

Abstract
Staphylococcus epidermidis expresses a 140-kDa cell wall-bound protein accumulation-associated protein (AAP) to adhere to and accumulate as a biofilm on a surface. Potentially blocking AAP with a monoclonal antibody (MAb) could reduce or eliminate S. epidermidis bacterial colonization of biomedical devices. Here, we report on our efforts to (i) isolate AAP, (ii) generate MAbs against AAP, and (iii) determine the efficacy of MAbs to inhibit S. epidermidis biofilm formation. An M7 S. epidermidis mutant, reportedly deficient in AAP expression, was used as a negative control. Postinoculation murine sera, containing polyclonal antibodies against AAP, were able to reduce S. epidermidis biofilm formation by 54%. Select MAbs against AAP were able to reduce S. epidermidis by no more than 66%. Two MAb mixtures, 12C6/12A1 and 3C1/12A1, reduced S. epidermidis accumulation up to 79 and 87%, respectively, significantly more than individual MAbs. Contrary to a previous report, biofilm-deficient S. epidermidis mutant M7 expressed a 200-kDa protein on its cell wall that specifically bound AAP MAbs. Peptide characterization of this M7 protein by microcapillary reversed-phase high-pressure liquid chromatography-nanoelectrospray tandem mass spectrometry resulted in 53% homology with AAP. Ongoing studies will elucidate the dynamic expression of AAP and the M7 200-kDa protein in order to define their roles in biofilm formation.
Staphylococcus epidermidis is one of the most commonly isolated bacterial pathogens in hospitals and the most frequent cause of nosocomial infections (26, 37, 38). Compared with Staphylococcus aureus, S. epidermidis does not produce as many toxins and tissue-damaging exoenzymes (38), but its virulence is related to its ability to form biofilms on inert surfaces of implanted medical devices (21, 26, 37, 38). Within biofilms, multilayers of S. epidermidis are embedded within extracellular matrices comprising mainly polysaccharides that the bacteria secrete (21). Biofilms impair the penetration of antibiotics, negate normal immune responses, and increase the difficulty of eradicating biofilm infections. Ultimately, infected biomedical implants require surgical removal (38).
The traditional approach to prevent biofilm formation in vivo is local administration of bactericidal agents (7). However, dead bacteria could cause a strong host defense response and severe tissue damage. Recent studies aimed at identifying the molecular mechanisms of biofilm formation indicate that the process is mediated by cell membrane-associated macromolecules (7). Antibodies generated against those membrane-bound molecules could disrupt cell-surface and cell-cell interaction, thus preventing biofilm formation without killing the bacteria (2, 3, 20, 39). Immunospecific probes including antibodies and antibody fragments are promising alternative approaches to prevent bacterial colonization on biomedical implants.
The formation of an S. epidermidis biofilm can be roughly divided into two phases: rapid primary adhesion to the synthetic surface followed by biofilm accumulation (21, 26, 37, 38). Various cell surface-associated macromolecules have been found to be involved in both steps.
Primary attachment of S. epidermidis to unmodified polymer surfaces is mediated by several protein and carbohydrate factors, including capsular polysaccharide adhesin (PS/A) (21, 34), S. epidermidis major autolysin AtlE (10, 11), and staphylococcal surface proteins SSP-1 and SSP-2 (36). After implantation, medical devices are quickly coated with an absorbed layer of blood plasma proteins, such as fibronectin, fibrinogen, and vitronectin. S. epidermidis cell surface factors (e.g., protein receptors and cell wall teichoic acids) (13, 24, 28) can interact with these absorbed proteins, mediating specific bacterial adhesion to the protein-coated implants.
Once attached to the device, S. epidermidis will proliferate, secrete extracellular products, and accumulate as multilayered cell clusters. The extracellular polysaccharide PIA (polysaccharide intercellular adhesin) has been found to be essential in this process because PIA mediates cell-cell adhesion of proliferating cells (23, 40). PIA is synthesized by the icaADBC operon of S. epidermidis. In addition, three other gene loci exhibit regulatory effects on PIA synthesis by influencing transcription of icaADBC (22). One of these genes, named rsbU, is a positive regulator of the alternative sigma factor σ B. Tn917 insertion into rsbU leads to a biofilm-negative phenotype (16).
In addition to polysaccharide controls of biofilm formation, proteins are also important for biofilm formation. A 140-kDa extracellular protein named accumulation-associated protein (AAP) was shown to be essential for the accumulation of S. epidermidis on polymer surfaces (14). A biofilm-negative mutant, S. epidermidis M7, generated from S. epidermidis RP62A by mitomycin mutagenesis, reportedly lacks the 140-kDa protein and is unable to accumulate as a biofilm. Rabbit antiserum raised against the AAP was shown to inhibit biofilm accumulation of S. epidermidis RP62A (14). However, the means by which the 140-kDa AAP mediated biofilm formation is still not known.
This study reports on the development of monoclonal antibodies (MAbs) specific to AAP intended to biologically negate biofilm formation and thereby inhibit S. epidermidis biofilm formation on medical implants. Our data show that MAbs specific to AAP and certain F(ab)2 fragments can inhibit the formation of S. epidermidis biofilms. Further, we demonstrate that mixtures of MAbs specific to different epitopes on AAP can inhibit S. epidermidis biofilms more significantly than each MAb alone.
MATERIALS AND METHODS
Bacterial strains and culture medium.
S. epidermidis RP62A and S. epidermidis M7 (AAP-deficient mutant) were kindly provided by Muzaffar Hussain, Universität Münster, Münster, Germany. S. epidermidis RP62A is well known as a strong biofilm producer (33). S. epidermidis M7 is an AAP-deficient RP62A mutant, reported by Hussain et al. to be a biofilm-negative strain. S. epidermidis strains were cultivated for inoculum batchwise at 37°C with 10 g of tryptic soy broth (TSB) medium/liter. Biofilm cultures of both strains were cultivated at 37°C in chemically defined medium [7.4 ml of glycerol, 5.2 g of (NH4)2SO4, 1.072 g of Na2HPO4 · 7H2O, 0.544 g of KH2PO4, 20 mg of thiamine, and 10 mg of biotin in 1 liter of Millipore water].
Preparation of secreted and cell wall proteins.
