

Abstract
Aims
Canagliflozin is a recently approved drug for use in the treatment of type 2 diabetes. The potential for canagliflozin to cause clinical drug–drug interactions (DDIs) was assessed.
Methods
DDI potential of canagliflozin was investigated using in vitro test systems containing drug metabolizing enzymes or transporters. Basic predictive approaches were applied to determine potential interactions in vivo. A physiologically‐based pharmacokinetic (PBPK) model was developed and clinical DDI simulations were performed to determine the likelihood of cytochrome P450 (CYP) inhibition by canagliflozin.
Results
Canagliflozin was primarily metabolized by uridine 5′‐diphospho‐glucuronosyltransferase 1A9 and 2B4 enzymes. Canagliflozin was a substrate of efflux transporters (P‐glycoprotein, breast cancer resistance protein and multidrug resistance‐associated protein‐2) but was not a substrate of uptake transporters (organic anion transporter polypeptide isoforms OATP1B1, OATP1B3, organic anion transporters OAT1 and OAT3, and organic cationic transporters OCT1, and OCT2). In inhibition assays, canagliflozin was shown to be a weak in vitro inhibitor (IC50) of CYP3A4 (27 μmol l –1, standard error [SE] 4.9), CYP2C9 (80 μmol l –1, SE 8.1), CYP2B6 (16 μmol l–1, SE 2.1), CYP2C8 (75 μmol l –1, SE 6.4), P‐glycoprotein (19.3 μmol l –1, SE 7.2), and multidrug resistance‐associated protein‐2 (21.5 μmol l –1, SE 3.1). Basic models recommended in DDI guidelines (US Food & Drug Administration and European Medicines Agency) predicted moderate to low likelihood of interaction for these CYPs and efflux transporters. PBPK DDI simulations of canagliflozin with CYP probe substrates (simvastatin, S‐warfarin, bupropion, repaglinide) did not show relevant interaction in humans since mean areas under the concentration‐time curve and maximum plasma concentration ratios for probe substrates with and without canagliflozin and its 95% CIs were within 0.80–1.25.
Conclusions
In vitro DDI followed by a predictive or PBPK approach was applied to determine DDI potential of canagliflozin. Overall, canagliflozin is neither a perpetrator nor a victim of clinically important interactions.
Keywords: canagliflozin, drug–drug interactions, drug metabolizing enzymes, physiologically‐based pharmacokinetics, transporters
What is Already Known about this Subject
Canagliflozin, a sodium‐dependent glucose transporter‐2 inhibitor, is a recently approved drug for the treatment of patients with type 2 diabetes mellitus.
The patient population is expected to take canagliflozin along with multiple concomitant medications that are metabolized or transported by common pathways, which may cause metabolic and/or transporter mediated drug–drug interactions (DDIs) of clinical relevance.
Prescription information and various publications cover key clinical DDI study results.
What this Study Adds
A systematic evaluation of cytochrome P450 (CYP), uridine 5′‐diphospho‐glucuronosyltransferase and transporter (efflux and uptake) DDI potential of canagliflozin as a perpetrator and victim was assessed in vitro, and interpretation of results was reported based on basic predictive approaches recommended by major regulatory guidelines.
A PBPK model for canagliflozin was developed, and simulated PK in healthy subjects was in agreement with observed PK.
PBPK DDI simulations with prototypical CYP probe substrates simvastatin (CYP3A4), bupropion (CYP2B6), S‐warfarin (CYP2C9) and repaglinide (CYP2C8) did not show interaction with canagliflozin.
Tables of Links
| TARGETS | |
|---|---|
| Enzymes 2 | Transporters 3 |
| CYP3A4 | SGLT2 |
| CYP2B6 | OAT3 |
| CYP2C9 | OCT2 |
| CYP2C8 | OATP1B1 |
| BCRP | |
| OCT1 | |
| OAT1 | |
| Pgp | |
| OATP1B3 | |
| MRP2 |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2, 3.
