

Abstract
Allenbach and colleagues have recently reported for the first time the results of an intriguing study of the histopathologic, immunopathologic and gene expression differences in muscle biopsy tissue from adult dermatomyositis (DM) patients who do and do not have circulating MDA5 autoantibodies (anti-MDA5). Anti-MDA5 were originally identified in a clinically-defined subset of DM patients whose disease was expressed predominately in the skin for unusually long periods of time without accompanying muscle weakness [i.e., “clinically-amyopathic DM” (CADM)] and were at risk for acute, rapidly-progressive form of interstitial lung disease (ILD). As an academic dermatologist in the United States of America (USA) having a career-long interest in the CADM subset, I would like to share my perspective on the results of the work by Allenbach and colleagues and offer some suggestions for additional study in this area. But to do so most effectively, I first would like to review the clinical concept of CADM and its association with anti-MDA5 antibody production and a potentially-fatal form of (ILD).
Keywords: Amyopathic dermatomyositis (ADM), classic dermatomyositis (CDM), clinically-amyopathic dermatomyositis (CADM, hypomyopathic dermatomyositis (HDM), interstitial lung disease (ILD), melanoma differentiation associated protein 5 (MDA5), MDA5 autoantibody (anti-MDA5), nitric oxide synthetase (NOS)
Introduction
In its classical form, dermatomyositis (DM) (referred to here as “classical DM”) has traditionally been thought of as a single clinical entity defined by the presence of the Hallmark cutaneous manifestations and skeletal muscle weakness resulting from a characteristic pattern of autoimmune myositis. The DM pattern of autoimmune inflammation can also produce disease in other organ systems in a variable fashion from patient to patient (e.g., arthritis/arthralgia, interstitial lung disease (ILD), fasciitis, calcinosis, vasculopathy, cardiomyopathy).
However, it known that adult-onset classical DM can also be a paraneoplastic phenomenon. Approximately 1 in 5 individuals who develop classical DM at 50 years of age or beyond will have an internal malignancy diagnosed within two years before or two years after the diagnosis of DM.
It is also recognized that the pattern of illness experienced by DM onset in children and adolescents is somewhat different clinically from that experienced by disease-onset in adults. Juvenile-onset DM presents an increased risk of multiorgan vasculopathy, fasciitis and soft tissue calcinosis but a decreased risk of associated internal malignancy and ILD.
A major challenge to caring for DM patients is the fact that this autoimmune process is expressed clinically in a highly variable fashion from patient to patient. Some patients die quickly from aggressive systemic disease manifestations such as rapidly-progressive ILD while others have skin-predominant disease manifestations throughout their disease course.
In a clinically heterogeneous disease process such as this, identifying subgroups (subsets) of patients affected by the disease process in a similar fashion (e.g., clinical features, laboratory abnormalities) can be of great therapeutic and prognostic benefit. Traditionally, subsets of DM patients have been defined by having clinical features in common such as ILD or associated internal malignancy.
More recently, serologically-defined subsets of DM patients have been identified by virtue of sharing a common autoantibody specificity. An example would be the “anti-synthetase syndrome”. DM and polymyositis patients that produce autoantibodies to histidyl transfer RNA synthetase (Jo-1) often share a similar clinical constellation including fever, myositis, polyarthritis, ILD, the Mechanic’s hand skin lesion and Raynaud’s phenomenon. In addition, it has been reported by several groups that adult-onset classical DM patients who are found to produce autoantibodies to transcription intermediary factor 1-gamma (TIF1-λ) have an increased risk for an associated internal malignancy (1,2).
Allenbach and colleagues have recently reported for the first time the results of an intriguing study of the histopathologic, immunopathologic and gene expression differences in muscle biopsy tissue from adult dermatomyositis (DM) patients who do and do not have circulating MDA5 autoantibodies (anti-MDA5) (3). Anti-MDA5 were originally identified in a CADM patients having an increased risk for rapidly-progressive, potentially-fatal ILD. As an academic dermatologist having a career-long interest in CADM, I would like to share my perspective on the results of the work by Allenbach and colleagues and offer suggestions for additional study in this area. But to do so most effectively, I would first like to review the progress that has been made in better understanding the clinical concepts of CADM, DM-associated ILD and their association with anti-MDA5 antibody production, focusing especially on the work in this area that has been reported over the past decade.
Clinically-amyopathic DM
It has been recognized that individuals can display the hallmark cutaneous manifestations of DM for abnormally long periods of time (≥6 months) without developing clinically-significant muscle involvement (i.e., the absence of muscle weakness and normal levels of muscle-specific blood enzymes including creatine kinase and aldolase). Astute clinicians in the past recognized the existence of such patients and referred to them anecdotally as “dermatomyositis sine myositis” (4,5). However, other than a few case reports at that time there were no published data describing this clinical variant of DM. The presumption of many at that time was that all such patients if followed long enough would ultimately develop muscle weakness as was the case with the patients reported by Krain in 1975 (4).
In 1991 the author and a younger colleague reported the clinical and laboratory features of six “skin disease-only” DM patients under the designation of “amyopathic DM” (6). This designation had been informally used in earlier writings by Carl Pearson who was a renowned American rheumatologist clinician-scholar in the field of dermatomyositis/polymyositis (7).
The case definition of “amyopathic DM” that we initially proposed for the purpose of clinical research included the presence of biopsy-confirmed Hallmark cutaneous changes of DM present for 24 months or longer without the development of clinically-significant muscle weakness nor elevated blood levels of muscle enzymes (creatine kinase, aldolase). However, in published case series of classical DM at that time it was very unusual to have clinical evidence of muscle involvement follow the initial onset of DM skin inflammation by more than 2–3 months. We therefore subsequently revised our case definition of amyopathic DM by reducing the lag interval from 24 to 6 months (8).
