

Abstract
Aim
Canagliflozin is an SGLT2 inhibitor approved for the treatment of type‐2 diabetes. A dynamic population pharmacokinetic–pharmacodynamic (PK/PD) model relating 24‐h canagliflozin exposure profiles to effects on glycosylated haemoglobin was developed to compare the efficacy of once‐daily and twice‐daily dosing.
Methods
Data from two clinical studies, one with once‐daily, and the other with twice‐daily dosing of canagliflozin as add‐on to metformin were used (n = 1347). An established population PK model was used to predict full 24‐h profiles from measured trough concentrations and/or baseline covariates. The dynamic PK/PD model incorporated an E max relationship between 24‐h canagliflozin exposure and HbA1c‐lowering with baseline HbA1c affecting the efficacy.
Results
Internal and external model validation demonstrated that the model adequately predicted HbA1c‐lowering for canagliflozin once‐daily and twice‐daily dosing regimens. The differences in HbA1c reduction between the twice‐daily and daily mean profiles were minimal (at most 0.023% for 100 mg total daily dose [TDD] and 0.011% for 300 mg TDD, up to week 26, increasing with time and decreasing with TDD) and not considered clinically meaningful.
Conclusions
Simulations using this model demonstrated the absence of clinically meaningful between‐regimen differences in efficacy, supported the regulatory approval of a canagliflozin‐metformin immediate release fixed‐dose combination tablet and alleviated the need for an additional clinical study.
Keywords: Canagliflozin, diabetes mellitus, modelling, pharmacodynamics, pharmacokinetics, simulation
What is Already Known About This Subject
A fixed‐dose combination tablet of metformin immediate release and canagliflozin may improve patient convenience and compliance to antihypertensive agent therapy. Because metformin immediate release is typically administered twice‐daily for patients with type‐2 diabetes mellitus, the canagliflozin component was divided to 50‐mg and 150‐mg twice‐daily to provide the same currently approved daily dose (100‐mg and 300‐mg).
What This Study Adds
This population pharmacokinetic–pharmacodynamic exposure‐response analysis establishes a quantitative relationship between canagliflozin exposure and glycosylated haemoglobin response, and demonstrates that differences in canagliflozin dosing regimens would have little to no impact on the HbA1c response.
Tables of Links
| TARGETS |
|---|
| SGLT2 |
| LIGANDS |
|---|
| Canagliflozin |
| Metformin |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2.
Introduction
Diabetes is gaining a pandemic status, with a global prevalence in adults of approximately 9% and affecting approximately 415 million people 3, 4. Metformin, an oral biguanide that reduces hepatic glucose production, 5, 6 is typically the first line of therapy for glycaemic control. Disease progression often necessitates use of combination therapy with other antihyperglycaemic agents (AHA) that can complement the primary treatment for management of hyperglycaemia 7. Canagliflozin, a selective oral inhibitor of sodium‐glucose transporter‐2 (SGLT2), has a different mechanism of action than metformin. Canagliflozin is approved in the USA and numerous other countries for the treatment of adult patients with type‐2 diabetes mellitus (T2DM). The recommended canagliflozin dose is 100 or 300 mg once‐daily (QD), and is indicated as an adjunct to diet and exercise to improve glycaemic control 8, 9, 10. Canagliflozin reduces plasma glucose (PG) in individuals with hyperglycaemia by lowering the renal threshold for glucose excretion (RTG), thus causing increased urinary glucose excretion 11, 12, 13.
The canagliflozin concentration resulting in half maximal reduction (EC50) in RTG was estimated to be 32 ng ml–1 (95% confidence interval [CI]: 19; 45) corresponding to a 90% effective concentration (EC90) of 289 ng ml–1 and maximal decrease in RTG of approximately 64%.9 In a phase 3 study in patients with T2DM, monotherapy with canagliflozin 100 and 300 mg compared with placebo demonstrated a significant reduction of glycosylated haemoglobin (HbA1c) from baseline (8.0% HbA1c) to week 26 (–0.77%, –1.03% and 0.14%, respectively) 13. With both canagliflozin doses, reductions in both fasting and postprandial PG levels were observed that were consistent with the observed reductions in HbA1c. The increased efficacy observed with 300 mg canagliflozin was expected, as it provided more sustained maximal decrease in RTG than 100 mg canagliflozin and also provided an additional, nonrenal effect to lower postprandial PG 14. Because the pharmacodynamic (PD) effects of canagliflozin‐related increases in urinary glucose excretion are dependent on estimated glomerular filtration rate (eGFR), efficacy was reduced in patients with renal impairment due to reduced filtered glucose load 15, 16.
For patients requiring combined treatment with metformin and canagliflozin, the use of a fixed‐dose combination (FDC) tablet (comprised of metformin immediate release [IR] and canagliflozin), may improve patient convenience and compliance to AHA therapy. Because metformin IR is typically administered twice a day for patients with T2DM,5 the FDC was developed to be dosed twice‐daily (BID), with the canagliflozin component divided to provide the same 100 and 300 mg total daily dose (TDD) as currently approved for QD dosing (i.e., 50 mg and 150 mg BID) 17. Comparison of steady‐state pharmacokinetics (PK)/PD of canagliflozin administered either QD or BID at the same TDD of 100 and 300 mg in a study in healthy subjects showed that canagliflozin plasma area under the concentration‐time curves (AUC0‐24h,ss) for the QD vs. BID dosing regimens were equivalent. Although, as expected, the steady‐state maximum plasma concentration (Cmax,ss) of QD dosing was higher than the corresponding morning Cmax,ss of the BID regimen in this study; the 24‐h mean RTG for QD and BID regimens at both 100 and 300 mg TDDs were similar 18.
