition is important for bacterial growth, downregulation of ferroportin by Hamp1 is beneficial for bacteria survival, as demonstrated for C. trachomatis, L. pneumophila and Salmonella (Chlosta et al., 2006; Paradkar et al., 2008). To evaluate whether the downregulation of Hamp1 in TLR4- and cGAS-deficient BMDMs affects the expression of ferroportin, we infected WT, TLR4−/−, and cGAS−/− BMDMs and measured ferroportin expression. When WT BMDMs were infected with GC, ferroportin expression was strongly downregulated, but when TLR4- and cGAS-deficient BMDMs were infected, almost no reduction was observed (Figure 7D). This result supports our hypothesis of a lower intracellular iron pool in both cGAS- and TLR4-deficient BMDMs, which is detrimental to the bacteria.
Discussion
Neisseria gonorrhoeae is an exclusively human pathogen transmitted by sexual contact and the risk of severe infection may increase due to the increasing number of antibiotic-resistant strains. Much remains to be learned about the immune response to GC. Because humans can repeatedly acquire the infection, with little change in either the severity or duration of disease, it is becoming clear that each episode of infection does not induce protective immunity against the bacteria (Fox et al., 1999). In the present work, we demonstrated that GC infection induces type I IFN production through both cGAS/STING and TLR4 pathways, raising the possibility that IFN-α/β may play a role in human infection.
N. gonorrhoeae has always been thought of as an illness in which the lipid A component of its LOS drives the immediate inflammatory response. Like E. coli, the gonococcus expresses a hexa-acylated lipid A (Zhou et al., 2014), and like E. coli, all six fatty acid substituents are required to drive a strong innate immune response via TLR4/MD2, including the production of proinflammatory cytokines, such as TNF-α, and the production of IFN-α/β (Liu et al., 2010). Indeed, given the high potency of gonococcal LOS, it was surprising to learn that there was more to the innate immune pathways during gonococcal infection than just TLR4 activation.
Exogenous DNA that gains access to the cytosol is a particularly potent and clear danger signal. For this reason, much effort has been spent both identifying and characterizing the response to DNA in the host cytosol. STING was identified as a critical molecule in the recognition of cytosolic nucleic acids. Infection by several pathogens, including herpes simplex virus type 1, L. monocytogenes, Chlamydia muridarum, M. tuberculosis, and dengue virus, have been reported to have STING-dependent IFN-β induction (Barber, 2014). While the production of type I IFN has long been known to be involved in anti-viral immunity, it is increasingly recognized as important in bacterial and parasitic immunity as well (Barber, 2014). The extent to which pathogens, especially prokaryotes, produce STING-activating cyclic dinucleotides remains to be determined. However, as many organisms presumably fail to express endogenous ligands for STING, it is important for the innate immune system to have other means to activate the type I IFN pathway as is seen, for example, by the products of cGAS activity, which create mammalian cyclic dinucleotides. The overall picture for gonococcus infection is that it induces type I IFN via TLR recognition, primarily by the recognition of LOS by TLR4/MD-2. In addition to TLR recognition, bacteria can induce type I IFN via intracellular DNA sensors and exogenous cyclic dinucleotides, which are both STING-dependent.
In some bacterial infections, such as L. monocytogenes, M. tuberculosis, and C. trachomatis, type I IFNs are detrimental to the host (Monroe et al., 2010). IFN-α/β receptor deficient mice infected with L. monocytogenes, exhibit lower bacterial loads in the liver and spleen (Carrero et al., 2004). In the case of Chlamydia infection, IFN-β impairs clearance of bacteria in the genital tract and lungs (Qiu et al., 2008). One explanation for why type I IFNs are sometimes detrimental to bacterial infection, is that IFN-β suppresses the immune response by inducing IL-10 (Carrero and Unanue, 2006). On the other hand, in the case of Group B streptococcus, Streptococcus pneumoniae, and E. coli, IFNAR-deficient mice exhibit decreased host survival and increased bacterial loads, probably due to the decreased levels of TNF-α and IFN-γ, both important to control bacterial growth (Mancuso et al., 2007). Our results showed that N. gonorrhoeae infection induced type I IFNs in human and murine cells via both TLR4 and cGAS, and that IFN-β induction was detrimental to in vitro killing of GC by murine BMDMs and human neutrophils.