AAP was isolated from an immobilized-cell culture. S. epidermidis RP62A and M7 cultures were grown overnight at 37°C on TSB (Sigma) agar plates. Colonies were suspended in sterile 0.9% NaCl, and the optical density at 600 nm was adjusted to 0.1. Two milliliters of cell suspension was placed on a sterile dialysis membrane (molecular mass cutoff [MMCO], 12 to 14 kDa) laid over a TSB agar plate. After overnight incubation at 37°C, the mixture of extracellular products and bacteria was collected and sonicated for 1 min. Supernatant was collected after the mixture was centrifuged at 8,000 rpm for 15 min. Secreted proteins were concentrated by a centrifugal filter device (Microcon; Millipore) with a 10-kDa MMCO. Cell wall proteins were prepared from the bacterial pellet by digestion with lysostaphin in 30% raffinose (Sigma) (5). After a 1-h incubation at 37°C, the lysate was centrifuged at 13,000 rpm for 10 min and the supernatant was collected and stored at −20°C.
Purification of AAP.
Supernatant from immobilized S. epidermidis cultures containing soluble secreted protein was concentrated by a centrifugal filter device (Microcon; Millipore) with 50-kDa MMCO. Sixty milliliters of Tris-HCl (25 mM, pH 7.8) was added to 10 ml of the concentrate, and the mixture was concentrated to 5 ml. The concentrated extracellular proteins were purified by an anion-exchange chromatography column (Macro-Prep High Q support; Bio-Rad) with a linear gradient of NaCl (0 to 1 M) in Tris-HCl (25 mM, pH 7.8). Peak fractions were tested by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; precast Tris-HCl gel, 4 to 15% separating gel, 4% stacking gel; Bio-Rad), and those fractions with AAP were collected and concentrated by a 50-kDa-MMCO centrifugal filter device.
Polyclonal antibodies and MAbs.
All polyclonal antibodies and MAbs were raised at the University of Alabama-Huntsville Hybridoma/Phage Display Core Resource Facility. Polyclonal antibodies and MAbs against purified AAP were raised in mice. Primary immunization consisted of 100 μg of AAP emulsified in complete Freund's adjuvant injected subcutaneously over the rear femur. On day 14, mice were given a second injection consisting of 50 μg of AAP emulsified in incomplete Freund's adjuvant (IFA) injected in a similar manner. Thirty days after primary injection, antisera were obtained by tail bleeding and examined for anti-AAP activity by enzyme-linked immunosorbent assay (ELISA) and Western blotting. Mice exhibiting positive serum titers were given a booster injection of 50 μg of AAP in IFA. Five days after the final injection, mice were sacrificed and popliteal lymph node cells were fused with P3X63-Ag8.653 myeloma cells. Hybridoma culture supernatants were screened for activity for AAP by ELISA and biofilm inhibition assay (see below). MAb 12C6 (immunoglobulin G2a [IgG2a]), MAb 12A1 (IgG3), and MAb 3C1 (IgM) were selected to be cloned by limited dilution. MAbs were purified by passage over Gamma Bind Plus Sepharose (Pharmacia) and then eluted with glycine-HCl buffer, pH 3.0, and quickly neutralized with 1.0 M Tris-HCl buffer, pH 9.0. F(ab′)2 fragments of MAb 12C6 were prepared by digesting the MAb with immobilized pepsin (Pierce) for 6 h at 37°C. Any undigested MAb was removed by passage through a protein A affinity column (Pierce), and the digested Fc portions were separated from the F(ab)2 fragment by passage through a 50-kDa-MMCO centrifugal filter.
Indirect protein ELISA.
Reactivities of the various antibodies against AAP immobilized on inert surfaces were quantified by indirect protein ELISA. Each well of a 96-well ELISA plate was exposed to 200 ng of purified AAP in 50 mM sodium carbonate buffer (pH 9.6) at room temperature overnight. After three washes with 0.05% Tween 20 in phosphate-buffered saline (PBST), the plate was blocked by 2% bovine serum albumin (BSA) in PBS for 1 h at 37°C. Either MAbs or F(ab′)2 fragments (1 ng/ml to 10 μg/ml) were added to a well and incubated for 3 h at 37°C. After three washes with PBST, a 1:2,500 dilution of goat anti-mouse IgG plus IgM (heavy and light chains) conjugated to alkaline phosphatase (Pierce) in PBST was added to each well and allowed to react with the bound MAbs or F(ab′)2 fragments for 2 h at 37°C. The substrate tablet p-nitrophenylphosphate disodium salt (PNPP) was dissolved in diethanolamine substrate buffer (Pierce) right before usage. Wells were then washed again, and 100 μl of PNPP substrate solution was added to each well. The color reaction was stopped by 1 M NaOH, and the optical density was read at 405 nm.
Competitive ELISA.
Reactivity of antibodies against soluble AAP was tested by competitive ELISA. Each well of a 96-well plate was coated with AAP and blocked with BSA as described above. Various concentrations of AAP (4.8 ng/ml to 20 μg/ml) were mixed with 100-ng/ml MAbs. The mixtures were then added to each well, and the plate was incubated at 37°C for 3 h. After three washes, bound MAbs were detected by goat anti-mouse IgG plus IgM (heavy and light chains) conjugated to alkaline phosphatase and PNPP as described for the indirect protein ELISA. The inhibition percentage was calculated from the formula (A405, positive − A405, AAP)/(A405, positive − A405, negative) × 100%, where A405 is the optical density reading at 405 nm.
Double-sandwich ELISA.
A double-sandwich ELISA method was used to verify that the various selected MAbs bound specifically to different epitopes of AAP. Each well of 96-well ELISA plate was exposed to 100 ng of purified MAb (the capture MAb) at room temperature overnight. After the plates were washed with PBST and blocked by BSA as described above, various concentrations of AAP (1 ng/ml to 10 μg/ml) were added to individual wells and wells were incubated for 3 h at 37°C. After three washes with PBST, a second MAb (the detection MAb) labeled with biotin was added to each well and allowed to react with the bound AAP for 2 h at 37°C. The bound biotin-labeled MAb was detected by streptavidin-alkaline phosphatase and PNPP. The binding of the detection MAb suggests that it does not bind to the same epitope as the capture MAb.
SDS-PAGE and Western blotting.