While the short‐term PK/PD data suggested similarity between QD and BID regimens given at the same TDD, no long‐term study was performed that directly compared the efficacy of different regimens. Three late‐stage studies were performed with canagliflozin treatment added on to metformin; two of these studies (12‐week study, NCT00642278 [Study1] 19, and 26‐week study, NCT01106677 [Study 2] 20 contained QD dosing of 100 and 300 mg vs. placebo, whereas a third study (18 weeks, NCT01340664 [Study 3] 21) contained 50 mg and 150 mg BID doses vs. placebo. The placebo‐subtracted least square mean reductions in baseline HbA1c seen with 100 mg and 300 mg QD doses in the first two studies (–0.76 and –0.62% for 100 mg QD; –0.92 and –0.77% for 300 mg QD, respectively) were somewhat greater than the corresponding reductions seen with 50 and 150 mg BID doses in the third study (–0.44% and –0.60%, respectively) 19, 20, 21. However, there were differences in other factors between the studies that limited the utility of such cross‐study comparisons (most notably baseline HbA1c was lower in the BID study than in the QD studies; Table S1). Therefore, the aim of the current analysis was to develop a robust model‐based solution that could account for differences in study populations and, in the absence of directly comparable long‐term study results, could assess whether there are differences in the efficacy between QD and BID regimens of canagliflozin at the same TDD. This solution included developing and validating a dynamic population PK/PD model using pooled data from all three studies to characterize the exposure–response relationship of canagliflozin as add‐on to metformin on HbA1c‐lowering, and performing simulations to compare the effect of QD and BID canagliflozin dosing regimens on HbA1c‐lowering using model‐based simulations.
Methods
Study populations and data
As this population PK/PD modelling analysis was performed to support the use of an FDC tablet of canagliflozin and metformin, only long‐term studies having comparable patient populations with metformin monotherapy as sole background AHA medication and including a placebo dosing arm were used. Studies meeting these criteria were the QD dosing canagliflozin studies (Study 1 19 and Study 2 20) and the BID canagliflozin study (Study 3 21) for which patients received canagliflozin or placebo as add‐on therapy to metformin monotherapy at randomization. In total, 5764 PD (HbA1c) observations were available for 1347 patients with T2DM, which included 352 patients from Study 1, 717 patients from Study 2 and 278 patients from Study 3.
In Study 1, patients with T2DM received 50, 100, 200 or 300 mg QD, or 300 mg BID canagliflozin, sitagliptin 100 mg QD, or placebo as add‐on to metformin for 12 weeks 19. Blood samples for PD were taken at baseline and at weeks 6, 9 and 12, while PK was sampled predose at baseline and at weeks 3, 6 and 12. In Study 2, patients with T2DM received 100 mg QD or 300 mg QD canagliflozin or sitagliptin (52 weeks), or placebo (26 weeks) as add‐on to metformin 20, with PD samples taken at baseline and at weeks 6, 12, 18 and 26. Study 3 included T2DM patients who received 50 mg BID and 150 mg BID canagliflozin or placebo as add‐on to metformin for 18 weeks 21, and sampling for PD was done at baseline and at weeks 6, 12 and 18. In all three studies, HbA1c estimation was carried out in the Diabetes Diagnostic Laboratory, University of Missouri School of Medicine using the boronate affinity chromatography method. The studies were conducted according to the Declaration of Helsinki, Good Clinical Practice guidelines, and other applicable regulatory requirements. The study protocol and amendments for all three studies included in this modelling were reviewed by an Independent Ethics Committee or Institutional Review Board, as appropriate, for each site. Written informed consent was obtained from all patients before enrolment.
A population PK model for canagliflozin was developed previously on pooled exposure data comprising a total of 9061 PK samples from 1616 patients, including 5715 PK samples from 245 richly sampled patients and 3346 trough samples from 1371 sparsely sampled patients including those from Study 1 22. Because this model was developed primarily on QD dosing data, it was externally validated to adequately predict the 24‐h BID dosing profiles in observed in a separate study (Supplementary Figure S1). The individual empirical Bayes estimates for PK parameters obtained from the population PK model for canagliflozin 22 were used to predict individual 24‐h canagliflozin exposure profiles in this study. As no canagliflozin PK samples were collected for Study 2 or Study 3, individual 24‐h canagliflozin plasma exposure profiles were predicted for patients in those two studies using their baseline covariates and the canagliflozin population PK model 22.
Model development was performed on an internal dataset that pooled the QD dosing exposure–response data from Studies 1 and 2 with baseline characteristics from all treatment arms in Study 3, as well as postrandomization data from the placebo arm in Study 3. The final model developed on the internal dataset was validated on an external dataset consisting of postrandomization exposure–response data from the BID dosing canagliflozin arms of Study 3. The final parameter estimates used for all simulation‐based analyses were obtained on the pooled internal and external datasets. The baseline demographics for the pooled internal/external dataset are listed in Table 1.
Table 1.
Baseline demographics (identical for both the internal and the pooled internal/external datasets)
| Placebo | 50 mg QD | 50 mg BID | 100 mg QD | 200 mg QD | 150 mg BID | 300 mg QD | 300 mg BID | Total | |
|---|---|---|---|---|---|---|---|---|---|
| n = 301 | n = 60 | n = 93 | n = 346 | n = 55 | n = 93 | n = 339 | n = 60 | n = 1347 | |
| Sex, n (%) | |||||||||
| Men | 146 (48.5) | 31 (51.7) | 40 (43) | 171 (49.4) | 29 (52.7) | 44 (47.3) | 158 (46.6) | 26 (43.3) | 645 (47.9) |
| Women | 155 (51.5) | 29 (48.3) | 53 (57) | 175 (50.6) | 26 (47.3) | 49 (52.7) | 181 (53.4) | 34 (56.7) | 702 (52.1) |
| Age (years) | |||||||||
| Median | 57.0 | 53.5 | 58.0 | 54.0 | 55.0 | 58.0 | 55.0 | 56.5 | 56.0 |
| Range | (26.0–80.0) | (33.0–65.0) | (33.0–80.0) | (27.0–78.0) | (31.0–65.0) | (29.0–79.0) | (21.0–77.0) | (32.0–65.0) | (21.0–80.0) |
| Weight (kg) | |||||||||
| Median | 85.0 | 86.0 | 87.0 | 86.0 | 84.0 | 89.6 | 83.0 | 81.9 | 85.0 |
| Range | (45.3–164) | (53.0–123) | (55.2–163) | (40.0–188) | (54.0–133) | (51.0–139) | (47.0–168) | (50.8–140) | (40.0–188) |
| Body mass index ( kg m –2 ) | |||||||||
| Median | 30.6 | 31.0 | 31.1 | 31.7 | 30.1 | 30.7 | 30.5 | 30.6 | 30.9 |
| Range | (19.7–46.6) | (24.9–41.8) | (21.6–55.4) | (19.3–55.3) | (24.9–44.4) | (20.4–53.4) | (18.1–73.0) | (24.2–43.7) | (18.1–73.0) |
| eGFR (ml min–1 1.73 m–2) | |||||||||
| Median | 86.0 | 92.0 | 85.0 | 90.0 | 88.0 | 86.0 | 89.0 | 100 | 89.0 |
| Range | (49.0–176) | (57.0–150) | (54.0–135) | (45.0–165) | (50.0–143) | (50.0–138) | (55.0–171) | (35.0–150) | (35.0–176) |
| HbA1c (%) | |||||||||
| Median | 7.6 | 8.0 | 7.5 | 7.7 | 7.4 | 7.4 | 7.8 | 7.5 | 7.6 |
| Range | (6.0–10.3) | (6.5–10.0) | (6.2–10.1) | (5.5–10.5) | (6.0–9.0) | (5.6–9.8) | (5.6–11.0) | (6.0–9.8) | (5.5–11.0) |
BID, Twice‐daily; eGFR, estimated glomerular filtration rate; QD, once‐daily.