In addition, we added the following exclusion criteria to the case definition of amyopathic DM: (I) Treatment with systemic immunosuppressive therapy for two consecutive months or longer within the first 6 months after skin disease onset, as such therapy could mask/prevent the development of clinically-significant myositis and (II) use of drugs known to be capable of producing isolated DM-like skin changes at the time of DM skin disease onset (e.g., hydroxyurea).
Subsequently, our definition of amyopathic DM was criticized by the argument that more extensive muscle testing had not been required for our case definition [e.g., electromyography (EMG), muscle biopsy, magnetic resonance imaging, muscle spectroscopy, muscle ultrasonography]. The implication was that had such testing been performed, early evolving, “subclinical” evidence of myositis would have been identified in all of patients meeting our definition of amyopathic DM.
We had previously seen some patients who displayed the hallmark cutaneous manifestations of DM but no muscle weakness for 6 months or longer but who were found upon laboratory testing to have abnormalities in muscle enzymes, electromyography, or muscle biopsy. As these patients were “clinically-amyopathic” we felt that they deserved to be recognized in some way, but not as amyopathic DM. Thus, we proposed the designation “hypomyopathic DM” for such patients and suggested that individuals with amyopathic DM and hypomyopathic DM could be recognized together under the umbrella designation “clinically-amyopathic DM” (CADM). This designation has gained traction reflected by the fact that a PubMed search on February 11, 2017 using the search phrase “clinically amyopathic dermatomyositis” returned 121 citations, with only eight coming from research groups with which the author has been affiliated. Figure 1 illustrates the clinically-defined subsets of DM that are widely recognized today.
Figure 1.
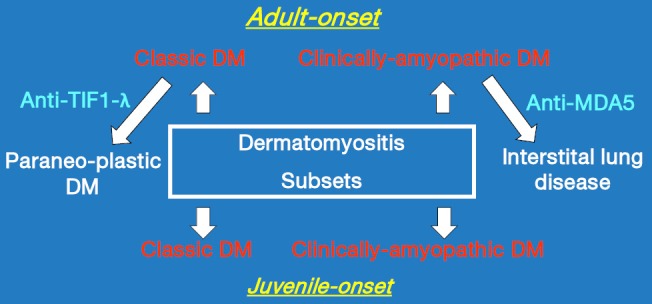
Currently recognized subsets of DM as defined by having similar constellations of clinical, pathologic and serologic features. Anti-TIF1-λ, transcriptional intermediary factor-1 gamma autoantibody; Anti-MDA5, melanoma differentiation association protein 5 autoantibody.
Some have questioned the concept of CADM with the argument that if an aggressive search for muscle inflammation is carried out [i.e., all five muscle enzymes (aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase, creatine kinase and aldolase)], as well as magnetic resonance imaging and muscle biopsy), virtually all juvenile-onset CADM patients will be found to have “sub-clinical” evidence of myositis (9). (Such patients would meet the criteria for the hypomyopathic subtype of CADM).
This argument has been voiced most ardently for patients with juvenile-onset CADM. This is understandable as compared to adult-onset classic DM, juvenile-onset classic DM patients tend to have more aggressive, potentially disabling muscle disease as well as higher rates of multiorgan vasculopathy and soft tissue dystrophic calcification. Also, early diagnosis and intervention with aggressive systemic immunomodulatory treatment has been shown to moderate disability and improve long-term prognosis in juvenile-onset classic DM patients. However, a key question here is does “subclinical” laboratory, biopsy or imaging evidence of muscle inflammation in the absence of clinical muscle weakness in the context of juvenile-onset CADM justify the risks of the aggressive, long-term, systemic immunosuppressive therapy approach that is used for patients with juvenile-onset classic DM.
Some children presenting with CADM have been observed to have their DM skin changes smolder or spontaneously remit with or without symptomatic treatment never having displayed muscle weakness or other systemic complications of classic DM (10,11). In addition, the author has cared for several such children in the past (personal unpublished observation). It will take long-term outcome studies involving larger numbers of patients to resolve these questions.
This conundrum can be viewed from the perspective of the recent pseudo-epidemics of cancer that have in part been attributed to overly aggressive screening with modern laboratory, pathologic and radiologic techniques. Some argue that the marked increase in the incidence of malignant melanoma in the USA over the past 20 years has been due largely to over diagnosis resulting from changes in skin biopsy indication and interpretation of skin biopsy findings (12-15). Similar arguments have led the United States Preventive Services Task Force (USPSTF) in 2012 to no longer recommend prostate cancer screening by prostate-specific antigen (PSA) blood testing (14,16,17).
ILD in CADM
Several clinical-pathologic subtypes ILD can be encountered in DM patients. Some subtypes are milder and carry a good prognosis (chronic ILD) while others are rapidly-progressive and potentially-fatal even with heroic subspecialty medical treatment and support (acute ILD). This latter life-threatening form of ILD can occur in both adult-onset classic DM as well adult-onset CADM but is very rare in juvenile-onset disease. ILD and internal malignancy are encountered much less frequently and juvenile-onset classic DM and CADM compared to adult-onset disease. The association of the more severe form of acute ILD with the presence of circulating anti-MDA5 antibodies will be discussed in the next section below.
The rate of occurrence of ILD in DM patients is a function of both ethnicity and how one defines ILD. Traditionally in the United States ILD has occurred in approximately 10% of adult-onset classic DM patients while in Japan it occurs in approximately 40%. This is in part due to an apparent genetic and/or environmental predisposition of Asian DM patients to ILD compared to USA and European DM patients. In addition, in Japan virtually all new DM patients appear to undergo a high-resolution chest computed tomography (CT) scan as part of their initial evaluation. A Japanese DM patient is considered to have ILD if the appropriate radiologic abnormalities are present even in the absence of pulmonary symptoms. However, in clinical practice in the USA it is relatively uncommon for DM patients to undergo a high-resolution chest CT exam as part of their initial evaluation unless they have some clinical evidence of ILD (chronic non-productive cough, shortness of breath, dyspnea upon exertion, abnormalities on chest auscultation, abnormalities on screening pulmonary function tests).