The role of IFN-α/β in gonococcal infection in vivo has yet to be determined, but as in Chlamydia infection, type I IFN might be detrimental to the host, which is supported by our in vitro killing assay data. This would be highly likely during co-infection with Chlamydia, a very common clinical occurrence. It has also been demonstrated that type I IFNs inhibit NLRP1 and NLRP3 activation, thus inhibiting IL-1β production (Guarda et al., 2011), and that GC induces NLRP3 and ASC-dependent cell death in THP-1 cells (Duncan et al., 2009). We speculate that IFN-β induction in GC infection could lead to NLRP3 inflammasome inhibition.
Recently, Hansen et al. reported that induction of IFN-β in THP-1 cells after L. monocytogenes infection was dependent on STING, cGAS, and IFI16 (Hansen et al., 2014). IFI16 expression was induced in THP-1 cells after phorbol-12-myristate-13-acetate (PMA) differentiation, but cGAS expression was highly reduced. IFN-β was induced by L. monocytogenes only in PMA-differentiated THP-1 cells, suggesting a major role for IFI16 in Listeria infection. The cyclic dinucleotide c-di-AMP produced by L. monocytogenes also plays an important role in type I IFN induction, in a cGAS-independent and STING-dependent manner (Collins et al., 2015; Woodward et al., 2010). In our hands, however, IFN-β was induced at similar levels in both PMA-differentiated and undifferentiated THP-1 cells after gonococcal infection (data not shown). This data is consistent with our observation that IFI16 does not appear to play a role in GC infection.
Several recent studies have identified cGAS as a broad-specificity DNA sensor. Our results demonstrated cGAS activation by a bacterium mostly known for its extracellular residence, in contrast to C. trachomatis. However, it was already reported that GC invade and survive inside human cells (Apicella et al., 1996; Chateau and Seifert, 2015; Post et al., 2002). We have presented multiple lines of evidence supporting our conclusion that cGAS is involved in gonococcal pathogenesis including the observations that transferred lysates from THP-1 cells that were either infected with GC or transfected with gonococcal DNA activated IFN-α/β in digitonin-permeabilized THP-1 cells, and MS analysis identified 2′3′-cGAMP in lysates from both GC-infected and GC DNA-transfected cells. So far, how gonococcal DNA reaches the cytosol to activate cGAS is unknown, but GC also activates the cytosolic sensor NLRP3, and the mechanism by which the agonist is delivered to the cytosol to engage NLRP3 activation has also not been described (Guarda et al., 2011). Recently it was reported that GC can access and replicate inside the cytosol of macrophages (Chateau and Seifert, 2015). Spontaneous bacteriolysis in L. monocytogenes that reaches the cytosol activates AIM2 through the recognition of its DNA (Sauer et al., 2011) and a similar mechanism could be responsible for cGAS activation by GC. The dual mechanism of IFN-a/b responses via TLRs and cGAS appears to be unique to gonococcal infection.
Bacterial pathogens have evolved a wide range of strategies to invade and survive in human cells despite the presence of multiple host defense mechanisms. N. gonorrhoeae, like many other human pathogens, must adapt to the new environment encountered to survive and proliferate. Iron is an essential nutrient for both host and microbes, being required for many metabolic processes. A tight control of intracellular and extracellular iron levels is important for the host to control bacterial infection (Nairz et al., 2010). Both the male and female genital tract are iron-depleted environments (Agarwal et al., 2005; Agarwal et al., 2008; McClure et al., 2015), thus GC requires different strategies to acquire iron, such as expression of outer membrane receptors that bind directly to host iron sources (i.e. transferrin) (Mickelsen and Sparling, 1981). Our results suggest that one effect of type I IFN-priming by cGAS and TLR4 in GC infection is to increase host Hamp1 (hepcidin) expression to increase iron retention in macrophages. Zughaier et al. have proposed that increased hepcidin expression along with downregulation of BDH2, during GC infection increases the labile iron pool in macrophages and facilitates gonoccocal iron acquisition. Our findings suggest that type I IFN may be an important mediator of GC-induced changes in iron metabolism in macrophages and that IFN is responsible for the enhanced survival and replication that we observed for WT but not cGAS- and TLR4-deficient cells.
The inflammation processes induced during N. gonorrhoeae infection are responsible for many of the clinical complications caused by this organism, including infertility associated with pelvic inflammation. The inflammatory process is generally considered an important component of the host immune response to pathogens, including the production of cytokines and chemokines as well as cell recruitment. N. gonorrhoeae is a highly adapted human pathogen, which has not evolved many anti-inflammatory mechanisms, but possesses many other immunosuppressive properties. Understanding the innate immune response to GC infection is important to the development of a vaccine. The data presented in this work indicate that GC infection induces type I IFN through both DNA sensors cGAS and TLR4, leading to 2′3′-cGAMP production. Whether type I IFN is involved in controlling gonorrhea pathogenesis, or is detrimental to the host, is still an open question, but it could provide strategies to control bacterial growth or avoid inflammation and cell death.