Population PK/PD model development
A dynamic population PK/PD model was developed by linking the complete 24‐h time profile of drug concentrations to the time‐profiles for HbA1c. The PK component consisted of a two‐compartment population PK model for canagliflozin described earlier 22. The PD component was based on a well‐established turnover model for HbA1c dynamics over time, with a zero‐order rate constant (k in) for HbA1c production through haemoglobin glycation (a nonenzymatic and irreversible reaction between haemoglobin and glucose), and a first‐order rate constant (k out) for elimination of HbA1c through erythrocyte cell‐death 23. The individual empirical Bayes PK parameter estimates from the population PK model were used to predict individual 24‐h plasma exposure profiles, which were linked to the PD component using an E max model.
The population PK/PD analysis was performed using nonlinear mixed effects modelling as implemented in NONMEM 7.2.0 using the FOCE INTERACTION and ADVAN13 algorithms 24. An efficient method (method of averaging) was developed to solve numerically the ordinary differential equations of the population PK/PD model. This method is described by Dunne et al., 25 and was used throughout the analysis. Data set exploration and visualization, as well as statistical and graphical analyses and diagnostics were performed using R for Windows (Version 3.0.1.).
Physiological considerations, graphical diagnostics and comparison of competing models using the objective function values (OFV) in a likelihood ratio test guided the model development, where a >10.83 points reduction of the OFV (α = 0.001) for one additional parameter in nested models was deemed significant. This significance level was chosen to account for repeated model testing during development. Different implementations of the various model components were tested, such as the implementation of the treatment effects of canagliflozin and placebo (including the effects of diet and exercise counseling) and their dependence on HbA1c at baseline.
Interindividual variability in parameters was regarded as random and was modelled using eta (η) variables (commonly referred to as random effects). The individual η‐values were assumed to be normally distributed with a mean of zero and an estimated variance (ω2). The distribution of the individual parameters around the typical population value was assumed to be log‐normal for parameters representing physiological properties that can only take positive values to be meaningful, such as HbA1c at baseline, and normal for parameters that can potentially attain negative values, such as the placebo effect. Correlations between random effects were evaluated by means of graphical assessment and tested by inclusion of covariance terms between Interindividual variability parameters in the model. Residual variability was assumed to be random and normally distributed. An additive error model was used to describe the error on log‐transformed data.
Covariate effects for age, body weight, body mass index, sex, race and eGFR (calculated according to the MDRD formula) were explored graphically on the empirical Bayes estimates of the η‐values in the final structural model, provided shrinkage was sufficiently low (<25%). Thus determined influential covariates were evaluated at α = 0.001 level by a forward inclusion and backward deletion procedure. In order to evaluate the impact of delayed glucose absorption on HbA1c lowering with 300 mg canagliflozin dose only, an additional PD effect for the 300 mg canagliflozin dose strength was also tested.
Model evaluation
The final population PK/PD model was evaluated using both internal and external validation procedures. A visual predictive check (VPC) was performed for internal validation to assess the ability of the model to predict the observed data of Dataset 1 adequately 26. The final population PK/PD model was externally validated by using it to predict the post‐treatment HbA1c observations of BID dosing from Study 3 included in Dataset 2. Prediction of the external HbA1c observations was performed and a VPC was used to assess the quality of the external predictions. Percent prediction errors were calculated as PE% = 100 × (exp (DV)‐exp(IPRED) / exp(IPRED), where DV represents the log‐transformed observed values and IPRED represents the log‐transformed individual model predictions. Absolute percent prediction errors (|PE|%) were computed to evaluate bias and precision of the model predictions 27. The compatibility of data and model were assessed by comparing the values of PE% and |PE|% with the 5th and/or 95th percentiles of their posterior predictive distributions (PPD) under the model 28. The PPD were estimated by repeated simulation and re‐estimation/prediction (300 repetitions).
Model‐based QD to BID bridging
Potential differences in HbA1c lowering effect between QD and BID canagliflozin dosing regimens were evaluated using the population PK model and final validated dynamic population PK/PD model, respectively, to simulate subject‐specific concentration and HbA1c vs. time profiles. Simulated subject‐specific HbA1c change from baseline profiles were derived from the latter and were used to evaluate the difference in effect between QD and BID canagliflozin dosing regimens for TDD of 100 and 300 mg. The population PK model 22 was used to simulate 24‐h steady‐state subject‐specific PK concentration–time profiles using baseline covariate values from the pooled internal/external dataset (100 simulations per individual subject, for each dose regimen) and by simulating random effects from their estimated distribution (between‐subject variation) without incorporating within‐subject variability. In this way, for each of the 1347 subjects in the pooled internal/external dataset, 200 different 24‐h steady‐state concentration–time profiles were simulated, 100 for QD dosing and 100 for BID dosing. The simulated concentration‐time profiles were then used in the final dynamic population PK/PD model (i.e. using the final estimated parameter values from Table 2) to produce simulated subject‐specific HbA1c profiles. These were simulated using as input, in addition to the simulated concentration–time profiles, subject‐specific baseline HbA1c values from the pooled internal/external dataset, and incorporated both between‐ and within‐subject variability, with random effects and within‐subject errors simulated from the respective estimated distributions. Moreover, to account for parameter estimation uncertainty in the population PK/PD model, an additional layer of randomness was added by generating for each subject‐specific simulated HbA1c profile, a set of model coefficients (fixed effects, only), from the corresponding asymptotic distribution of parameter estimates in the population PK/PD model. This can be regarded as a parametric bootstrap approach to account for parameter estimation uncertainty in simulations. We used only the fixed effects from the population PK/PD model in the parametric bootstrap, as model sensitivity analysis results suggested that the HbA1c predictions were relatively insensitive to variation in the population PK parameters.