The prevalence of pulmonary function test abnormalities in USA cohorts of academic dermatology department-ascertained classic DM and CADM patients has recently been reported by George and coworkers (18). They found abnormal pulmonary function tests in 20 of 47 (43%) patients with CADM and 34 of 69 (49%) with classic DM (these differences were not statistically significant). In their patients having abnormal pulmonary function test results who also had chest CT exams performed, ILD by radiographic findings was present in only 38% with CADM and 39% with classic DM. Thus, the clinical significance of abnormal pulmonary function test results in the majority of both CADM and classic DM patients in this study could be questioned.
It is known that the individual tests of pulmonary function are subject to a 10% false positive result rate (19). Thus, if three individual tests of pulmonary function were performed (e.g., spirometry, diffusing capacity and lung volume) there could be a false positive rate of 30% presuming that pulmonary function test abnormality was defined by there being a single abnormal test.
As the patients in the study of George and coworkers were ascertained by visits to a dermatology department, it is understandable that the rapidly-progressive, potentially-fatal form of ILD was under represented in their study. In addition, neither anti-MDA5 screening results nor the presence or absence of the atypical cutaneous lesions that have been reported to occur in anti-MDA5 antibody positive CADM patients were presented in this report.
Concurrent with this publication, Moghadam-Kia and coworkers reported the results of a survey of anti-MDA5 autoantibody production in matched cohorts of classic DM and CADM patients identified in a USA academic rheumatology division (20,21). They found the anti-MDA frequency to be identical in both classic DM and CADM patients (~13%). Anti-MDA5 was significantly associated with cutaneous ulcers, digital tip ulcerations and puffy fingers as well as ILD. This confirms the earlier work of Fiorentino and coworkers who had reported an association of anti-MDA5 in DM patients with cutaneous ulcers, tender palmar papules and ILD (22,23). These atypical cutaneous features are illustrated in Figure 2.
Figure 2.

Examples of acral skin ulceration that has been associated with anti-MDA 5 and rapidly progressive ILD in CADM patients. The upper left and right images display solitary deep punched-out ulcers overlying the dorsal aspect of metacarpophalangeal joints of two different DM patients. The lower image shows papules of the palmar creases that have undergone ulceration in a DM patient. The photos in this figure were obtained from DM patients that the author of this review had cared for clinically during his earlier career. Unfortunately, the clinical and laboratory information concerning these patients are not available to determine their classification status as classic DM versus CADM. ILD, interstitial lung disease; CADM, clinically-amyopathic DM.
In the study by Moghadam-Kia and coworkers, ILD was defined as radiographic pulmonary fibrosis noted on chest radiography or high resolution chest CT exam. The rates of ILD in classic DM and CADM patients in this study were not significantly different (26–31%). ILD was significantly associated with anti-MDA5. A large majority of the ILD patients in this study had the acute, rapidly-progressive subtype. There was not a significant difference in the rates of acute, rapidly-progressive ILD between classic DM and CADM patients in this study (5–8%).
The above work argues that as many as 1 in 3 adult-onset CADM patients seen in USA medical school settings have radiologic evidence of ILD with anti-MDA5 serving as a biomarker for the rapidly-progressive, potentially-fatal subgroup of patients. In addition, approximately 1 in 4 CADM patients with radiologic evidence of ILD have the rapidly-progressive, life-threatening variety of ILD. As an academic dermatologist, I personally was less likely to have been in a position to diagnose and primarily care for such acutely ill DM patients as they would typically have been triaged to the care of rheumatologists and pulmonary medicine specialists.
Thus, the above analysis would argue that it at least some of the adult-onset CADM patients under my care should have developed clinical manifestations of chronic ILD. However, in 40 years of caring for CADM patients on a weekly subspecialty clinic basis in four different USA academic dermatology departments, this never happened. In addition, since the concept of CADM is now approximately 25 years old, it is surprising that there would not be more published reports of adult-onset CADM patients developing clinical symptoms of ILD in a subacute or chronic fashion. Thus, the clinical and prognostic significance of pulmonary function test abnormalities and high-sensitivity radiologic imaging evidence of ILD in adult-onset CADM patients in a non-acute dermatologic setting could be questioned.
As discussed in the Treatment Philosophies section below, I have personally used a symptomatic approach to treating adult-onset CADM skin disease activity over my four-decade clinical career. My approach has not included screening asymptomatic patients for ILD by pulmonary function test or radiologic imaging. When systemic therapy has been needed I have first used aminoquinoline antimalarial therapy (hydroxychloroquine and if needed hydroxychloroquine plus compounded quinacrine for resistant patients).
Both the cutaneous and muscular manifestations of DM have been shown to be associated with a class I interferon gene signature. The aminoquinoline antimalarial drugs such as hydroxychloroquine are known to inhibit toll-like receptor-7 signaling (TLR-7) and thereby dampen the upregulated TLR-7 induced interferon production that has been reported in DM skin and muscle inflammation.
Several groups have reported an upregulated interferon gene signature in peripheral blood cells of CADM patients with anti-MDA5 positive, rapidly-progressive ILD (24,25). However, it has been difficult to find direct published evidence that the inflamed pulmonary tissue of such patients display evidence of upregulated class I interferon. If this is found to be the case in future studies, it might be possible that the chronic antimalarial therapy used to treat DM skin inflammation in my CADM patients over the years might have had a dampening effect on the development of clinically-apparent ILD as well. This perhaps could account for a lower-than-predicted rate of symptomatic ILD occurring in the antimalarial-treated adult-onset CADM patients that I have seen.
Alternatively, perhaps the reason I have not personally encountered clinical evidence of ILD in any of my adult-onset CADM patients has been the result of luck alone. It will be interesting to learn from future studies whether anti-MDA5 screening might help identify CADM patients at increased risk for developing chronic ILD as well as the acute, rapidly progressive form of ILD.