Introduction
Canagliflozin (CAS#: 842 133–18‐0; (1S)‐1,5‐anhydro‐1‐[3‐[[5‐(4‐fluorophenyl)‐2‐thienyl] methyl]‐4‐methylphenyl]‐D‐glucitol), a selective sodium‐dependent glucose transporter‐2 (SGLT2, a member of SLC5 family) inhibitor, has been shown to reduce the renal threshold for glucose reabsorption in the kidney, increase urinary glucose excretion, reduce plasma glucose, and promote weight loss in preclinical and clinical studies 4, 5, 6, 7. Canagliflozin is a recently approved drug that offers a new option for the treatment of patients with type 2 diabetes mellitus. During its development, the metabolism and disposition of canagliflozin were characterized in mice, rats, dogs and humans 8. In humans, direct glucuronidation of canagliflozin is the primary pathway (accounting for at least 34% of an administered dose as two pharmacologically inactive O‐glucuronide conjugates of canagliflozin, M5 and M7) contributing to metabolic clearance with a limited contribution from phase‐I metabolism (7.6% of the dose as a pharmacologically inactive hydroxylated metabolite, M9) 8. In general, susceptibility of the glucuronidation pathway to potential drug interactions is considered low 9. Evaluation of the drug interaction potential of a new drug is an integral part of risk assessment during drug development and regulatory review, and provides guidance to clinicians and pharmacists.
To assess interaction potential with coadministered drugs, canagliflozin was tested in a battery of in vitro test preparations containing human liver microsomes or recombinant drug metabolizing enzymes (DMEs), or various cell systems expressing drug transporters. The goal of these studies was to determine whether canagliflozin is an inhibitor or substrate of a given DME (cytochrome P450 [CYP] and uridine 5′‐diphospho‐glucuronosyltransferase [UGT] enzymes) or transporter (uptake transporters: organic anion transporter polypeptide isoforms OATP1B1 and OATP1B3; organic anion transporters OAT1 and OAT3; organic cationic transporters OCT1 and OCT2, efflux transporters: P‐glycoprotein [Pgp], breast cancer resistant protein [BCRP], and multidrug resistance‐associated protein‐2 [MRP2]). In addition, DME induction potential of canagliflozin was investigated in primary human hepatocytes. If canagliflozin was determined to be an inhibitor, substrate, or inducer of a given DME or transporter in vitro, further assessment was done using basic models recommended by US Food & Drug Administration (FDA) and European Medicines Agency (EMA) 10, 11 or physiologically‐based pharmacokinetic (PBPK) modelling to define the clinical relevance of in vitro findings. Due to lack of industry standardized models, whether canagliflozin was a victim of UGT inhibition or induction was tested in vitro; however, potential for victim‐based interaction was discussed based on available clinical pharmacology studies conducted during development.
Materials and methods
Materials
Canagliflozin and 14C‐canagliflozin (>98%) were synthesized at Janssen Research & Development (Spring House, PA, USA or Beerse, Belgium). All other chemicals and reagents were obtained from commercial sources.
Study methods
All study procedures followed sound scientific principles and were conducted according to applicable guidance documents 10, 11, 12. Assay performance for all test systems was confirmed by conducting experiments with positive control inhibitors/inducers or probe substrates for both metabolic enzymes and transporters.