Table 2.
Parameter estimates for the final population PK/PD model as fitted to the pooled internal and external datasets
| Parameter | Estimate | Std. Error |
|---|---|---|
| t ½ HbA1c (day) | 28.2 | 2.24 |
| Baseline HbA1c (%) | 7.72 | 0.024 |
| Variance of random effect on baseline HbA1c | 0.011 | 0.00044 |
| Ef p (%HbA1c @ steady‐state, Study1) | –0.483 | 0.062 |
| Ef p (%HbA1c @ steady‐state, Study 2) | –0.330 | 0.051 |
| Ef p (%HbA1c @ steady‐state, Study 3) | –0.137 | 0.057 |
| Variance of random effect on Ef p | 0.369 | 0.026 |
| E max (%HbA1c @ steady‐state) | –0.738 | 0.070 |
| Log(EC 50 ) (Log(ng ml –1 )) | 4.12 | 0.54 |
| Residual error variance | 0.00182 | 0.00014 |
Study1: NCT00642278; Study 2: NCT01106677; Study 3: NCT01340664; HbA1c: glycosylated haemoglobin
t½HbA1c = half‐life of glycosylated haemoglobin turnover = log(2)/kout
Ef p = effect of placebo + diet and exercise on HbA1c at steady‐state for a typical patient (HbA1c at baseline 8.0%)
E max = maximum placebo‐corrected HbA1c‐lowering effect of canagliflozin at steady‐state for a typical patient with HbA1c at baseline of 8.0%
EC50 = exposure (C(t)) at which half‐maximal effect is reached, the estimate for log(EC50) corresponds to an estimate of 61.6 ng ml–1 on the normal scale.
The simulated subject‐specific HbA1c change from baseline profiles were summarized per dose regimen and time by their respective means and standard deviations. Graphical analyses were used to evaluate the impact of dosing regimen on the mean and variability of the model‐derived HbA1c change from baseline. A potential impact of baseline HbA1c on dosing regimen effect differences was evaluated by grouping simulated HbA1c change from baseline profiles according to simulated baseline value intervals.
Results
Dynamic population PK/PD model
Of all the models tested during model development, the dynamic population PK/PD model described below provided best fit for the observed HbA1c data of the internal dataset used for model development (see methods section). This model integrated a turnover model for HbA1c 23 with an E max model relating the HbA1c‐lowering effect of canagliflozin to the canagliflozin plasma exposure at time t using the following set of structural equations:
| (1) |
| (2) |
| (3) |
Equation (1) describes the turnover model for HbA1c, where H(t) is the HbA1c (%) at time t, k in and k out are rate parameters related to haemoglobin glycation and red blood cell turnover, respectively, and Ef describes the combined HbA1c‐lowering effects of canagliflozin and placebo treatment. The rate parameter k out was estimated as a half‐life by scaling it over log(2) and the rate parameter k in was estimated as the HbA1c (%) at time 0 (baseline) by scaling it over k out (i.e. k in = H(0) / k out, see also Table 1).
The term Ef was derived in Equation (2), where Ef c represents the exposure–response relationship of canagliflozin on HbA1c (Equation (3)) and Ef p represents the effect of placebo treatment (including diet and exercise counselling) as a step function activated for t > 0; these treatment effects were found to be additive. Similar to k in, Ef c and Ef p were estimated as scaled by k out in order to render their estimates in Table 1 more readily interpretable in terms of change in %HbA1c from baseline at steady‐state. Because baseline glycaemia is known to affect the magnitude of glucose‐lowering in response to AHAs, 29 the combined effect parameter Ef was scaled in Equation (2) by the estimated individual HbA1c at baseline, H(0), normalized by a reference baseline HbA1c of 8.0%. In addition, because normoglycaemia is typically associated with HbA1c values of ~5.0% and virtually no reductions in PG are observed in subjects with normoglycaemia who are treated with canagliflozin, this term was corrected for a lower boundary for HbA1c‐lowering of 5.0%. This baseline scaling of the treatment effects resulted in a highly significant reduction of the OFV by 305 points (135 points for baseline scaling as such, plus 170 points for correction by a lower HbA1c boundary of 5%).
Equation (3) describes the exposure–response relationship between the HbA1c‐lowering effect of canagliflozin, Ef c, and the canagliflozin plasma exposure at time t, C(t), where E max represented the maximal HbA1c‐lowering effect of canagliflozin at steady‐state for a subject with a baseline HbA1c of 8.0%, and EC50 was the canagliflozin plasma exposure at which the half‐maximal effect was reached. Estimating a Hill factor for this E max model did not result in an improvement of the model fit. The final dynamic population PK/PD model as described above by Equations (1)–(3) provided a satisfactory fit to the observed HbA1c data of the internal dataset as per criteria listed under Methods. Figure 1 shows that the dose groups were predicted without significant evidence of bias, most notably 100 mg QD and 300 mg QD doses.
Figure 1.