Anti-MDA5 as a serologic marker for rapidly-progressive, potentially-fatal ILD complicating CADM
In 1990, Tokiyama and coworkers reported two female Japanese amyopathic DM patients who developed rapidly-progressive ILD eventuating in death of both patients from pulmonary insufficiency despite aggressive systemic immunosuppressive therapy (26). Others subsequently confirmed that Japanese CADM patients had an increased risk for acute, rapidly-progressive ILD compared to classical DM patients.
In 2003, Sato and coworkers reported a new autoantibody in Japanese CADM patient sera that precipitated a 140 kD polypeptide in a radiolabeled immunoprecipitation protocol. This autoantibody could not be detected in control serum specimens from patients with other autoimmune connective tissue disorders. Sato and coworkers designated this autoantibody as “CADM-140” (27-29).
The CADM-140 autoantigen was subsequently shown to be identical to the melanoma differentiation-associated protein 5 (MDA5) that had previously been identified independently by others (30). MDA5 is a RIG-I-like receptor dsRNA helicase enzyme that is encoded in humans by the IFIH1 gene (31,32). MDA5 functions as a virus sensor through its actions as a pattern recognition receptor for dsRNA. This is congruent with the molecular mimicry paradigm for human autoimmunity. While one could make a case for IFIH1 being the most appropriate designation for this autoantibody specificity, MDA5 has now replaced CADM-140 as the consensus designation.
The presence of anti-MDA5 is now generally accepted as being a risk factor for the development of potentially-fatal ILD in DM patients, especially those with the CADM sub-phenotype. In Japan the primary clinical association of anti-MDA5 in DM patient populations has been an increased risk for the rapidly-progressive, potentially-fatal form of ILD in patients presenting with the CADM phenotype. In both Japanese and North American DM patients, anti-MDA5 positive DM patients have also been shown to have an increased frequency of rare, acral ulcerative skin changes that are different from the consensus Hallmark skin changes of DM (22,23).
We are greatly indebted to all of the dedicated, collaborative Japanese investigators who have taken the lead on developing new insight and therapeutic strategies in this area. Space here does not allow the recognition of all such contributions.
Figure 3 illustrates the relationships that exist between classic DM, CADM, ILD, atypical skin features and anti-MDA 5 autoantibody production.
Figure 3.
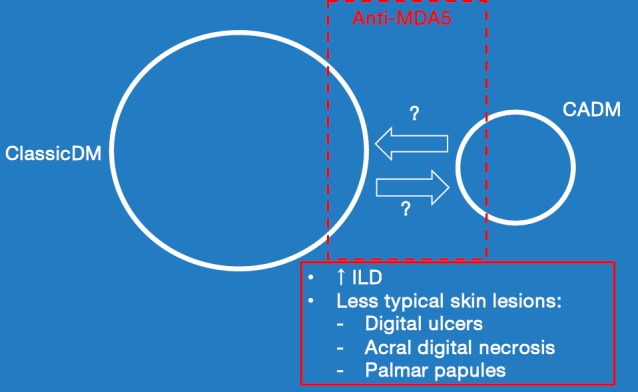
Clinical associations of anti-MDA5 autoantibody production in DM patients. The circles representing classic DM and CADM are drawn in proportion to their relative prevalence in a population-based study. Recent studies have demonstrated that anti-MDA5 as determined by solid-phase immunoassay, occurs in low percentages of both North American classic DM and CADM patients (~13%). CADM patients who produce anti-MDA5 display a characteristic set of atypical, acral upper extremity cutaneous findings that are listed here. The arrow pointing from the CADM circle to the classic DM circle represents those CADM patients who over the first decade after diagnosis develop overt muscle weakness thereby allowing them to be re-classified as classic DM. The arrow pointing from the classic DM circle to the CADM circle was inserted here to point out a misconception in the literature. After some classic DM patients have been treated aggressively to the point of remission, their skin disease activity can return without a return of muscle disease activity. Some workers have incorrectly referred to such patients as having CADM at this stage of their illness. However, it is inappropriate to use the designation “amyopathic” for a patient who has previously displayed overt weakness from “clinically-significant” DM muscle disease activity. One designation that has been suggested for such patients is “post-myopathic DM”. The skin disease activity in such patients is characteristically very difficult to treat. CADM, clinically-amyopathic DM.
CADM treatment philosophies
There is no debate about the need for patients having either adult-onset or juvenile-onset CADM who develop acute, rapidly-progressive ILD to be treated aggressively with systemic immunosuppressive therapy in an intensive care support setting. The recently confirmed observation that anti-MDA5 levels may reflect clinical ILD activity and fall or disappear with treatment-induced ILD remission promises to improve management of this life-threatening complication (33,34). However, there are differences of opinion on how adult-onset CADM patients without ILD should be managed over time.
During medical school in the USA in the late 1960’s and early 1970’s, I was taught that whenever in doubt treat the patient (i.e., their symptoms and physical findings) rather than confusing, inconsistent laboratory or imaging results. And, during my dermatology clinical training in the mid-1970s I was taught that DM patients which we would later designate as having adult-onset CADM should be treated aggressively with high-dose systemic corticosteroids and steroid-sparing immunosuppressives with the hope that we could abort the progression from the skin disease activity-only sub-phenotype to the development of the systemic manifestations of DM (35). However, the personal experience of watching an adult female with classic DM having only mild muscle weakness die after a total dose of 52.5 mg of oral methotrexate from what at autopsy was felt to be methotrexate-induced pulmonary hypersensitivity gave me reason to question this uniformly aggressive treatment approach to all DM patients, even those with no demonstrable weakness.
Later in my own clinical practice I decided to adopted a “just in time” approach to treating symptomatic skin changes in adult-onset CADM patients. I was in the position to follow such patients carefully over time and therefore felt that if systemic disease manifestations did develop in my CADM patients we would recognize that at an early stage and switch to a more aggressive systemic immunosuppressive treatment strategy. It appears that over the past 25 years many dermatologists have adopted the same approach (36,37).