Assay specific details, along with outline of sample analysis and end points measured in enzyme identification (CYP and UGT), inhibition (CYP reversible and time‐dependent [TDI]; UGT), and induction (CYP) studies, are presented in Table S1. Information on positive control inducers and inhibitors, marker substrates, and metabolites that were used in these assays are listed in Table S2. All incubations were conducted at 37°C. In enzyme identification studies, canagliflozin metabolism was examined in individual CYP or UGT enzymes under defined conditions (Table S1), and metabolite formation rate was calculated. In CYP and UGT inhibition studies, canagliflozin inhibition potential was examined over a range of concentrations (0–100 μmol l–1), and inhibition of probe substrate metabolism was assessed. If applicable, IC50 was determined in both CYP and UGT inhibition studies. In a CYP induction study, canagliflozin induction of CYPs was expressed as fold‐increase in activity compared to solvent control and % induction of the respective positive control inducer. Similarly, assay specific details for efflux (Pgp, MRP2, BCRP) and uptake (OATP1B1, OATP1B3, OCT1, OCT2, OAT1, OAT3) transporter studies to investigate whether canagliflozin is a substrate or inhibitor are presented in Table S3. Information on positive control transporter inhibitors and probe substrates used in the assays are listed in Table S4. To test whether canagliflozin was a substrate in efflux transporters, transport (apparent permeability expressed as Papp = cm s–1 × 10−7 or 10−6) of 14C‐canagliflozin was measured bidirectionally (basolateral to apical [B‐A and A‐B]), in triplicate, and the efflux ratio (B‐A/A‐B) was calculated from the mean Papp values across the overexpressed transporter systems. If the efflux ratio was >2, the compound was considered to be a substrate of the efflux transporter. To test whether canagliflozin was an inhibitor of the efflux transporter, the % change in efflux ratio of 14C‐labelled protypical substrates in the absence or presence of canagliflozin (0–100 μmol l–1) was determined. If applicable, IC50 was calculated for each efflux transporter.
Uptake studies were conducted in transporter injected Xenopus laevis oocytes (human OAT1, OAT3, OATP1B1, OCT1, or OCT2). 14C‐canagliflozin was measured in transporter‐injected and water‐injected (control) oocytes (n = 10 oocytes per experiment), and the mean uptake activity was expressed as pmol/oocyte h–1. If the ratio of the mean substrate uptake activity in the transporter‐injected to control oocytes was >2, the compound was considered to be a substrate of the transporter. In inhibition assays, reduction in uptake of probe substrate was measured with and without canagliflozin at a single probe concentration (100 μmol l–1), and IC50 values were determined when >50% inhibition was observed. Due to unavailability of an OATP1B3 oocyte expressed system, uptake transporter, substrate and inhibition studies were conducted in HEK293 cells expressing OATP1B3.
Data analysis
In vitro predictive models
Where applicable, inhibition constants (Ki) for canagliflozin were calculated from the IC50 value derived from the inhibition assay with an assumption of competitive inhibition (Ki = IC50/2). For prediction of DME (CYP or UGT) or transporter inhibition based interactions, basic models that are recommended by USFDA 10 and EMA 11 were used to assess the potential of in vivo interaction. Relevant equations and calculation of r values for the assessment of DDI from the DDI guidance documents 10, 11 are provided in the results tables of respective sections. If the basic models predicted a likelihood of in vivo interaction, further interpretation was done either by PBPK DDI modelling (see PBPK section below) or clinical studies conducted during development of canagliflozin. If the compound was identified as a substrate of a specific DME or transporter, interpretation of likelihood of victim‐based DDIs was provided based on compound disposition mechanisms or published canagliflozin clinical pharmacology information. In vivo CYP induction potential was assessed based on a predefined threshold of induction (>20% of the induction observed with positive control inducer or >2‐fold induction of the vehicle control) observed with canagliflozin in the in vitro hepatocyte induction study.
PBPK modelling
Physicochemical and PK data for canagliflozin generated during nonclinical and clinical development were used to derive the input parameters for modelling. PBPK simulation software (Symcyp v11.1, Simcyp Ltd, UK) was employed to simulate canagliflozin PK in humans following a 300 mg once daily (QD) oral dose using the input parameters that are shown in Table 1. Verified PBPK models for the DDI substrates simvastatin, S‐warfarin, bupropion and repaglinide were available in the Simcyp v11.1 compound library and were used in conjunction with canagliflozin PBPK model in simulating DDI to test whether canagliflozin is a perpetrator of DDIs with identified CYP isoforms. The trial designs for DDI simulations are shown in Table 2.