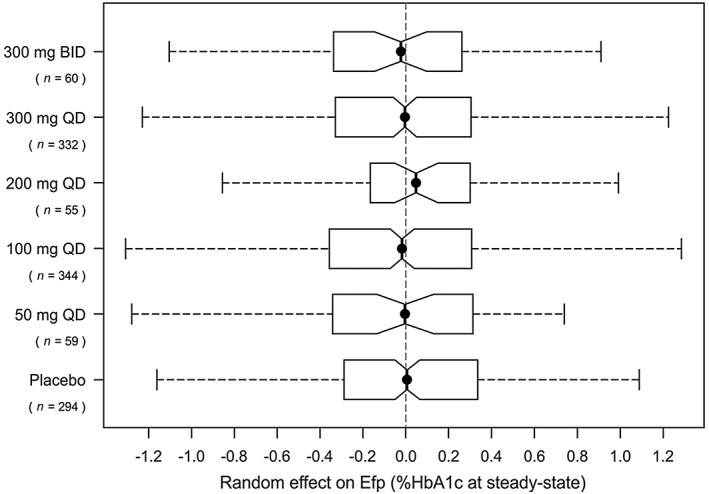
Box plots of the distributions of the random effect on Ef p per dose group for the fit of the dynamic population PK/PD model on the internal dataset. BID, twice‐daily; Ef p, effect of placebo + diet and exercise on HbA1c at steady‐state for a typical patient (HbA1c at baseline 8.0%); HbA1c, glycosylated haemoglobin; PK, pharmacokinetics; PD, pharmacodynamics; QD, once‐daily
Other than a highly significant effect of baseline HbA1c on efficacy, no covariate effects could be identified on the internal dataset that satisfied the prespecified significance level of P < 0.001 (see Methods). The strongest covariate effect found was a power function of eGFR on canagliflozin efficacy as described in Equation (4), yielding a drop of 5.6 points OFV (P = 0.018) where E max90 (representing the maximum canagliflozin effect for a typical subject with an eGFR of 90 ml min–1 1.73 m–2) was estimated at –0.75% HbA1c and γ was estimated at 0.46.
| (4) |
Because this term did not meet the prespecified significance level, it was not retained in the model.
Model validation
The final population PK/PD model (Equations (1)–(3)) was validated on the internal dataset by a VPC. The VPC plot in Figure 2 shows no major systemic deviation between simulated and observed data, and demonstrated that the model adequately described in variability in the observed HbA1c data. The model was externally validated by using it to predict the post‐randomization the HbA1c observations of the 18‐week BID dosing study in the external dataset (see Methods). Figure 3 shows that the model accurately predicted the trends in the external dataset for both 50 mg and 150 mg BID dose levels. The percent prediction errors (PE%) obtained for external validation of the final population PK/PD model remained well within the 5th and 95th percentiles of their corresponding PPD 28 and the absolute percent prediction errors (|PE|%) remained well below the 95th percentile of their corresponding PPD (Table 2).
Figure 2.
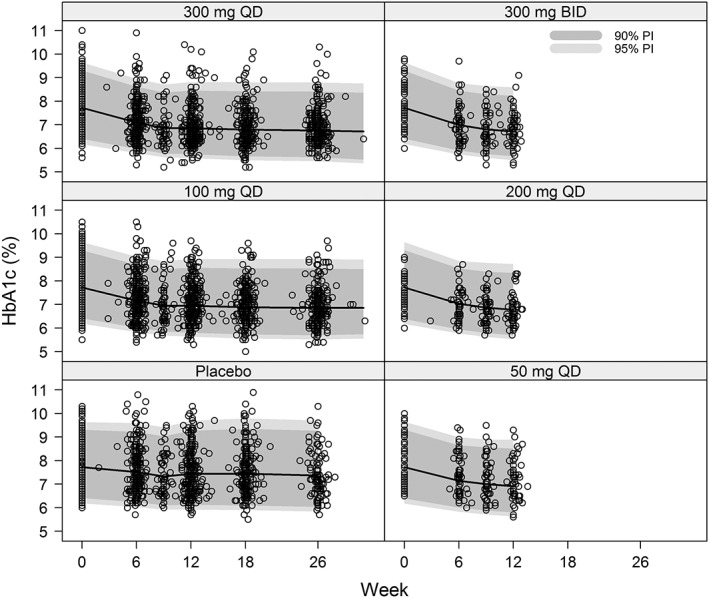
Visual predictive check of the final population PK/PD model on the internal dataset. BID, twice‐daily; HbA1c, glycosylated haemoglobin; PD, pharmacodynamics; PI, predicted interval; PK, pharmacokinetics; QD, once‐daily
Figure 3.
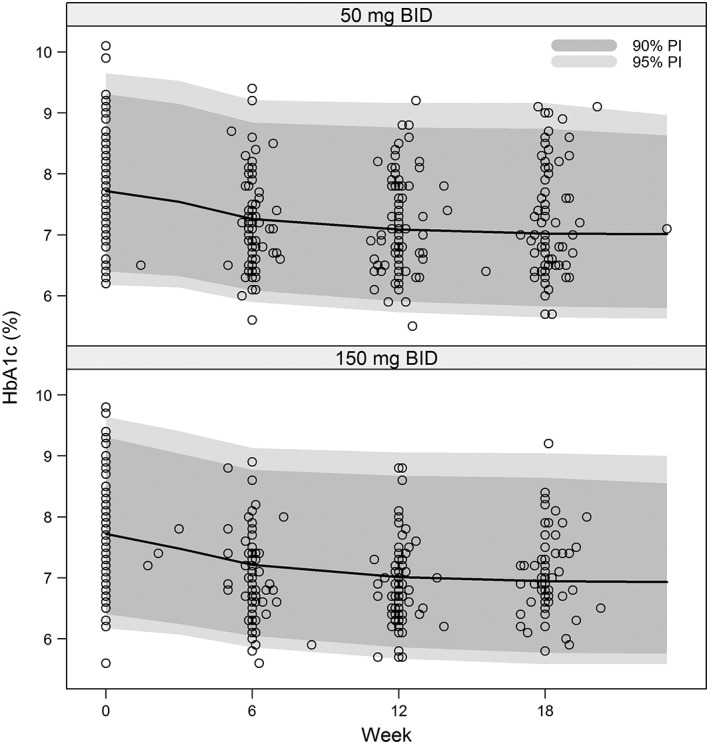
Visual predictive check of the final population PK/PD model on the 50 mg BID and 150 mg BID canagliflozin dosage arms of the external dataset. BID, twice‐daily; HbA1c, glycosylated haemoglobin; PD, pharmacodynamics; PI, predicted interval; PK, pharmacokinetics; QD, once‐daily
It follows that the canagliflozin BID dosing HbA1c observations from the external dataset could be predicted with negligible bias and acceptable precision by the model as estimated on the mostly QD dosing data from the internal dataset. Therefore, the dynamic population PK/PD model described by Equations (1)–(3) was retained for all subsequent analyses and model‐based simulations. Table 3 lists the final parameter estimates for this model as fitted to the pooled internal and external datasets (comprised of all available observations from studies 1, 2, and 3).