Experimental Procedures
Ethics Statement
The protocols and consent forms for experiments with human cells were approved by the Institutional Research Board from UMass Medical School (IRB H-10368)
Neisseria gonorrhoeae strains
Strains used in this study were Neisseria gonorrhoeae FA1090 wt (PorB.1B, streptomycin resistant, serum resistant) (Fox et al., 1999), MS11 and ND500. Gonococcal strains HH549-traH (T4SS mutant) and HH557-traH/+traH (T4SS complemented) were generated as described before (Hamilton et al., 2005). The two clinical gonococcal isolates (#39 and #61) were a gift of P.A. Rice (UMass Medical School, Worcester, MA) and are deidentified specimens from patients seen at an STI clinic in China (Li et al., 2014).
Cell culture, stimulation, and ELISA
WT, TLR4−/−, MyD88/TRIF−/− immortalized macrophages and WT, cGAS, and STING−/− BMDMs were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco-BRL) containing 4 mM glutamine and 10% fetal bovine serum (FBS). Undifferentiated WT, cGAS KO, and IFI16 KO THP-1 cells were grown in RPMI 1640/glutamine supplemented with 10% FBS. Transient transfections with GC DNA were performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cells were infected or stimulated as stated and supernatant collected after 16 hr. ELISA for mouse IFN-β was performed as described (Roberts et al., 2007). TNF-α and IL-6 ELISAs (R&D Systems) were performed following the manufacturer’s instructions
Preparation of endogenous cGAMP and STING activation assay
After transfection with GC DNA or GC infection, 2 × 107 cells were lysed in hypotonic buffer (10 mM Tris-HCl [pH 7.4], 10 mM KCl, 1.5 mM MgCl2). The lysates were homogenized by douncing, heated at 95°C for 5 min and centrifuged at 17,000 g for 15 min to remove denatured proteins. The heat-resistant supernatant/buffer ratio 1:1 (40 mM Hepes [pH 7.2], 10 mM MgCl2, 4 mM ATP, 4 mM GTP, digitonin 20 μg/mL) were incubated with 106 THP-1 cells.
Bactericidal assay on bone marrow-derived macrophages and human neutrophils
To determine the efficiency of gonococci killing by WT, TLR4−/−, and cGAS−/− BMDMs or human neutrophils, we used the killing assay protocol described before (Zughaier et al., 2014). Briefly, 1 × 106 BMDMs were seeded in a tissue culture plate and allowed to adhere overnight prior to infection with GC FA1090 at an MOI 5. After 1 hour of infection at 37 °C to allow for phagocytosis, cells were washed four times with antibiotic-free medium containing 10% heat-inactivated FBS. To quantify viable adherent and intracellular gonococci at 1, 6, 12, 18, 24 and 36 hr post phagocytosis for BMDMs, and at 1, 2, 6, and 12 hr for human neutrophils, cells were resuspended in 0.5% saponin in sterile antibiotic-free medium containing 10% heat-inactivated FBS and incubated for 5 min at 37 °C, then vortexed thoroughly to permeate cell membranes. The lysed cell mixture was serially diluted in sterile medium containing 0.5% saponin and cultured on chocolate agar plates followed by 24 hr incubation at 37 °C with 5% CO2, after which viable GC colonies were counted. When needed, BMDMs were primed overnight with murine recombinant IFN-β (100U/ml). PMNs were primed with recombinant human type I IFN-α (500U/ml).
Statistical Analysis
All data were analyzed using an unpaired, two-tailed Student’s t-test with a 95% confidence interval, a non-parametric Mann-Whitney or a non-parametric ANOVA (Kruskal-Wallis) (Prism; GraphPad Software, Inc.). Data are represented as means ± SD.
Supplementary Material
Highlights.
Induction of type I IFN by Neisseria gonorrhoeae depends on the cGAS/STING axis
Complete induction of type I IFN is dependent on both cGAS/STING and TLR4 pathways
Neisseria gonorrhoeae infection induces the production of noncanonical 2′3′-cGAMP
Type I IFN is detrimental for Neisseria gonorrhoeae killing by murine macrophages
Acknowledgments
We thank Eisai Inc. (Japan) for providing E5564. This work was supported by NIH grant AI084048 (PAR, DTG), U19AI084048 (EKJ, DTG) and R37GM540060 (DTG). W.A.A. was supported by a fellowship from CNPq (Brazil). The authors declare no competing financial interests.