Table 1.
Input parameters for the canagliflozin physiologically‐based pharmacokinetic model
| Input parameter | Value | Comment | |
|---|---|---|---|
| Molecular weight | 444.52 g mol–1 | CAS # 842 133–18‐0 | |
| Canagliflozin Structure |
 (1S)‐1,5‐anhydro‐1‐[3‐[[5‐(4‐fluorophenyl)‐2‐thienyl]methyl]‐4‐methylphenyl]‐D‐glucitol hemihydrate
(1S)‐1,5‐anhydro‐1‐[3‐[[5‐(4‐fluorophenyl)‐2‐thienyl]methyl]‐4‐methylphenyl]‐D‐glucitol hemihydrate |
||
| Molecular weight | 444.52 g mol–1 | Molecular weight of free base (i.e. without hemihydrate) | |
| LogP | 3.8 | Internal dataa | |
| Compound Type | Neutral | Internal dataa | |
| pKa1 | unionized | Internal dataa | |
| B/P | 0.69 | Reference: 8 | |
| Fraction of drug unbound (Fu) plasma | 0.017 | Internal dataa | |
| Absorption model | ADAM | ||
| Solubility (Fessif at pH 5) | 5.01 mg ml−1 | Predicted value from GastroPlus | |
| Peff, man | 3.66 10−4 cm s–1 | Predicted value from GastroPlus | |
| Particle size (monodispersed) | 17 μm | Internal dataa | |
| Dosage form | 300 mg QD | ||
| Fraction of drug unbound in gut (Fugut) | 0.06 | Fitted parameter in Simcyp v11.1 | |
| Distribution | Full PBPK | Method 2, Simcyp v11.1 | |
| Observed Rat tissue/plasma Kp (partition coefficient) values at Cmax after oral dose of 5 mg kg–1 | Radiolabelled rat tissue distribution study (Internal dataa) | ||
| Adipose | 0.268 | Radiolabelled rat tissue distribution study (Internal dataa) | |
| Liver | 7.68 | ||
| Muscle | 2.10 | ||
| Skin | 1.02 | ||
| Other tissue Kps | 0.05 | Fitting of Kp values on the basis of observed canagliflozin intravenous pharmacokinetic profile 13. | |
| Percentage UD available for reabsorption | 0% | No evidence of enterohepatic recirculation 4 | |
| Renal clearance (ClR) | 0 l h–1 | l h–1 | In human mass balance study <1% of the dose was excreted in urine as unchanged 8 |
| Ki for CYP2B6, CYP3A4, CYP2C8, CYP2C9 | 8, 13.5, 37.5, 40 μmol l–1 | CYP inhibition experiments in human liver microsomes (data from Table 4) | |
| Fraction of drug unbound in microsomes‐ Fumic b (0.2 mg) | 0.735 | Predicted from Simcyp v 11.1 | |
Internal data on file, Janssen Research & Development. Data can be provided on request to the corresponding author
Predictive performance of the Fumic calculator in the Simcyp has been published by Turner et al. 14.
CYP, cytochrome P450
Table 2.