Table 3.
PE% and |PE|% for the external validation of the final population pharmacokinetics/pharmacodynamics model on the post‐randomization glycosylated haemoglobin observations from the 50 mg and 150 mg BID dosing arms of Study 3(NCT01340664) and the 5th and/or 95th percentiles of their posterior predictive distributions
| Regimen | Median | 5 th percentile of PPD | 95 th percentile of PPD | |
|---|---|---|---|---|
| PE% | 50 mg BID | –1.1 | –2.0 | 2.1 |
| 150 mg BID | 0.0 | –2.2 | 2.2 | |
| |PE|% | 50 mg BID | 4.2 | – | 6.6 |
| 150 mg BID | 4.5 | – | 6.4 |
BID, twice‐daily; PPD, posterior predictive distributions; PE, prediction error.
Percent prediction errors calculated as PE% = 100 × (exp(DV)‐exp(IPRED)) / exp(IPRED), where DV = dependent variable and IPRED = individual prediction; Absolute percent prediction errors (|PE|%) was computed as the absolute value of %PE.
Model‐based QD to BID bridging
Based on the HbA1c change from baseline data simulated from the population PK and population PK/PD models (under similar baseline covariate values, including HbA1c, and same study effect), BID and QD dosing at the same TDD were found to be similar in terms of mean HbA1c lowering effect (Figure 4). The differences in HbA1c reduction between BID and QD mean profiles were minimal (at most 0.023% for 100 mg TDD and 0.011% for 300 mg TDD, up to week 26, increasing with time and decreasing with TDD) and not considered clinically meaningful. Similar results were obtained after conditioning on baseline HbA1c.
Figure 4.
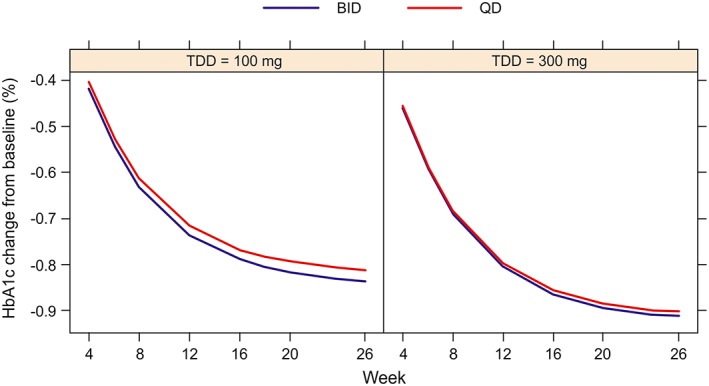
Mean HbA1c change from baseline profiles per regimen and total daily dose derived by averaging out the simulated subject‐specific HbA1c change from baseline profiles simulated from the population PK and population PK/PD models. BID, twice‐daily; HbA1c, glycosylated haemoglobin; PD, pharmacodynamics; PI, predicted interval; PK, pharmacokinetics; QD, once‐daily; TDD, total daily doses
Discussion
The purpose of this study was to establish a robust relationship between canagliflozin exposure and HbA1c response to compare the effect of BID vs. QD canagliflozin dosing regimens in support of the registration of the canagliflozin‐metformin IR FDC tablet 30. To this end, a robust dynamic model was developed by integrating a turnover model for HbA1c 23 with an E max model relating the HbA1c‐lowering effect of canagliflozin to its plasma exposure at time t. This dynamic model was used to compare the predicted HbA1c response time‐courses between QD and BID canagliflozin dosing regimens, and demonstrated that HbA1c change from baseline was similar between QD and BID canagliflozin dosing.
For model development, QD dosing HbA1c data were obtained for the patient population that was comparable to the patient population from Study 3 (18‐week BID dosing study) 21. This dataset included patients from Study 1 (12‐week QD dosing study) 19 and Study 2 (26‐week QD dosing study) 20 with metformin monotherapy as sole background AHA medication at screening, as well as baseline data from Study 3 (internal dataset). The model was externally validated on postbaseline BID dosing observations from Study 3 (external dataset). Full 24‐h plasma PK concentration–time profiles were simulated per patient using a separately developed and validated population PK model for canagliflozin 22 based on its baseline covariates and, where available, observed plasma trough concentrations (only for Study 1). The final parameter estimates used for simulations were obtained on the pooled internal and external datasets. Table S1 shows adequate consistency between the observed and model‐predicted placebo‐subtracted least square mean changes from baseline in HbA1c at study visits (last observation carried forward), which is the standard method for comparing study results in diabetes drug development.
The placebo response in diabetes trials is known to be highly variable, including variability within subjects over time, variability between subjects within the same study population, and variability between study populations. Because the mean change in HbA1c in placebo groups is usually small relative to the placebo‐subtracted differences in the active treatment arms, it is generally accepted that the placebo‐subtracted efficacy is the best measure to use for assessing the efficacy of AHAs. In this model‐based analysis we have conformed to this accepted practice by assuming an additive relationship between the placebo effect and the canagliflozin effect in Equation (2).
The HbA1c‐lowering efficacy of canagliflozin and placebo treatment was found to be highly dependent on the predicted HbA1c levels at baseline. This is consistent with the finding that patients with higher baseline HbA1c had larger reductions in HbA1c with canagliflozin 100 and 300 mg than those with lower baseline HbA1c 26. Indeed, it has been demonstrated that, irrespective of drug class, the baseline glycaemic status of patients who have been recruited into clinical trials strongly influence the fasting PG and HbA1c reductions following pharmacological intervention 29. Other than this highly significant effect of baseline HbA1c on efficacy, no significant covariate effects could be identified on the internal dataset.