After topical immunosuppressive therapy has been tried and failed (e.g., corticosteroids, pimecrolimus), single agent or combination oral antimalarials therapy is given (hydroxychloroquine or hydroxychloroquine plus compounded quinacrine). A short-term burst and taper of systemic corticosteroids (e.g., oral prednisone or intramuscular triamcinolone) can be used for symptom control while awaiting the slower-acting aminoquinoline antimalarials to each fully-therapeutic equilibrium blood levels (6–8 weeks). Oral dapsone therapy and oral methotrexate therapy can be of value in some CADM patients (37-39). However, it has been our experience that high-dose intravenous immunoglobulin (IVIG) given on a monthly basis is most consistently able to control otherwise-refractory cutaneous manifestations of DM.
As mentioned in the section below it is the standard of care in USA pediatric rheumatology to treat juvenile-onset CADM patients from the outset like juvenile-onset classic DM patients with high-dose systemic steroids and steroid-sparing immunosuppressive therapy.
Prognosis of patients presenting with CADM
As the clinical concept of CADM is a rather new one, we do not yet have reliable long-term disease outcome data on such patients. However, over the past decade the clinical picture of CADM has started to come into sharper focus as a result of additional descriptive case reports, case series, summative systematic literature reviews and one population-based study (40-44).
Previously thought to be an extremely rare occurrence, there are now population-based epidemiologic data to suggest that incidence of CADM is approximately 20% of that of classic DM in the United States (44). This study by Bendewald and coworkers also supports previous work suggesting that a higher percentage of CADM patients are female compared to classic DM and that CADM carries a somewhat lower risk for association with internal malignancy compared to classic DM. It is of interest that this population-based study found that none of the six CADM patients that they identified (one juvenile-onset case, five adult-onset cases) were found to have lung disease.
Adult-onset CADM patients have several possible outcomes. A relatively small percentage have the risk of succumbing from their disease early as a result of an associated incurable internal malignancy or rapidly-progressive ILD. However as previously noted, adult-onset CADM patients appear to have a reduced risk of developing an associated internal malignancy compared to adult-onset classic DM patients. Most including this author still recommend that adult-onset CADM patients be screened annually for internal malignancy for at least two years after their diagnosis of CADM.
Some adult-onset CADM patients have been observed to go for very long periods of time without developing systemic manifestations of DM or internal malignancy. The author has personally cared for several patients who had gone for three decades or longer with intermittent Hallmark DM skin disease activity but no muscle weakness nor pulmonary disease (personal unpublished observation). In addition, one of the adult-onset CADM patients reported by Bendewald and coworkers had been followed for 20 years without developing systemic manifestations of DM (44).
As was originally reported by Krain, some patients presenting with adult-onset CADM will develop overt muscle weakness within 10 years after skin disease onset (4). The specific level of risk is not certain at this time. However, it would appear that patients who have gone 10 years or longer after presenting with CADM have an extremely low risk for developing systemic manifestations of DM thereafter (44).
The hypomyopathic DM sub-phenotype could be viewed as a lynchpin for the ongoing debate concerning the validity of the concept of CADM. These are patients who have otherwise met the criteria for CADM but who upon initial evaluation are found to have some “sub-clinical” muscle enzyme, electrophysiologic, biopsy or imaging evidence of muscle inflammation. Because of this, it might be assumed that patients with hypomyopathic DM would be at higher risk than patients with amyopathic DM for subsequently developing overt muscle weakness. However, to date this has not been found to be the case. In our extensive review of the literature in this area, patients with adult-onset hypomyopathic DM often continued to be asymptomatic with respect to muscle disease after prolonged periods of follow up (45-47).
And, there are even fewer systematically-ascertained outcome data on juvenile-onset CADM. In the USA it is currently the standard of care in pediatric rheumatology to treat juvenile-onset CADM with the same aggressive systemic immunomodulatory treatment regimens used in juvenile-onset classic DM patients. As such, it could be challenging to identify subjects presenting with isolated Hallmark cutaneous manifestations of DM for comparative clinical studies. However, as in adults there is published experience arguing that the prolonged presence of isolated skin manifestations of DM can spontaneously remit in children who might be considered to have CADM with only conservative symptomatic treatment (10,11).
Further perspective on the studies of Allenbach and coworkers
A group of European workers recently reported that subgroups of adult DM patients who did and did not produce circulating anti-MDA5 displayed different histologic and immunopathologic patterns of muscle biopsy inflammation. In addition, they observed a difference in the expression of interferon-stimulated genes in muscle biopsy tissue from the two subgroups. Finally, they reported that as opposed to the anti-MDA5 negative DM patients, the anti-MDA5 positive patients displayed the cytokine-inducible form of NOS, NOS2, on involved muscle fibers. As they observed NOS2 expression to co-localize with markers of muscle regeneration and markers of cell stress, they interpreted the presence of NOS2 as being a protective mechanism (3).
There are elements of the design of this study that impact interpretation of its results and conclusions. Muscle biopsy tissues from only 16 patients were examined in this study (6 from the anti-MDA5 positive DM patients and 7 from anti-MDA5 negative DM patients). Larger numbers of anti-MDA5 positive and negative DM patients will need to be examined to confirm the findings in this study.
In several places in this report is implied that the anti-MDA5 positive DM patient subgroup could be viewed as being equivalent to the previously described CADM subset. However, it is stated in this report that 72% of the anti-MDA5 positive DM patient subgroup in this study had no muscle weakness while only 45% had normal creatine kinase levels. However, as previously discussed the case definition of CADM excludes the presence of any clinical evidence of muscle weakness. Thus, approximately one quarter anti-MDA5 positive patients in this report would not meet the case definition of CADM. This view is supported by the fact that the mean blood level of creatine kinase in international units in the anti-MDA5 positive subgroup in this study was 500% above the upper normal limit in international units. In addition, recent studies of North American CADM patients indicate that only 13% of them are anti-MDA5 positive (20). It is unfortunate that this this issue was not more clearly addressed for the readers of this report. Perhaps in their future studies, Allenbach and coworkers might consider repeating their studies on DM patients meeting the above noted criteria for CADM who do and do not produce anti-MDA5 autoantibodies.