Footnotes
Author contributions
W.A.A., K.A.F., E.K.J., and D.T.G. conceived and designed experiments. W.A.A., S.A., S.M., S.A.S., and T.S. performed experiments. J.P.D. and V.H. provided reagents. W.A.A., K.A.F., E.K.J., and D.T.G. analyzed data. W.A.A., E.K.J. and D.T.G. wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agarwal S, King CA, Klein EK, Soper DE, Rice PA, Wetzler LM, Genco CA. The gonococcal Fur-regulated tbpA and tbpB genes are expressed during natural mucosal gonococcal infection. Infect Immun. 2005;73:4281–4287. doi: 10.1128/IAI.73.7.4281-4287.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Sebastian S, Szmigielski B, Rice PA, Genco CA. Expression of the gonococcal global regulatory protein Fur and genes encompassing the Fur and iron regulon during in vitro and in vivo infection in women. J Bacteriol. 2008;190:3129–3139. doi: 10.1128/JB.01830-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apicella MA, Ketterer M, Lee FK, Zhou D, Rice PA, Blake MS. The pathogenesis of gonococcal urethritis in men: confocal and immunoelectron microscopic analysis of urethral exudates from men infected with Neisseria gonorrhoeae. J Infect Dis. 1996;173:636–646. doi: 10.1093/infdis/173.3.636. [DOI] [PubMed] [Google Scholar]
- Backert S, Meyer TF. Type IV secretion systems and their effectors in bacterial pathogenesis. Curr Opin Microbiol. 2006;9:207–217. doi: 10.1016/j.mib.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annu Rev Immunol. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- Barber GN. Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr Opin Immunol. 2011;23:10–20. doi: 10.1016/j.coi.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber GN. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 2014;35:88–93. doi: 10.1016/j.it.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Barker JR, Koestler BJ, Carpenter VK, Burdette DL, Waters CM, Vance RE, Valdivia RH. STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during Chlamydia trachomatis infection. MBio. 2013;4:e00018–00013. doi: 10.1128/mBio.00018-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrero JA, Calderon B, Unanue ER. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med. 2004;200:535–540. doi: 10.1084/jem.20040769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrero JA, Unanue ER. Lymphocyte apoptosis as an immune subversion strategy of microbial pathogens. Trends Immunol. 2006;27:497–503. doi: 10.1016/j.it.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Charrel-Dennis M, Latz E, Halmen KA, Trieu-Cuot P, Fitzgerald KA, Kasper DL, Golenbock DT. TLR-independent type I interferon induction in response to an extracellular bacterial pathogen via intracellular recognition of its DNA. Cell Host Microbe. 2008;4:543–554. doi: 10.1016/j.chom.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chateau A, Seifert HS. Neisseria gonorrhoeae survives within and modulates apoptosis and inflammatory cytokine production of human macrophages. Cell Microbiol. 2015 doi: 10.1111/cmi.12529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlosta S, Fishman DS, Harrington L, Johnson EE, Knutson MD, Wessling-Resnick M, Cherayil BJ. The iron efflux protein ferroportin regulates the intracellular growth of Salmonella enterica. Infect Immun. 2006;74:3065–3067. doi: 10.1128/IAI.74.5.3065-3067.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR, Scharn CR, Stamm CE, Levine B, Chen ZJ, et al. Cyclic GMP-AMP Synthase Is an Innate Immune DNA Sensor for Mycobacterium tuberculosis. Cell Host Microbe. 2015;17:820–828. doi: 10.1016/j.chom.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criss AK, Seifert HS. A bacterial siren song: intimate interactions between Neisseria and neutrophils. Nat Rev Microbiol. 2012;10:178–190. doi: 10.1038/nrmicro2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez NM, Hackett KT, Dillard JP. XerCD-mediated sitespecific recombination leads to loss of the 57-kilobase gonococcal genetic island. J Bacteriol. 2011;193:377–388. [Google Scholar]
- Duncan JA, Gao X, Huang MT, O’Connor BP, Thomas CE, Willingham SB, Bergstralh DT, Jarvis GA, Sparling PF, Ting JP. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J Immunol. 2009;182:6460–6469. doi: 10.4049/jimmunol.0802696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox KK, Thomas JC, Weiner DH, Davis RH, Sparling PF, Cohen MS. Longitudinal evaluation of serovar-specific immunity to Neisseria gonorrhoeae. Am J Epidemiol. 1999;149:353–358. doi: 10.1093/oxfordjournals.aje.a009820. [DOI] [PubMed] [Google Scholar]