Although a covariate effect of renal function (eGFR) on canagliflozin efficacy was expected, 15, 16 it failed to reach statistical significance at the prespecified level (α = 0.001), possibly reflecting the patients with moderately impaired renal function in the internal dataset. Given that patients with moderate renal impairment were excluded from the patient population for which the canagliflozin/metformin FDC is indicated, and that the differences in eGFR between the corresponding QD and BID dosing groups were small (Table 1), this was not considered to be a relevant limitation for this study.
In the external validation of the population PK/PD model, the model‐predicted that HbA1c profiles agreed well with the observed profiles from the 50 mg and 150 mg BID dosing arms of the external dataset (Study 3). Together with the previous internal model validation, this external validation confirmed the ability of the developed dynamic population PK/PD model to accurately predict HbA1c‐lowering for QD as well as BID canagliflozin dosing regimens. A final fit of the dynamic population PK/PD model on the pooled internal and external datasets was performed to obtain parameter estimates that could be used for simulations to compare the predicted efficacy of QD vs. BID canagliflozin dosing regimens.
Based on HbA1c change from baseline data simulated from the population PK and PK/PD models, negligible greater than average HbA1c‐lowering was predicted for BID over QD dosing regimens (at most 0.023% for 100 mg TDD and 0.011% for 300 mg TDD after 26 weeks of treatment). Such small predicted differences in HbA1c‐lowering between QD and BID canagliflozin dosing regimens were not clinically meaningful.
In conclusion, the dynamic population PK/PD model adequately predicted the HbA1c profiles observed in the 18‐week canagliflozin BID dosing study (NCT01340664), 21 and showed good agreement between model‐predicted and observed reductions in HbA1c for 100 and 300 mg TDDs given as QD and BID dosing regimens. Model‐based simulations predicted no clinically meaningful differences in efficacy between QD or BID canagliflozin dosing regimens at either 100 or 300 mg TDD. Therefore, patients on a stable background medication of metformin who switch from canagliflozin taken QD as individual tablets to a single FDC tablet containing metformin and canagliflozin taken as BID should experience similar glycaemic control. The results of this modelling and simulation analysis supported the regulatory approval of the canagliflozin‐metformin IR FDC tablet and alleviated the need for an additional clinical trial to directly compare the efficacy of BID vs. QD dosing 30.
Competing Interests
All authors are (former) employees of Janssen Research & Development, LLC, and are stock holders of Johnson & Johnson.
The study was sponsored by Janssen Research & Development LLC. Authors thank Eef Hoeben for her work on predicting the PK exposures and Martine Neyens for data management. Shruti Shah, PhD (SIRO Clinpharm Pvt. Ltd.) and Bradford Challis, PhD (Janssen Research & Development, LLC) provided editorial support for this manuscript.
Contributors
W.d.W. was the primary author of both the population PK and PK/PD models for canagliflozin and participated with A.D., X.W.d.T., C.H.H., J.P. and D.P. in the strategic planning and implementation of the modeling and simulation activities presented here. D.D. participated in its design and coordination, and helped with acquisition of data. W.d.W. wrote the initial draft of the manuscript and all authors provided contributions to the text and approved the final manuscript.
All authors meet ICMJE criteria and all those who fulfilled those criteria are listed as authors. All authors had access to the study data, provided direction and comments on the manuscript, made the final decision about where to publish these data and approved submission to this journal. All authors contributed to the data interpretation of the results, development, and review of this manuscript, and confirm that they have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Figure S1 Visual predictive check for the external validation of the canagliflozin population PK model on the twice‐daily dosing study stratified per dose and per dose regimen. The population PK model for canagliflozin as described in 22 was used to simulate 200 databases using the study design and covariate distributions of the external twice‐daily dosing study population. Dark grey solid line: median of simulations; light grey solid lines: percentiles of 90% prediction interval; white symbols: observed canagliflozin concentrations. Plasma concentrations are presented on the log scale
Table S1 Comparison of observed and model‐predicted placebo‐subtracted least square mean changes from baseline in glycosylated haemoglobin at study visits (last observation carried forward)
Figure S1 Supporting info item
Table S1 Supporting info item
de Winter, W. , Dunne, A. , de Trixhe, X. W. , Devineni, D. , Hsu, C.‐H. , Pinheiro, J. , and Polidori, D. (2017) Dynamic population pharmacokinetic–pharmacodynamic modelling and simulation supports similar efficacy in glycosylated haemoglobin response with once or twice‐daily dosing of canagliflozin. Br J Clin Pharmacol, 83: 1072–1081. doi: 10.1111/bcp.13180.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 2015; 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. International Diabetes Federation. Diabetes: facts and figures . (2015) [online] Available at http://www.idf.org/WDD15‐guide/facts‐and‐figures.html (last accessed 20 December 2015).
- 4. van Dieren S, Beulens JW, van der Schouw YT, Grobbee DE, Neal B. The global burden of diabetes and its complications: an emerging pandemic. Eur J Cardiovasc Prev Rehabil 2010; 17 (Suppl. 1): S3–S8. [DOI] [PubMed] [Google Scholar]
- 5. Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014; 510: 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Natali A, Ferrannini E. Effects of metformin and thiazolidinediones on suppression of hepatic glucose production and stimulation of glucose uptake in type 2 diabetes: a systematic review. Diabetologia 2006; 49: 434–441. [DOI] [PubMed] [Google Scholar]
- 7. Nathan DM, Buse JB, Davidson MB, Ferrannini E, Holman RR, Sherwin R, et al. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009; 32: 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Elkinson S, Scott LJ. Canagliflozin: first global approval. Drugs 2013; 73: 979–988. [DOI] [PubMed] [Google Scholar]
- 9. Invokana, Prescribing information Titusville, NJ: Janssen Pharmaceuticals, Inc., (2013) [online] Available at http://www.accessdata.fda.gov/drugsatfda_docs/label /2013/204042s000lbl.pdf (last accessed 20 December 2015).
- 10. Invokana, Summary of product characteristics. Beerse, Belgium: Janssen‐Cilag International NV (2013).[online] Available at http://www.ema.europa.eu/docs /en_GB/document_library/EPAR_‐_Product_Information/human/002649/WC500156456.pdf (last accessed on December 2015).