In addition, the anti-MDA5 positive and negative patient subgroups in this study were not stratified with respect to the type and intensity of immunomodulatory therapy that the study subjects had received prior to their muscle biopsies. Considering that all of the anti-MDA5 negative patients in this study had muscle weakness and as a group had very high mean creatine kinase blood levels (2,693 international units), it would be safe to assume that patients in this group would more likely have received more intense immunosuppressive therapy prior to and at the time of their muscle biopsies were performed compared to those in the anti-MDA5 positive subgroup. Had this been taken into account in the study design, the reported differences between the anti-MDA5 positive and negative groups might have been even more significant than was reported.
One of the more intriguing ideas raised in the report by Allenbach and coworkers is the possibility that variant inducible NOS expression might be a biomarker for a milder pattern of myositis associated with anti-MDA5 production. They observed the presence of NOS2 expression on anti-MDA positive muscle biopsy fibers in association with markers of muscle regeneration. NOS2 expression was not observed on anti-MDA5 negative muscle biopsy fibers. This led the authors to hypothesize that the inducible NOS2 expression in this clinical setting represented a protective mechanism inhibiting autoimmune-mediated muscle injury.
While it has been suggested that inducible NOS expression might play a role in healing healthy muscle tissue that has been damaged (48), its role in modulating autoimmune mediated myositis is not clear. It has been reported that inducible NOS2 has been indiscriminately expressed in the muscle tissue from myositis patients without association with clinical subtypes of autoimmune myositis (49,50). However, if the results of Allenbach and coworkers suggesting that the inducible NOS/nitric acid axis plays a role in the repair of damaged muscle in myositis patients are confirmed, this observation could be truly exciting with respect to new therapeutic strategies. Recent work by others suggesting that novel new strategies for therapeutically delivering nitrous oxide to damaged tissue in a clinical setting may be on the horizon (51-53).
Conclusions
Over the past 25 years, the clinical concept of adult-onset CADM has come into better focus. However, the clinical significance of juvenile-onset CADM continues to be debated. Autoantibodies to the MDA5 autoantigen, a protein involved in the interferon signaling pathway, have been shown to occur in a small percentage of adult-onset CADM patients who have an increased risk for acute rapidly-progressive, potentially-fatal ILD that when present requires aggressive systemic immunosuppressive therapy. This subset of patients also displays a characteristic set of atypical skin findings that includes acral skin ulcers and palmar papules on the upper extremities.
The author’s personal experience in clinically caring for a large number of adult-onset CADM patients over four decades leads him to question the clinical significance of pulmonary function tests and high-resolution chest CT imaging abnormalities in such patients in the absence of pulmonary symptoms and/or abnormalities on physical examination of the lung.
Allenbach and coworkers have reported that DM patients who are anti-MDA5 positive display a variant pattern of inflammation and gene expression on muscle biopsy tissue compared to DM patients who do not produce this autoantibody. This variant pattern of muscle inflammation was associated with the presence of the cytokine-inducible form of nitric oxide synthase, NOS2, on muscle fibers. As Allenbach and coworkers observed NOS2 expression to co-localize with markers of muscle regeneration and markers of cell stress, they interpreted the presence of NOS2 as being a protective mechanism. As NOS2 has been suggested to play a role in healing healthy muscle tissue that has been damaged, this observation could be exciting with respect to new therapeutic strategies. Recent work by others suggests that novel new approaches for therapeutically delivering nitric oxide to damaged tissue in a clinical setting may be on the horizon.
Acknowledgements
None.
Footnotes
Conflicts of Interest: The author has no conflicts of interest to declare.
References
- 1.Fiorentino DF, Chung LS, Christopher-Stine L, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1gamma. Arthritis Rheum 2013;65:2954-62. 10.1002/art.38093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fiorentino D, Casciola-Rosen L. Autoantibodies to transcription intermediary factor 1 in dermatomyositis shed insight into the cancer-myositis connection. Arthritis Rheum 2012;64:346-9. 10.1002/art.33402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allenbach Y, Leroux G, Suarez-Calvet X, et al. Dermatomyositis With or Without Anti-Melanoma Differentiation-Associated Gene 5 Antibodies: Common Interferon Signature but Distinct NOS2 Expression. Am J Pathol 2016;186:691-700. 10.1016/j.ajpath.2015.11.010 [DOI] [PubMed] [Google Scholar]
- 4.Krain L. Dermatomyositis in six patients without initial muscle involvement. Arch Dermatol 1975;111:241-5. 10.1001/archderm.1975.01630140099013 [DOI] [PubMed] [Google Scholar]