- Ganz T. The role of hepcidin in iron sequestration during infections and in the pathogenesis of anemia of chronic disease. Isr Med Assoc J. 2002;4:1043–1045. [PubMed] [Google Scholar]
- Gratz N, Hartweger H, Matt U, Kratochvill F, Janos M, Sigel S, Drobits B, Li XD, Knapp S, Kovarik P. Type I interferon production induced by Streptococcus pyogenes-derived nucleic acids is required for host protection. PLoS Pathog. 2011;7:e1001345. doi: 10.1371/journal.ppat.1001345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34:213–223. doi: 10.1016/j.immuni.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Hamilton HL, Dominguez NM, Schwartz KJ, Hackett KT, Dillard JP. Neisseria gonorrhoeae secretes chromosomal DNA via a novel type IV secretion system. Mol Microbiol. 2005;55:1704–1721. doi: 10.1111/j.1365-2958.2005.04521.x. [DOI] [PubMed] [Google Scholar]
- Hansen K, Prabakaran T, Laustsen A, Jorgensen SE, Rahbaek SH, Jensen SB, Nielsen R, Leber JH, Decker T, Horan KA, et al. Listeria monocytogenes induces IFNbeta expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J. 2014;33:1654–1666. doi: 10.15252/embj.201488029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan PW, Snyder LA, Saunders NJ. Strain-specific differences in Neisseria gonorrhoeae associated with the phase variable gene repertoire. BMC Microbiol. 2005;5:21. doi: 10.1186/1471-2180-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Li S, Su X, Le W, Jiang F, Wang B, Rice PA. Antimicrobial susceptibility of Neisseria gonorrhoeae isolates from symptomatic men attending the Nanjing sexually transmitted diseases clinic (2011 inverted question mark2012): genetic characteristics of isolates with reduced sensitivity to ceftriaxone. BMC Infect Dis. 2014;14:622. doi: 10.1186/s12879-014-0622-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, John CM, Jarvis GA. Phosphoryl moieties of lipid A from Neisseria meningitidis and N. gonorrhoeae lipooligosaccharides play an important role in activation of both MyD88- and TRIF-dependent TLR4-MD-2 signaling pathways. J Immunol. 2010;185:6974–6984. doi: 10.4049/jimmunol.1000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XB, Nguyen NB, Marquess KD, Yang F, Haile DJ. Regulation of hepcidin and ferroportin expression by lipopolysaccharide in splenic macrophages. Blood Cells Mol Dis. 2005;35:47–56. doi: 10.1016/j.bcmd.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, Tomasello F, Gambuzza M, Macri G, Ruggeri A, et al. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol. 2007;178:3126–3133. doi: 10.4049/jimmunol.178.5.3126. [DOI] [PubMed] [Google Scholar]
- Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe. 2012;11:469–480. doi: 10.1016/j.chom.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure R, Nudel K, Massari P, Tjaden B, Su X, Rice PA, Genco CA. The Gonococcal Transcriptome during Infection of the Lower Genital Tract in Women. PLoS One. 2015;10:e0133982. doi: 10.1371/journal.pone.0133982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickelsen PA, Sparling PF. Ability of Neisseria gonorrhoeae, Neisseria meningitidis, and commensal Neisseria species to obtain iron from transferrin and iron compounds. Infect Immun. 1981;33:555–564. doi: 10.1128/iai.33.2.555-564.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe KM, McWhirter SM, Vance RE. Induction of type I interferons by bacteria. Cell Microbiol. 2010;12:881–890. doi: 10.1111/j.1462-5822.2010.01478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nairz M, Schroll A, Sonnweber T, Weiss G. The struggle for iron - a metal at the host-pathogen interface. Cell Microbiol. 2010;12:1691–1702. doi: 10.1111/j.1462-5822.2010.01529.x. [DOI] [PubMed] [Google Scholar]
- Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- Paradkar PN, De Domenico I, Durchfort N, Zohn I, Kaplan J, Ward DM. Iron depletion limits intracellular bacterial growth in macrophages. Blood. 2008;112:866–874. doi: 10.1182/blood-2007-12-126854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne SM. Iron acquisition in microbial pathogenesis. Trends Microbiol. 1993;1:66–69. doi: 10.1016/0966-842x(93)90036-q. [DOI] [PubMed] [Google Scholar]