Extracellular proteins and cell wall proteins of S. epidermidis RP62A and S. epidermidis M7 were separated on 6% separating gel and transferred to a polyvinylidene difluoride (PVDF) membrane for 2 h. The membrane was blocked by 5% skim milk in PBS overnight and then probed with MAbs (100 ng/ml) and F(ab′)2 fragments (200 ng/ml) for 1 h. After the membrane was washed, the bound MAb or F(ab′)2 fragments were detected by goat anti-mouse IgG (heavy and light chains)-horseradish peroxidase (1:10,000) and ECL chemiluminescence reagent.
Mass spectrometry.
Amino acid sequence analysis was carried out to compare the homology between the wild-type RP62A's AAP and a 200-kDa protein discovered on the cell wall of the M7 mutant. The amino acid sequence of the 200-kDa protein isolated from S. epidermidis M7 was analyzed by tandem mass spectrometry. Sequence analysis was performed at the Harvard Microchemistry Facility by microcapillary reversed-phase high-pressure liquid chromatography-nanoelectrospray tandem mass spectrometry (μLC/MS/MS) on a ThermoFinnigan LCQ DECA XP quadrupole ion trap mass spectrometer. Resultant MS/MS spectra were correlated with known amino acid sequences by using the algorithm Sequenst, developed at the University of Washington (9), and programs developed at the Harvard University Microchemistry Facility.
Biofilm inhibition assay.
The amount of biofilm formed was determined by a semiquantitive microtiter plate method. S. epidermidis RP62A, grown on a TSB agar plate, was suspended in chemically defined media and mixed with either MAbs or F(ab′)2 fragments. The mixture was added to each well of a 96-well tissue culture plate and incubated at 37°C for 24 h. After three washes with PBS, any remaining biofilm was stained with safranin O dye for 1 min and washed with PBS again. Optical density at 492 nm was determined with a 96-well plate spectrometer reader. S. epidermidis RP62A without antibodies was the positive control, and S. epidermidis M7 without antibodies was the negative control. Percent inhibition of biofilm accumulation was determined from the formula (A492, positive − A492, antibody)/(A492, positive − A492, negative) × 100%.
RESULTS
As reported by Hussain et al. (14), the parent strain S. epidermidis RP62A produces a 140-kDa extracellular protein (AAP), which is not produced by the biofilm-negative mutant M7. We have confirmed that the AAP is not only secreted into extracellular fluids by biofilm-bound cells but also resides on bacterial cell walls (Fig. 1, lanes A and C). Although mutant M7 does not produce AAP, it does produce a 200-kDa protein, which is not present in S. epidermidis RP62A but which binds to selected MAbs against AAP (see below). AAP was purified by low-pressure anion-exchange chromatography, and fractions were eluted by a linear gradient of NaCl in Tris-HCl buffer. The purity of AAP was confirmed by SDS-PAGE with silver stain. No minor band was visible, only the 140-kDa major band (data not shown).
FIG. 1.

Coomassie blue-stained 7.5% Tris-HCl polyacrylamide gel. Lanes: A, extracellular proteins of S. epidermidis RP62A (sessile growth); B, extracellular proteins of S. epidermidis M7 (sessile growth); C, cell wall protein of S. epidermidis RP62A (sessile growth); D, cell wall protein of S. epidermidis M7 (sessile growth). Arrow, 200-kDa protein; E, molecular weight markers.
Antiserum reactivity with AAP was tested by ELISA and Western blotting. ELISA results show that antiserum reacts with the AAP. The titer was only 1:2,560 (antiserum titer was defined as the highest serum dilution resulting in 10% of maximum optical density). In a Western blot assay, a 1:750 dilution of antiserum revealed a 140-kDa band, indicating that the antiserum specifically recognized the AAP (data not shown; available upon request). Biofilm formation was reduced by 54% by a 1:40 dilution of the antiserum.
After repeated immunization, hybridoma cells were produced (University of Alabama-Huntsville) by cell fusion between mouse spleen cells and a myeloma cell line. Hybridoma cell media were screened by protein ELISA first. The 25 hybridoma supernatants with a positive response in ELISA were screened by the biofilm inhibition assay. The negative control for these two assays was stock hybridoma medium. Although stock hybridoma medium did not react with AAP, as shown in ELISA, it inhibited biofilm formation by ∼44% at 24 h (data available upon request). Supplemented with bovine serum, the hybridoma medium contains bovine IgG and complement molecules, which may inhibit biofilm formation (15, 29, 35). To eliminate this background biofilm inhibition by the hybridoma medium, hybridoma supernatant containing MAbs was purified prior to use by passage through a protein L spin column. Protein L, which specifically binds to the κ light chain of immunoglobulins, can distinguish between bovine and mouse IgG. After purification, background biofilm inhibition by hybridoma medium alone was dramatically reduced from 44 to 6.5%. Once the four MAbs in hybridoma supernatants that exhibited the highest biofilm inhibition capacity were purified by the protein L spin column, they maintained their antibiofilm activity (Table 1).
TABLE 1.
Biofilm inhibition by hybridoma media before and after protein L purification
| Hybridoma | Biofilm inhibition (%)
|
|
|---|---|---|
| Before purification | After purification | |
| 12C6 | 54.8 ± 3.0 | 42.4 ± 4.3 |
| 12A1 | 56.3 ± 3.2 | 40.6 ± 6.3 |
| 3A5 | 52.8 ± 2.1 | 47.7 ± 1.7 |
| 3C1 | 50.9 ± 1.0 | 43.2 ± 3.4 |
| Hybridoma media | 43.6 ± 3.4 | 6.5 ± 0.4 |
Hybridoma 12C6 (IgG2a) was chosen to be cloned and MAb MAb 12C6 was purified by a protein G chromatography column. The reactivity of purified MAb 12C6 against AAP was confirmed by both indirect protein ELISA (Fig. 2) and competitive ELISA (Fig. 3). Results of indirect ELISA show that purified MAb 12C6 binds to AAP immobilized on a polymer surface in a dose-response fashion. Results of competitive ELISA confirm the highly sensitive MAb-specific binding to the AAP antigen in free solution.
FIG. 2.

Indirect protein ELISA of MAb 12C6 with 140-kDa AAP. A 96-well ELISA plate was coated with AAP. MAb 12C6 was added to each well and incubated for 3 h at 37°C. Bound MAb were detected with alkaline phosphatase-conjugated goat anti-mouse IgG. □, MAb 12C6; ▪, MAb 1934 (an irrelevant MAb against fibronectin).