- 5.Callen JP, Jorizzo JL. Amyopathic dermatomyositis (dermatomyositis siné myositis). J Am Acad Dermatol 1992;26:505-6. 10.1016/S0190-9622(08)80590-7 [DOI] [PubMed] [Google Scholar]
- 6.Euwer RL, Sontheimer RD. Amyopathic dermatomyositis (dermatomyositis siné myositis). Presentation of six new cases and review of the literature. J Am Acad Dermatol 1991;24:959-66. 10.1016/0190-9622(91)70153-S [DOI] [PubMed] [Google Scholar]
- 7.Pearson CM. Polymyositis and dermatomyositis. In: McCarty DJ. editor. Arthritis (and allied conditions). 9 ed. Philadelphia: Lea & Febiger, 1979:742-61. [Google Scholar]
- 8.Euwer RL, Sontheimer RD. Amyopathic dermatomyositis: a review. J Invest Dermatol 1993;100:124S-127S. 10.1038/jid.1993.35 [DOI] [PubMed] [Google Scholar]
- 9.Oberle EJ, Bayer ML, Chiu YE, et al. How Often are Pediatric Patients with Clinically Amyopathic Dermatomyositis Truly Amyopathic? Pediatr Dermatol 2017;34:50-7. 10.1111/pde.13013 [DOI] [PubMed] [Google Scholar]
- 10.Plamondon S, Dent PB. Juvenile amyopathic dermatomyositis: Results of a case finding descriptive survey. J Rheumatol 2000;27:2031-4. [PubMed] [Google Scholar]
- 11.Peloro TM, Miller OF, 3rd, Hahn TF, et al. Juvenile dermatomyositis: a retrospective review of a 30-year experience. J Am Acad Dermatol 2001;45:28-34. 10.1067/mjd.2001.113686 [DOI] [PubMed] [Google Scholar]
- 12.Weyers W. The 'epidemic' of melanoma between under- and overdiagnosis. J Cutan Pathol 2012;39:9-16. 10.1111/j.1600-0560.2011.01831.x [DOI] [PubMed] [Google Scholar]
- 13.Glusac EJ. The melanoma 'epidemic', a dermatopathologist's perspective. J Cutan Pathol 2011;38:264-7. 10.1111/j.1600-0560.2010.01660.x [DOI] [PubMed] [Google Scholar]
- 14.Glusac EJ. The melanoma 'epidemic': lessons from prostate cancer. J Cutan Pathol 2012;39:17-20. 10.1111/j.1600-0560.2011.01848.x [DOI] [PubMed] [Google Scholar]
- 15.Swerlick RA, Chen S. The melanoma epidemic. Is increased surveillance the solution or the problem? Arch Dermatol 1996;132:881-4. 10.1001/archderm.1996.03890320029004 [DOI] [PubMed] [Google Scholar]
- 16.Cabarkapa S, Perera M, McGrath S, et al. Prostate cancer screening with prostate-specific antigen: A guide to the guidelines. Prostate Int 2016;4:125-9. 10.1016/j.prnil.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fleshner K, Carlsson SV, Roobol MJ. The effect of the USPSTF PSA screening recommendation on prostate cancer incidence patterns in the USA. Nat Rev Urol 2017;14:26-37. 10.1038/nrurol.2016.251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.George MD, Shah R, Kreider M, et al. Pulmonary function tests, interstitial lung disease and lung function decline in outpatients with classic and clinically amyopathic dermatomyositis. Br J Dermatol 2017;176:262-4. 10.1111/bjd.14771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vedal S, Crapo RO. False positive rates of multiple pulmonary function tests in healthy subjects. Bull Eur Physiopathol Respir 1983;19:263-6. [PubMed] [Google Scholar]
- 20.Moghadam-Kia S, Oddis CV, Sato S, et al. Antimelanoma Differentiation-associated Gene 5 Antibody: Expanding the Clinical Spectrum in North American Patients with Dermatomyositis. J Rheumatol 2017;44:319-25. 10.3899/jrheum.160682 [DOI] [PubMed] [Google Scholar]
- 21.Moghadam-Kia S, Oddis CV, Sato S, et al. Anti-Melanoma Differentiation-Associated Gene 5 Is Associated With Rapidly Progressive Lung Disease and Poor Survival in US Patients With Amyopathic and Myopathic Dermatomyositis. Arthritis Care Res (Hoboken) 2016;68:689-94. 10.1002/acr.22728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol 2011;65:25-34. 10.1016/j.jaad.2010.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koga T, Fujikawa K, Horai Y, et al. The diagnostic utility of anti-melanoma differentiation-associated gene 5 antibody testing for predicting the prognosis of Japanese patients with DM. Rheumatology (Oxford) 2012;51:1278-84. 10.1093/rheumatology/ker518 [DOI] [PubMed] [Google Scholar]
- 24.Sun WC, Sun YC, Lin H, et al. Dysregulation of the type I interferon system in adult-onset clinically amyopathic dermatomyositis has a potential contribution to the development of interstitial lung disease. Br J Dermatol 2012;167:1236-44. 10.1111/j.1365-2133.2012.11145.x [DOI] [PubMed] [Google Scholar]
- 25.Horai Y, Koga T, Fujikawa K, et al. Serum interferon-α is a useful biomarker in patients with anti-melanoma differentiation-associated gene 5 (MDA5) antibody-positive dermatomyositis. Mod Rheumatol 2015;25:85-9. 10.3109/14397595.2014.900843 [DOI] [PubMed] [Google Scholar]
- 26.Tokiyama K, Tagawa H, Yokota E, et al. Two cases of amyopathic dermatomyositis with fatal rapidly progressive interstitial pneumonitis. Ryumachi 1990;30:204-9; discussion 209-11. [PubMed] [Google Scholar]
- 27.Satoh S, Hirakata M, Suwa A, et al. Antibodies to 140 kDa proteins in Japanese patients with amyopathic dermatomyositis. Arthritis Rheumat 2001;44:S205. [Google Scholar]
- 28.Sato S, Suwa A, Kuwana M, et al. Clinical and immunological associations of autoantibodies to the 140 kDa polypeptide (the US autoantigen) in patients with clinically amyopathic dermatomyositis. Arthritis Rheum 2003;48:S102. [DOI] [PubMed] [Google Scholar]
- 29.Sato S, Hirakata M, Kuwana M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum 2005;52:1571-6. 10.1002/art.21023 [DOI] [PubMed] [Google Scholar]