- Post DM, Phillips NJ, Shao JQ, Entz DD, Gibson BW, Apicella MA. Intracellular survival of Neisseria gonorrhoeae in male urethral epithelial cells: importance of a hexaacyl lipid A. Infect Immun. 2002;70:909–920. doi: 10.1128/iai.70.2.909-920.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prantner D, Darville T, Nagarajan UM. Stimulator of IFN gene is critical for induction of IFN-beta during Chlamydia muridarum infection. J Immunol. 2010;184:2551–2560. doi: 10.4049/jimmunol.0903704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pridmore AC, Jarvis GA, John CM, Jack DL, Dower SK, Read RC. Activation of toll-like receptor 2 (TLR2) and TLR4/MD2 by Neisseria is independent of capsule and lipooligosaccharide (LOS) sialylation but varies widely among LOS from different strains. Infect Immun. 2003;71:3901–3908. doi: 10.1128/IAI.71.7.3901-3908.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H, Fan Y, Joyee AG, Wang S, Han X, Bai H, Jiao L, Van Rooijen N, Yang X. Type I IFNs enhance susceptibility to Chlamydia muridarum lung infection by enhancing apoptosis of local macrophages. J Immunol. 2008;181:2092–2102. doi: 10.4049/jimmunol.181.3.2092. [DOI] [PubMed] [Google Scholar]
- Roberts ZJ, Goutagny N, Perera PY, Kato H, Kumar H, Kawai T, Akira S, Savan R, van Echo D, Fitzgerald KA, et al. The chemotherapeutic agent DMXAA potently and specifically activates the TBK1-IRF-3 signaling axis. J Exp Med. 2007;204:1559–1569. doi: 10.1084/jem.20061845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, Hyodo M, Hayakawa Y, Woodward JJ, Portnoy DA, et al. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun. 2011;79:688–694. doi: 10.1128/IAI.00999-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens DS. Biology and pathogenesis of the evolutionarily successful, obligate human bacterium Neisseria meningitidis. Vaccine. 2009;27(Suppl 2):B71–77. doi: 10.1016/j.vaccine.2009.04.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storek KM, Gertsvolf NA, Ohlson MB, Monack DM. cGAS and Ifi204 cooperate to produce type I IFNs in response to Francisella infection. J Immunol. 2015;194:3236–3245. doi: 10.4049/jimmunol.1402764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahle L, Marq JB, Brini A, Hausmann S, Kolakofsky D, Garcin D. Activation of the beta interferon promoter by unnatural Sendai virus infection requires RIG-I and is inhibited by viral C proteins. J Virol. 2007;81:12227–12237. doi: 10.1128/JVI.01300-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, Schmid-Burgk JL, Schmidt T, Hornung V, Cole ST, et al. Mycobacterium tuberculosis Differentially Activates cGAS- and Inflammasome-Dependent Intracellular Immune Responses through ESX-1. Cell Host Microbe. 2015;17:799–810. doi: 10.1016/j.chom.2015.05.003. [DOI] [PubMed] [Google Scholar]
- Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, Vance RE, Stallings CL, Virgin HW, Cox JS. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe. 2015;17:811–819. doi: 10.1016/j.chom.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward JJ, Iavarone AT, Portnoy DA. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science. 2010;328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shi H, Wu J, Sun L, Chen C, Chen ZJ. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell. 2013;51:226–235. doi: 10.1016/j.molcel.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yeruva L, Marinov A, Prantner D, Wyrick PB, Lupashin V, Nagarajan UM. The DNA sensor, cyclic GMP-AMP synthase, is essential for induction of IFN-beta during Chlamydia trachomatis infection. J Immunol. 2014;193:2394–2404. doi: 10.4049/jimmunol.1302718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Gao X, Broglie PM, Kebaier C, Anderson JE, Thom N, Apicella MA, Sempowski GD, Duncan JA. Hexa-acylated lipid A is required for host inflammatory response to Neisseria gonorrhoeae in experimental gonorrhea. Infect Immun. 2014;82:184–192. doi: 10.1128/IAI.00890-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zughaier SM, Kandler JL, Shafer WM. Neisseria gonorrhoeae modulates iron-limiting innate immune defenses in macrophages. PLoS One. 2014;9:e87688. doi: 10.1371/journal.pone.0087688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.