FIG. 3.

Competitive ELISA. Each well of a 96-well plate was coated with 200 ng of AAP. Various concentrations of AAP (4.8 ng/ml to 20 μg/ml were mixed with 100 ng of MAb 12C6/ml. The mixtures were added to each well, and the plate was incubated at 37°C for 3 h. Bound MAb were detected with alkaline phosphatase-conjugated goat anti-mouse IgG and PNPP. MAb binding inhibition is defined in the text.
Antigenic specificity of MAb 12C6 was also verified by Western blot assay (Fig. 4). Secreted proteins and cell wall proteins of both S. epidermidis RP62A and M7 were separated by 6% polyacrylamide gel and transferred to a PVDF membrane. Proteins on the PVDF membrane were probed by MAb 12C6. As anticipated, MAb 12C6 bound to the 140-kDa protein band in both the secreted and cell wall proteins of the S. epidermidis RP62A. Also, as anticipated, no 140-kDa protein was detected in S. epidermidis M7 cultures. However, unexpectedly MAb 12C6 reacted with a 200-kDa protein both secreted into supernatant and bound to the cell wall of the M7 biofilm-deficient mutant. MAb 12C6 also weakly reacted with a 150-kDa cell wall-bound protein on M7. One explanation for MAb reactivity could be the nonspecific binding of MAb 12C6 to the 200-kDa protein and other M7 cell wall proteins (Fig. 4, lane D). Many surface proteins (e.g., protein G) of S. aureus and S. epidermidis are able to nonspecifically bind immunoglobulins by way of IgG's Fc portion.
FIG. 4.
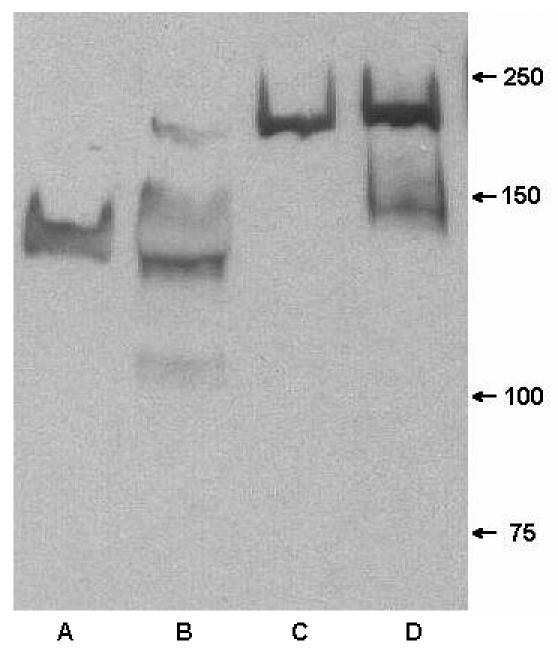
Western blot of extracellular proteins and cell wall protein from S. epidermidis RP62A and M7 probed with MAb 12C6. Lane A, extracellular proteins from RP62A; lane B, cell wall protein of RP62A; lane C, extracellular proteins from M7; lane D, cell wall protein from M7.
To distinguish whether the binding of MAb 12C6 to the M7 200-kDa protein is specific binding through antigen binding sites or nonspecific through the Fc fragment, F(ab′)2 fragments of MAbs were produced by pepsin digestion. F(ab′)2 fragments are MAbs with their Fc portions removed. Undigested MAbs were separated from F(ab′)2 fragments by a protein A column, and small fragments of Fc protein were removed by passage through a 50-kDa-MMCO ultrafiltration unit. The purity of the F(ab′)2 fragments was verified by SDS-PAGE, indicating that no heavy-chain Fc portions remained. In a Western blot assay, both secreted and cell wall proteins from S. epidermidis RP62A and M7 were again probed with a F(ab′)2 fragment of MAb 12C6. F(ab′)2 fragments bind strongly to a single 140-kDa band in secreted and cell wall proteins of S. epidermidis RP62A, which indicates that F(ab′)2 fragments maintain their antigen-recognizing ability after losing the Fc portion (Fig. 5, lanes A and B). Further, bands extraneous to the 140-kDa band in Fig. 4, lanes A and B, are not seen in Fig. 5, lanes A and B, suggesting that MAb binding to those proteins through the Fc portion was nonspecific. Although they did not produce strong bands, F(ab′)2 fragments did bind faintly to both secreted and cell wall-bound 200-kDa proteins (Fig. 5, lanes C and D). Results confirmed that the binding of MAb 12C6 to the 200-kDa protein is specific through antigen binding sites. Again, bands other than the 200-kDa band that reacted with MAb in Fig. 4, lanes C and D, are missing in Fig. 5.
FIG. 5.
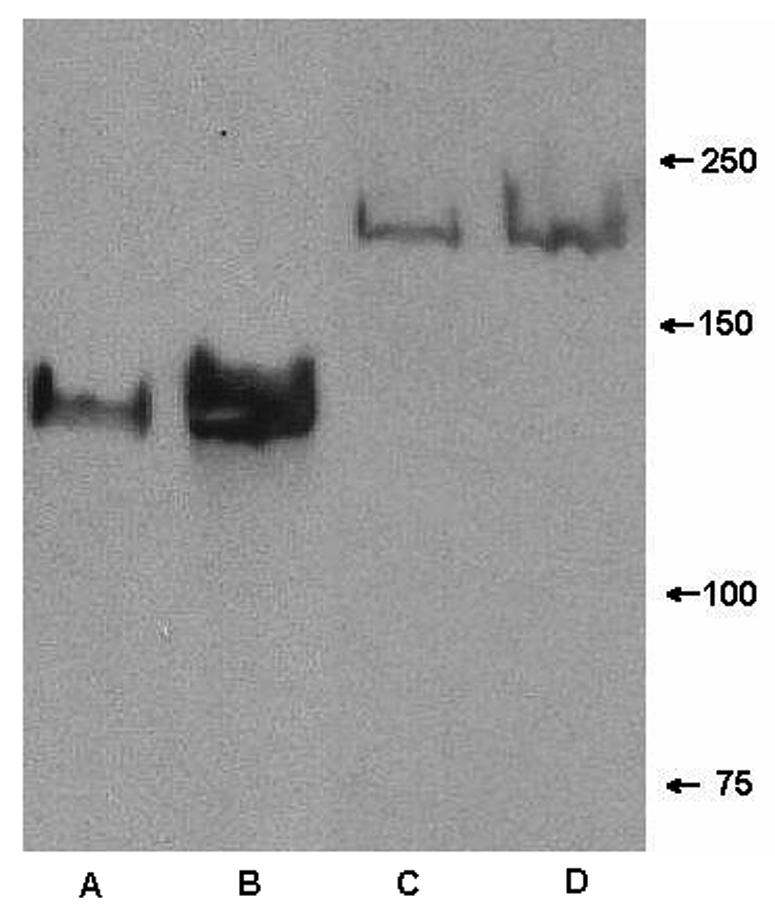
Western blot of extracellular proteins and cell wall protein from S. epidermidis RP62A and M7 probed with F(ab′)2 fragments of MAb 12C6. Lane A, extracellular proteins from RP62A; lane B, cell wall protein of RP62A; lane C, extracellular proteins from M7; lane D, cell wall protein from M7.