- 30.Kang DC, Gopalkrishnan RV, Wu Q, et al. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci U S A 2002;99:637-42. 10.1073/pnas.022637199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: Association with rapidly progressive interstitial lung disease. Arthritis Rheum 2009;60:2193-200. 10.1002/art.24621 [DOI] [PubMed] [Google Scholar]
- 32.Nakashima R, Imura Y, Kobayashi S, et al. The RIG-I-like receptor IFIH1/MDA5 is a dermatomyositis-specific autoantigen identified by the anti-CADM-140 antibody. Rheumatology (Oxford) 2010;49:433-40. 10.1093/rheumatology/kep375 [DOI] [PubMed] [Google Scholar]
- 33.Muro Y, Sugiura K, Hoshino K, et al. Disappearance of anti-MDA-5 autoantibodies in clinically amyopathic DM/interstitial lung disease during disease remission. Rheumatology (Oxford) 2012;51:800-4. 10.1093/rheumatology/ker408 [DOI] [PubMed] [Google Scholar]
- 34.Matsushita T, Mizumaki K, Kano M, et al. Antimelanoma differentiation-associated protein 5 antibody level is a novel tool for monitoring disease activity in rapidly progressive interstitial lung disease with dermatomyositis. Br J Dermatol 2017;176:395-402. 10.1111/bjd.14882 [DOI] [PubMed] [Google Scholar]
- 35.Sontheimer RD. Clinically amyopathic dermatomyositis: what can we now tell our patients? Arch Dermatol 2010;146:76-80. 10.1001/archdermatol.2009.323 [DOI] [PubMed] [Google Scholar]
- 36.Callander J, Robson Y, Ingram J, et al. Treatment of clinically amyopathic dermatomyositis in adults: a systematic review. Br J Dermatol 2016. [Epub ahead of print]. 10.1111/bjd.14726 [DOI] [PubMed] [Google Scholar]
- 37.Ishibuchi H, Motegi S, Amano H, et al. Successful treatment with dapsone for skin lesions of amyopathic dermatomyositis. J Dermatol 2015;42:1019-21. 10.1111/1346-8138.12999 [DOI] [PubMed] [Google Scholar]
- 38.Cohen JB. Cutaneous involvement of dermatomyositis can respond to Dapsone therapy. Int J Dermatol 2002;41:182-4. 10.1046/j.1365-4362.2002.01409.x [DOI] [PubMed] [Google Scholar]
- 39.Konohana A, Kawashima J. Successful treatment of dermatomyositis with dapsone. Clin Exp Dermatol 1994;19:367. 10.1111/j.1365-2230.1994.tb01220.x [DOI] [PubMed] [Google Scholar]
- 40.Ikeda S, Arita M, Misaki K, et al. Incidence and impact of interstitial lung disease and malignancy in patients with polymyositis, dermatomyositis, and clinically amyopathic dermatomyositis: a retrospective cohort study. Springer Plus 2015;4:240. 10.1186/s40064-015-1013-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghazi E, Sontheimer RD, Werth VP. The importance of including amyopathic dermatomyositis in the idiopathic inflammatory myositis spectrum. Clin Exp Rheumatol 2013;31:128-34. [PMC free article] [PubMed] [Google Scholar]
- 42.Gerami P, Walling HW, Lewis J, et al. A systematic review of juvenile-onset clinically amyopathic dermatomyositis. Br J Dermatol 2007;157:637-44. 10.1111/j.1365-2133.2007.08055.x [DOI] [PubMed] [Google Scholar]
- 43.Gerami P, Schope JM, McDonald L, et al. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis sine myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol 2006;54:597-613. 10.1016/j.jaad.2005.10.041 [DOI] [PubMed] [Google Scholar]
- 44.Bendewald MJ, Wetter DA, Li X, et al. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol 2010;146:26-30. 10.1001/archdermatol.2009.328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.el-Azhary RA, Pakzad SY. Amyopathic dermatomyositis: retrospective review of 37 cases. J Am Acad Dermatol 2002;46:560-5. 10.1067/mjd.2002.120620 [DOI] [PubMed] [Google Scholar]
- 46.Mukamel M, Brik R. Amyopathic dermatomyositis in children: a diagnostic and therapeutic dilemma. J Clin Rheumatol 2001;7:191-3. 10.1097/00124743-200106000-00012 [DOI] [PubMed] [Google Scholar]
- 47.Walling HW, Gerami P, Sontheimer RD. Juvenile-onset clinically amyopathic dermatomyositis: an overview of recent progress in diagnosis and management. Paediatr Drugs 2010;12:23-34. 10.2165/10899380-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 48.Rigamonti E, Touvier T, Clementi E, et al. Requirement of inducible nitric oxide synthase for skeletal muscle regeneration after acute damage. J Immunol 2013;190:1767-77. 10.4049/jimmunol.1202903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tews DS, Goebel HH. Cell death and oxidative damage in inflammatory myopathies. Clin Immunol Immunopathol 1998;87:240-7. 10.1006/clin.1998.4527 [DOI] [PubMed] [Google Scholar]
- 50.Wanchu A, Khullar M, Sud A, et al. Nitric oxide production is increased in patients with inflammatory myositis. Nitric Oxide 1999;3:454-8. 10.1006/niox.1999.0261 [DOI] [PubMed] [Google Scholar]
- 51.Elnaggar MA, Subbiah R, Han DK, et al. Lipid-based carriers for controlled delivery of nitric oxide. Expert Opin Drug Deliv 2017:1-13. [Epub ahead of print]. 10.1080/17425247.2017.1285904 [DOI] [PubMed] [Google Scholar]
- 52.Pinto RV, Antunes F, Pires J, et al. Vitamin B3 metal-organic frameworks as potential delivery vehicles for therapeutic nitric oxide. Acta Biomater 2017;51:66-74. 10.1016/j.actbio.2017.01.039 [DOI] [PubMed] [Google Scholar]
- 53.Kim J, Yung BC, Kim WJ, et al. Combination of nitric oxide and drug delivery systems: tools for overcoming drug resistance in chemotherapy. J Control Release 2016. [Epub ahead of print]. 10.1016/j.jconrel.2016.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]