- 11. Devineni D, Curtin CR, Polidori D, Gutierrez MJ, Murphy J, Rusch S, et al. Pharmacokinetics and pharmacodynamics of canagliflozin, a sodium glucose co‐transporter 2 inhibitor, in subjects with type 2 diabetes mellitus. J Clin Pharmacol 2013; 53: 601–610. [DOI] [PubMed] [Google Scholar]
- 12. Sha S, Devineni D, Ghosh A, Polidori D, Hompesch M, Arnolds S, et al. Pharmacodynamic effects of canagliflozin, a sodium glucose co‐transporter 2 inhibitor, from a randomized study in patients with type 2 diabetes. PLoS One 2014; 9: e105638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stenlöf K, Cefalu WT, Kim KA, Alba M, Usiskin K, Tong C, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab 2013; 15: 372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Devineni D, Polidori D. Clinical pharmacokinetic, pharmacodynamic, and drug–drug interaction profile of canagliflozin, a sodium‐glucose co‐transporter 2 inhibitor. Clin Pharmacokinet 2015; 54: 1027–1041. [DOI] [PubMed] [Google Scholar]
- 15. Devineni D, Curtin CR, Marbury TC, Smith W, Vaccaro N, Wexler D, et al. Effect of hepatic or renal impairment on the pharmacokinetics of canagliflozin, a sodium glucose co‐transporter 2 inhibitor. Clin Ther 2015; 37: 610–628. [DOI] [PubMed] [Google Scholar]
- 16. Yale JF, Bakris G, Cariou B, Yue D, David‐Neto E, Xi L, et al. Efficacy and safety of canagliflozin in subjects with type 2 diabetes and chronic kidney disease. Diabetes Obes Metab 2013; 15: 463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Invokamet™ (canagliflozin and metformin hydrocloride) US FDA prescribing information. Titusville, NJ: Janssen Pharmaceuticals, Inc. (2014) [online] Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204353s000lbl.pdf (last accessed 20 December 2015).
- 18. Devineni D, Polidori D, Curtin CR, Murphy J, Wang SS, Stieltjes H, et al. Pharmacokinetics and pharmacodynamics of once‐ and twice‐daily multiple‐doses of canagliflozin, a selective inhibitor of sodium glucose co‐transporter 2, in healthy subjects. Int J Clin Pharmacol Ther 2015; 53: 438–446. [DOI] [PubMed] [Google Scholar]
- 19. Rosenstock J, Aggarwal N, Polidori D, Zhao Y, Arbit D, Usiskin K, et al. Dose‐ranging effects of canagliflozin, a sodium‐glucose cotransporter 2 inhibitor, as add‐on to metformin in subjects with type 2 diabetes. Diabetes Care 2012; 35: 1232–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lavalle González FJ, Januszewicz A, Davidson J, Tong C, Qiu R, Canovatchel W, et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomised trial. Diabetologia 2013; 56: 2582–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qiu R, Capuano G, Meininger G. Efficacy and safety of twice‐daily treatment with canagliflozin, a sodium glucose co‐transporter 2 inhibitor, added on to metformin monotherapy in patients with type 2 diabetes mellitus. J Clin Transl Endocrinol 2014; 1: 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hoeben E, De Winter W, Neyens M, Devineni D, Vermeulen A, Dunne A. population pharmacokinetic modeling of canagliflozin in healthy volunteers and patients with type 2 diabetes mellitus. Clin Pharmacokinet 2014; 55: 209–223. [DOI] [PubMed] [Google Scholar]
- 23. de Winter W, DeJongh J, Post T, Ploeger B, Urquhart R, Moules I, et al. A mechanism‐based disease progression model for comparison of long‐term effects of pioglitazone, metformin and gliclazide on disease processes underlying Type 2 Diabetes Mellitus. J Pharmacokinet Pharmacodyn 2006; 33: 313–343. [DOI] [PubMed] [Google Scholar]
- 24. Bauer RJ. NONMEM Users Guide (Version 7.2.0). Icon Development Solutions: Ellicott City, Maryland, 2011. [Google Scholar]
- 25. Dunne A, de Winter W, Hsu CH, Mariam S, Neyens M, Pinheiro J, et al. The method of averaging applied to pharmacokinetic/pharmacodynamic indirect response models. J Pharmacokinet Pharmacodyn 2015; 42: 417–426. [DOI] [PubMed] [Google Scholar]
- 26. Wilding JP, Blonde L, Leiter LA, Cerdas S, Tong C, Yee J, et al. Efficacy and safety of canagliflozin by baseline HbA1c and known duration of type 2 diabetes mellitus. J Diabetes Complications 2015; 29: 438–444. [DOI] [PubMed] [Google Scholar]
- 27. Sheiner LB, Beal SL. Some suggestions for measuring predictive performance. J Pharmacokinet Biopharm 1981; 9: 503–512. [DOI] [PubMed] [Google Scholar]
- 28. Yano Y, Beal SL, Sheiner LB. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J Pharmacokinet Pharmacodyn 2001; 28: 171–192. [DOI] [PubMed] [Google Scholar]
- 29. Bloomgarden ZT, Dodis R, Viscoli CM, Holmboe ES, Inzucchi SE. Lower baseline glycemia reduces apparent oral agent glucose‐lowering efficacy: a meta‐regression analysis. Diabetes Care 2006; 29: 2137–2139. [DOI] [PubMed] [Google Scholar]
- 30. Food and Drug Administration Center for Drug Evulation and Research. Division Director Review of Canagliflozin/metformin immediate release . (2014) [online] Available at http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/204353Orig1s000MedR.pdf (last accessed on 20 Dec 2015).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Visual predictive check for the external validation of the canagliflozin population PK model on the twice‐daily dosing study stratified per dose and per dose regimen. The population PK model for canagliflozin as described in 22 was used to simulate 200 databases using the study design and covariate distributions of the external twice‐daily dosing study population. Dark grey solid line: median of simulations; light grey solid lines: percentiles of 90% prediction interval; white symbols: observed canagliflozin concentrations. Plasma concentrations are presented on the log scale
Table S1 Comparison of observed and model‐predicted placebo‐subtracted least square mean changes from baseline in glycosylated haemoglobin at study visits (last observation carried forward)
Figure S1 Supporting info item
Table S1 Supporting info item