To further investigate the structural similarity between AAP and the 200-kDa protein, the amino acid sequence of the 200-kDa protein was analyzed by μLC/MS/MS. The 200-kDa protein in SDS-PAGE bands was fragmented into small polypeptides, and MS/MS spectra of each polypeptide fragment were correlated with known peptide sequence spectra. From the layout of the peptide sequences determined, peptides from the 200-kDa protein match a 100-kDa portion of the AAP sequence. The 200-kDa segments correlated with 662 amino acids within the 1,245-amino-acid sequence of AAP. This result indicates that the primary structure of the 200-kDa protein is significantly homologous to AAP and suggests that the M7 200-kDa protein may be a dimer of a significant portion of AAP that fails to cleave. Further characterization of the 200-kDa protein may help to indirectly reveal the biological function of AAP.
Both the intact MAb 12C6 molecule and its F(ab′)2 fragment inhibit S. epidermidis RP62A biofilm formation (Fig. 6). At low concentrations, increasing the amount of either the MAb or F(ab′)2 fragment increases biofilm inhibition. Once MAb 12C6 concentration reaches 6 μg/ml, biofilm inhibition reaches a maximum of 42%; increasing MAb bulk concentration any further does not increase biofilm inhibition. When F(ab′)2 fragment concentrations reached or exceeded ∼4 μg/ml, biofilm inhibition plateaued at ∼30%.
FIG. 6.

S. epidermidis RP62A biofilm inhibition by MAb 12C6 in a 96-well polystyrene plate. Bacteria were suspended in chemically defined medium containing the indicated concentrations of MAb and incubated for 2 h at 4°C and then overnight at 37°C. Formation of the biofilm was measured with safranin O stain. □, F(ab′)2; ▪, MAb 12C6. Biofilm inhibition is defined in the text.
Reiterating, polyclonal antibodies against AAP exhibit modest biofilm inhibition ability (54%). Since polyclonal antibodies are actually a mixture of numerous MAbs specific to different epitopes on the AAP molecule, hypothetically the mixtures of different MAbs should exhibit higher biofilm inhibition than individual MAbs. Besides MAb 12C6, we cloned and purified two additional antibodies, MAbs 12A1 (IgG3) and 3C1 (IgM).
A double-sandwich ELISA technique was used to confirm that these three MAbs bond to different epitopes of AAP. Basically, AAP molecules were first immobilized on the surface by a “capture” antibody. The second, “detection,” antibody will not bind to the immobilized AAP unless the detection MAb is specific to a different epitope on AAP. Figure 7 illustrates that MAbs 12A1, 12C6, and 3C1 all bind to AAP at different epitopes.
FIG. 7.

Double-sandwich ELISA. Each well of a 96-well ELISA plate was exposed to 100 ng of purified capture MAb at room temperature overnight. AAP at 1 μg/ml was added to each well and incubated for 3 h at 37°C. After three washes with PBST, a detection antibody labeled with biotin was added to an appropriate well and allowed to react with the bound AAP for 2 h at 37°C. Bound biotin-labeled MAb were detected with streptavidin-alkaline phosphatase and PNPP. Other data for an AAP concentration range from 1 to 250 ng/ml are available.
MAbs 12C6, 12A1, and 3C1 individually inhibit S. epidermidis RP62A biofilm formation in a dosage-dependent pattern (Fig. 8). The maximum percentages of biofilm inhibition by MAbs 12C6, 12A1, and 3C1 observed were 42, 39, and 66%, respectively. However, mixtures of MAbs 12A1 and 3C1 and of MAbs 12C6 and 12A1 at a 1:1 mass ratio increased the inhibition of S. epidermidis RP62A biofilm formation to 87 and 79%, respectively. Significantly higher biofilm inhibition was obtained from mixtures of MAbs than from individual MAbs. A mixture of MAbs 12C6 and 3C1 inhibited S. epidermidis RP62A biofilm by only 59%, approximately the same level as observed for the two individual components, MAb 12C6 and MAb 3C1.
FIG. 8.

S. epidermidis RP62A biofilm inhibition in a 96-well polystyrene plate. Bacteria were suspended in chemically defined medium containing the indicated concentration of MAb and incubated for 2 h at 4°C and then overnight at 37°C. Formation of the biofilm was measured with safranin O stain. (a) Biofilm inhibition by individual MAbs. (b) Biofilm inhibition by MAb mixtures.
DISCUSSION
Accumulation of S. epidermidis at a surface and production of extracellular polysaccharide can lead to the formation of dense biofilm on polymer surfaces. The success of S. epidermidis as a pathogen is mainly attributed to its biofilm formation ability. Compared to S. aureus, S. epidermidis does not produce as much bacterial toxin and tissue-damaging exoenzymes (37, 38). Planktonic S. epidermidis usually does not cause infection unless the host is immunocompromised (immunosuppressive therapy, AIDS, or premature birth). Traditional antibiotic treatment of S. epidermidis biofilm faces an increasing challenge of antibiotic-resistant strains (37). Should antibiotic treatment be successful in killing the adherent bacteria, the debris of dead bacteria could still cause an inflammatory response around a biomedical implant, thus possibly damaging the surrounding tissue (30). Methods interrupting S. epidermidis adhesion and accumulation will keep the bacteria in less-virulent planktonic phase and reduce the risk of foreign-body-related infection. Thus, antibodies targeting S. epidermidis adhesion and biofilm formation factors have potential as therapeutic agents against infections of biomedical implants.
Several recent studies showed that pooled human immunoglobulins reduce bacterial adhesion on biomaterials. Rediske et al. (31) reports that a 25-mg/ml human immunoglobulin solution reduced Pseudomonas aeruginosa adhesion on contact lenses from 1.5 × 105 CFU/lens to 4.4 × 103 CFU/lens. Poelstra et al. (29) also reports that pooled human immunoglobulins significantly reduce adhesion of Pseudomonas aeruginosa in a parallel-plate flow cell study. After preopsonizing Pseudomonas aeruginosa with 0.2% (wt/wt) pooled antibodies, initial deposition rates and surface growth rates were reduced 53 and 54%, respectively. Interestingly, the initial bacterial deposition rate and surface growth rate were decreased to only 28 and 39%, respectively, once the Pseudomonas aeruginosa-specific IgGs were removed from the IgG pool (29). Results indicate that specific and nonspecific adsorption of antibodies to a bacterial surface can inhibit adhesion. Because the human antibodies in these other studies were not specific to biofilm-associated molecules, inhibition of bacterial adhesion was achieved only when the antibody concentration was extremely high. Limited supply of human immunoglobulins would affect their potential as a therapeutic reagent for bacterial infection. Their low avidity and the requirement for high concentrations would also preclude pooled IgGs from being dispensed directly from the medical device itself. However, the efficacy of mixed MAb epitope blocking could be greatly improved by generating bivalent or trivalent single-chain “dia-” or “tribodies” (12, 17). The elegance of such single-chain multivalent antibodies is that their size is greatly reduced compared to that of MAbs, they contain no Fc portion, and their binding avidity is increased. Such small binding molecules could be realistically dispensed directly from the biomaterial in a sustained- or controlled-release manner (18).
During the bacterial biofilm formation process, both cell-surface interaction and cell-cell adherence are regulated by various cell surface factors. MAbs targeting those factors inhibit bacterial adhesion and accumulation far more efficiently than the same concentration of pooled immunoglobulins. Leininger et al. raised three MAbs against filamentous hemagglutinin, which is a major Bordetella pertussis cell membrane adhesin. The ability of B. pertussis to adhere to the CHO cell monolayers was reduced 48% by 0.1 mg of MAb/ml (20). Pei and Flock (27) report that an S. epidermidis surface-located fibrinogen binding protein, termed Fbe, mediates bacterial adherence to a fibrinogen-coated surface in vitro. Antibodies against Fbe reduced bacterial adhesion on a fibrinogen-coated surface in vitro by 80% and subcutaneously in implanted catheters in rats by 50%.
Because MAbs were mostly of murine origin, administration in humans will lead to a host defense response against those MAbs, which would diminish their therapeutic effect (1, 32). The level of immune response is associated with the dose of MAbs (32). We have demonstrated that selected MAbs and their mixtures both inhibit S. epidermidis biofilm formation in a dosage-dependent manner in a very low concentration range. Mixtures of MAbs 12C6 and 12A1 exhibit biofilm inhibition activity at a concentration as low as 0.2 μg/ml. At 25 μg/ml, the MAb mixture inhibits S. epidermidis biofilm formation by ∼80%. Low MAb concentrations or use of F(ab′)2 fragments (omitting the immunogenic Fc portions) could reduce the risk of host defense response and tissue damage. Although a mixture of MAbs 12C6 and 3C1 exhibited higher biofilm inhibition (87%), in vivo usage of MAb 3C1 could pose a potential problem since it is an IgM isotype. IgMs are more likely than IgG to generate immune complexes and activate the complement system (15). The alternative approach to reduce the immunogenicity of MAbs would be to remove the Fc portion or create single-chain antibodies discussed above. Schroff et al. have reported that little or no anti-mouse IgG was detected in patients that received F(ab′)2 fragments of mouse IgG, in contrast to what was found for patients receiving the whole molecule of IgG (32). Our study showed that F(ab′)2 fragments of MAb 12C6 inhibit S. epidermidis biofilm by 32% at 4.0 μg/ml. With the advantage of low immunogenicity, F(ab′)2 12C6 alone, or in mixture of other F(ab′)2 fragments, has the potential for biofilm inhibition in vivo.
The fact that the MAbs tested here would have any effect on biofilm formation suggests that AAP instigates S. epidermidis biofilms through some type of receptor-ligand binding interaction. Since our results show that individual MAbs only partially blocked the function of AAP, we then studied the effect of MAb combinations on S. epidermidis biofilm formation. None of the three MAbs alone inhibited S. epidermidis biofilm formation more than 66%. We speculate that either AAP has multiple binding sites for its yet unknown target ligand or the binding epitopes on AAP for the MAbs are not close to the unknown ligand binding site(s). As a consequence, regardless of the MAb concentration, AAP is still able to partially function.
However, two out of three MAb combinations significantly improved S. epidermidis biofilm inhibition. Mixtures 12C6/12A1 and 3C1/12A1 reduced S. epidermidis biofilm formation by 79 and 87%, respectively; significantly higher inhibition than that produced by any MAb individually. While the binding of MAb 12A1 alone inhibits S. epidermidis biofilm formation by only 39%, its addition to either MAb 12C6 or MAb 3C1 increases S. epidermidis biofilm inhibition to 79 and 87%, respectively. This might suggest that MAb 12A1 may bind close to the ligand-binding active site(s) of AAP but alone is insufficient to affect significant inhibition. However, upon the binding of the second MAb (either 12C6 or 3C1), there is perhaps a conformational change in AAP that allows bound MAb 12A1 to block the active site on AAP. There is sufficient precedent indicating that antigens and antibodies can significantly change their molecular conformation upon binding (4, 6, 8, 19, 25). The MAb mixture 12C6/3C1 did not inhibit S. epidermidis biofilm formation any more than MAb 3C1 alone. Conceptually, MAb 12C6 and MAb 3C1 may bind to AAP at epitopes that are close to each other and yet distant from the unknown ligand-binding site; as a consequence their mutual binding provides no additive benefit.
In the report by Hussain et al., AAP was isolated from an extracellular protein mixture (14). We discovered that AAP is both secreted into extracellular fluid and expressed on the S. epidermidis cell wall. The amino acid sequence of AAP showed the presence of a common Staphylococcus cell wall protein anchor sequence, LPXTG, which confirms that AAP is covalently bound to the S. epidermidis cell. Hussain et al. also reported that the AAP is not expressed by S. epidermidis RP62A in planktonic growth (14). However, we found that AAP is present on the cell wall of S. epidermidis RP62A grown in suspension but is not secreted (data not shown). The presence of AAP in extracellular fluid is observed only during biofilm formation.
The mechanism by which AAP mediates S. epidermidis RP62A biofilm formation is still not clear. Hussain speculated that AAP is involved with bacterial accumulation instead of primary adhesion because the biofilm-negative mutant S. epidermidis M7 maintains an adhesion ability (14). Although S. epidermidis M7 does not produce AAP, it expresses a 200-kDa protein on its cell wall and secretes it into extracellular fluids. ELISA and Western blotting showed that all three MAbs developed against AAP (MAbs 12C6, 12A1, and 3C1) and F(ab)2 fragments of MAb 12C6 cross-react with the 200-kDa protein. μLC/MS/MS characterization of the 200-kDa protein showed that peptides from the 200-kDa protein matched a 100-kDa portion of the AAP sequence.
In conclusion, we confirm that MAbs 12C6, 12A1, and 3C1 raised against S. epidermidis AAP specifically bind to AAP and partially inhibit its role in biofilm formation. Meanwhile, F(ab′)2 fragments of MAb 12C6 maintained AAP binding and biofilm inhibition ability after Fc portions are removed. A “cocktail” of MAbs recognizing different epitopes of AAP significantly decreased the biofilm formation compare to applications of individual MAbs. Mixtures of MAbs and F(ab′)2 fragments appear to have promising potential as therapeutic reagents for the prevention of S. epidermidis infection. However, it is still unclear how AAP is actually involved in biofilm formation. Further studies of AAP expression, both on the bacterial surface and in extracellular fluid, during the biofilm accumulation process are in progress to reveal the function of AAP.
REFERENCES
- 1.Abiko, Y. 2000. Passive immunization against dental caries and periodontal disease: development of recombinant and human monoclonal antibodies. Crit. Rev. Oral Biol. Med. 11:140-158. [DOI] [PubMed] [Google Scholar]
- 2.Azghani, A. O., S. Idell, M. Bains, and R. E. Hancock. 2002. Pseudomonas aeruginosa outer membrane protein F is an adhesin in bacterial binding to lung epithelial cells in culture. Microb. Pathog. 33:109-114. [DOI] [PubMed] [Google Scholar]
- 3.Booth, V., F. P. Ashley, and T. Lehner. 1996. Passive immunization with monoclonal antibodies against Porphyromonas gingivalis in patients with periodontitis. Infect. Immun. 64:422-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown, J. C., and M. E. Koshland. 1977. Evidence for a long-range conformational change induced by antigen binding to IgM antibody. Proc. Natl. Acad. Sci. USA 74:5682-5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheung, A. L., and V. A. Fischetti. 1988. Variation in the expression of cell wall proteins of Staphylococcus aureus grown on solid and liquid media. Infect. Immun. 56:1061-1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chumanevich, A. A., Z. I. Kravchuk, A. P. Vlasov, O. V. Zhorov, and S. P. Martsev. 1998. Thermodynamic stability of immunoglobulins and allosteric interactions with ferritin and protein A: distinct properties of the two antibodies of IgG2a subclass. Biochemistry 63:476-484. [PubMed] [Google Scholar]
- 7.Danese, P. N. 2002. Antibiofilm approaches: prevention of catheter colonization. Chem. Biol. 9:873-880. [DOI] [PubMed] [Google Scholar]
- 8.Einhauer, A., and A. Jungbauer. 2003. Complex formation of a calcium-dependent antibody: a thermodynamical consideration. J. Chromatogr. A 1009:81-87. [DOI] [PubMed] [Google Scholar]
- 9.Eng, J., A. L. McCormick, and J. R. Yates III. 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5:976-989. [DOI] [PubMed] [Google Scholar]
- 10.Heilmann, C., C. Gerke, F. Perdreau-Remington, and F. Gotz. 1996. Characterization of Tn917 insertion mutants of Staphylococcus epidermidis affected in biofilm formation. Infect. Immun. 64:277-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heilmann, C., M. Hussain, G. Peters, and F. Gotz. 1997. Evidence for autolysin-mediated primary attachment of Staphylococcus epidermidis to a polystyrene surface. Mol. Microbiol. 24:1013-1024. [DOI] [PubMed] [Google Scholar]
- 12.Hudson, P. J., and C. Souriau. 2003. Engineered antibodies. Nat. Med. 9:129-134. [DOI] [PubMed] [Google Scholar]
- 13.Hussain, M., C. Heilmann, G. Peters, and M. Herrmann. 2001. Teichoic acid enhances adhesion of Staphylococcus epidermidis to immobilized fibronectin. Microb. Pathog. 31:261-270. [DOI] [PubMed] [Google Scholar]
- 14.Hussain, M., M. Herrmann, C. von Eiff, F. Perdreau-Remington, and G. Peters. 1997. A 140-kilodalton extracellular protein is essential for the accumulation of Staphylococcus epidermidis strains on surfaces. Infect. Immun. 65:519-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janeway, C. A. J., P. Travers, M. Walport, and D. J. Capra. 1999. Immunobiology: the immune system in health and disease, 4th ed. Elsevier Science Ltd./Garland Publishing, New York, N.Y.
- 16.Knobloch, J. K., K. Bartscht, A. Sabottke, H. Rohde, H. H. Feucht, and D. Mack. 2001. Biofilm formation by Staphylococcus epidermidis depends on functional RsbU, an activator of the sigB operon: differential activation mechanisms due to ethanol and salt stress. J. Bacteriol. 183:2624-2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kwok, C. S., T. A. Horbett, and B. D. Ratner. 1999. Design of infection-resistant antibiotic-releasing polymers. II. Controlled release of antibiotics through a plasma-deposited thin film barrier. J. Control Release 62:301-311. [DOI] [PubMed] [Google Scholar]
- 18.Kwok, C. S., P. D. Mourad, L. A. Crum, and B. D. Ratner. 2001. Self-assembled molecular structures as ultrasonically-responsive barrier membranes for pulsatile drug delivery. J. Biomed. Mater. Res. 57:151-164. [DOI] [PubMed] [Google Scholar]
- 19.Leid, J. G., D. A. Steeber, T. F. Tedder, and M. A. Jutila. 2001. Antibody binding to a conformation-dependent epitope induces L-selectin association with the detergent-resistant cytoskeleton. J. Immunol. 166:4899-4907. [DOI] [PubMed] [Google Scholar]
- 20.Leininger, E., P. G. Probst, M. J. Brennan, and J. G. Kenimer. 1993. Inhibition of Bordetella pertussis filamentous hemagglutinin-mediated cell adherence with monoclonal antibodies. FEMS Microbiol. Lett. 106:31-38. [DOI] [PubMed] [Google Scholar]
- 21.Mack, D. 1999. Molecular mechanisms of Staphylococcus epidermidis biofilm formation. J. Hosp. Infect. 43(Suppl.):S113-S125. [DOI] [PubMed] [Google Scholar]
- 22.Mack, D., H. Rohde, S. Dobinsky, J. Riedewald, M. Nedelmann, J. K. Knobloch, H. A. Elsner, and H. H. Feucht. 2000. Identification of three essential regulatory gene loci governing expression of Staphylococcus epidermidis polysaccharide intercellular adhesin and biofilm formation. Infect. Immun. 68:3799-3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKenney, D., J. Hubner, E. Muller, Y. Wang, D. A. Goldmann, and G. B. Pier. 1998. The ica locus of Staphylococcus epidermidis encodes production of the capsular polysaccharide/adhesin. Infect. Immun. 66:4711-4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nilsson, M., L. Frykberg, J. I. Flock, L. Pei, M. Lindberg, and B. Guss. 1998. A fibrinogen-binding protein of Staphylococcus epidermidis. Infect. Immun. 66:2666-2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oda, M., H. Kozono, H. Morii, and T. Azuma. 2003. Evidence of allosteric conformational changes in the antibody constant region upon antigen binding. Int. Immunol. 15:417-426. [DOI] [PubMed] [Google Scholar]
- 26.O'Gara, J. P., and H. Humphreys. 2001. Staphylococcus epidermidis biofilms: importance and implications. J. Med. Microbiol. 50:582-587. [DOI] [PubMed] [Google Scholar]
- 27.Pei, L., and J. I. Flock. 2001. Lack of fbe, the gene for a fibrinogen-binding protein from Staphylococcus epidermidis, reduces its adherence to fibrinogen coated surfaces. Microb. Pathog. 31:185-193. [DOI] [PubMed] [Google Scholar]
- 28.Pei, L., M. Palma, M. Nilsson, B. Guss, and J. I. Flock. 1999. Functional studies of a fibrinogen binding protein from Staphylococcus epidermidis. Infect. Immun. 67:4525-4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poelstra, K. A., H. C. van der Mei, B. Gottenbos, D. W. Grainger, J. R. van Horn, and H. J. Busscher. 2000. Pooled human immunoglobulins reduce adhesion of Pseudomonas aeruginosa in a parallel plate flow chamber. J. Biomed. Mater. Res. 51:224-232. [DOI] [PubMed] [Google Scholar]
- 30.Ratner, B. D., A. S. Hoffman, F. J. Schoen, and J. E. Lemons. 1996. Biomaterials science: an introduction to materials in medicine. Academic Press, San Diego, Calif.
- 31.Rediske, A. M., A. L. Koenig, N. Barekzi, L. C. Ameen, J. B. Slunt, and D. W. Grainger. 2002. Polyclonal human antibodies reduce bacterial attachment to soft contact lens and corneal cell surfaces. Biomaterials 23:4565-4572. [DOI] [PubMed] [Google Scholar]
- 32.Schroff, R. W., K. A. Foon, S. M. Beatty, R. K. Oldham, and A. C. Morgan, Jr. 1985. Human anti-murine immunoglobulin responses in patients receiving monoclonal antibody therapy. Cancer Res. 45:879-885. [PubMed] [Google Scholar]
- 33.Schumacher-Perdreau, F., C. Heilmann, G. Peters, F. Gotz, and G. Pulverer. 1994. Comparative analysis of a biofilm-forming Staphylococcus epidermidis strain and its adhesion-positive, accumulation-negative mutant M7. FEMS Microbiol. Lett. 117:71-78. [DOI] [PubMed] [Google Scholar]
- 34.Shiro, H., E. Muller, N. Gutierrez, S. Boisot, M. Grout, T. D. Tosteson, D. Goldmann, and G. B. Pier. 1994. Transposon mutants of Staphylococcus epidermidis deficient in elaboration of capsular polysaccharide/adhesin and slime are avirulent in a rabbit model of endocarditis. J. Infect. Dis. 169:1042-1049. [DOI] [PubMed] [Google Scholar]
- 35.Singh, P. K., M. R. Parsek, E. P. Greenberg, and M. J. Welsh. 2002. A component of innate immunity prevents bacterial biofilm development. Nature 417:552-555. [DOI] [PubMed] [Google Scholar]
- 36.Veenstra, G. J., F. F. Cremers, H. van Dijk, and A. Fleer. 1996. Ultrastructural organization and regulation of a biomaterial adhesin of Staphylococcus epidermidis. J. Bacteriol. 178:537-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.von Eiff, C., G. Peters, and C. Heilmann. 2002. Pathogenesis of infections due to coagulase-negative staphylococci. Lancet Infect. Dis. 2:677-685. [DOI] [PubMed] [Google Scholar]
- 38.Vuong, C., and M. Otto. 2002. Staphylococcus epidermidis infections. Microbes Infect. 4:481-489. [DOI] [PubMed] [Google Scholar]
- 39.Williams, R. J., B. Henderson, L. J. Sharp, and S. P. Nair. 2002. Identification of a fibronectin-binding protein from Staphylococcus epidermidis. Infect Immun. 70:6805-6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ziebuhr, W., C. Heilmann, F. Gotz, P. Meyer, K. Wilms, E. Straube, and J. Hacker. 1997. Detection of the intercellular adhesion gene cluster (ica) and phase variation in Staphylococcus epidermidis blood culture strains and mucosal isolates. Infect Immun. 65:890-896. [DOI] [PMC free article] [PubMed] [Google Scholar]