Simulation trial design to evaluate clinical drug–drug interaction (DDI) cytochrome P450 (CYP) inhibition of canagliflozin
| Trial description and purpose | Study design outline of simulation trials |
|---|---|
| Simulation of canagliflozin pharmacokinetics after single oral administration of 300 mg canagliflozin to healthy subjects. Purpose is to verify the performance of PBPK model | Ten trials of nine virtual male healthy subjects were simulated with a body weight between 62 and 94 kg to match the population of a clinical study in healthy subjects 13. Covariates of weight, age and sex were taken into consideration. |
| Simulation of DDI between 300 mg oral canagliflozin and a single dose of 40 mg simvastatin (CYP3A4 substrate) | Trial design consisted of 10 trials of 10 virtual healthy subjects who received an oral dose of 40 mg simvastatin with or without canagliflozin 300 mg QD. Simvastain was dosed without canagliflozin on day 1. Canagliflozin was dosed once daily from day 2 to day 7. One dose of 40 mg simvastatin was coadministered with canagliflozin on day 7. Simulated pharmacokinetic profiles were obtained for simvastatin with and without canagliflozin. |
| Simulation of DDI between 300 mg oral canagliflozin and a single dose of 15 mg S‐warfarin (CYP2C9 substrate) | Trial design consisted of 2 trials of 10 virtual healthy subjects who received an oral dose of 15 mg S‐warfarin with or without canagliflozin 300 mg QD. S‐warfarin was dosed without canagliflozin on day 1. Canagliflozin was dosed once daily from day 2 to day 12. One dose of 15 mg S‐warfarin was coadministered with canagliflozin on day 12. Simulated pharmacokinetic profiles were obtained for S‐warfarin with and without canagliflozin. |
| Simulation of DDI between 300 mg oral canagliflozin and a single dose of 150 mg bupropion (CYP2B6 substrate) | Trial design consisted of 10 trials of 10 virtual healthy subjects who received an oral dose of 150 mg with or without canagliflozin 300 mg QD. Bupropion was dosed without canagliflozin on day 1. Canagliflozin was dosed once daily from day 2 to day 6. One dose of 150 mg bupropion was coadministered with canagliflozin on day 6. Simulated pharmacokinetic profiles were obtained for bupropion with and without canagliflozin. |
| Simulation of DDI between 300 mg oral canagliflozin and a single dose of 0.25 mg repaglinide (CYP2C8 substrate) | Trial design consisted of 10 trials of 10 virtual healthy subjects who received an oral dose of 0.25 mg with or without canagliflozin 300 mg QD. Repaglinide was dosed without canagliflozin on day 1. Canagliflozin was dosed once daily from day 2 to day 6. One dose of 0.25 mg repaglinide was coadministered with canagliflozin on day 6. Simulated pharmacokinetic profiles were obtained for repaglinide with and without canagliflozin. |
PBPK, physiologically‐based pharmacokinetic; QD, once daily
PK parameters (area under the concentration–time curve [AUC0‐t], time to reach peak plasma concentration [Tmax] and peak plasma concentration [Cmax]) of simvastatin, S‐warfarin, bupropion and repaglinide were obtained alone and in the presence of canagliflozin. The overlay of mean simulated PK profiles of each CYP probe substrate alone and with canagliflozin was obtained in Simcyp v 11.1. The simulated AUC and Cmax ratios of a given substrate when given alone and with canagliflozin were used to measure the magnitude of the interaction. The simulated trial results are reported as median ± SD of individual studies and overall median and 5th and 95th percentiles of the total population.
Results
CYP enzyme phenotyping
Among the 10 recombinant CYP isoforms investigated, CYP3A4 modestly metabolized canagliflozin to form an oxidative metabolite M9, as indicated by ~39% of the sample radioactivity. A low amount of M9 was formed by CYP2D6 and CYP2C9 isoforms with sample radioactivity of 3.8% and 1.4%, respectively. These results suggest that CYP3A4 and to a lesser extent CYP2D6 and CYP2C9 may contribute to the oxidative metabolism of canagliflozin. The calculated rate of metabolism for these 3 isoforms is presented in Table 3.
Table 3.
Enzyme identification in the metabolism of canagliflozin by cytochrome P450 (CYP) and uridine 5′‐diphospho‐glucuronosyltransferase (UGT) enzymes
| Metabolism Rate | ||||
|---|---|---|---|---|
| CYPa (pmol min–1 nmol CYP) | UGTb (pmol min–1 mg UGT) | |||
| M9 | M5 | M7 | ||
| CYP2C9 | 2.57 | UGT1A3 | 0 | 2 |
| CYP2D6 | 5.45 | UGT1A7 | 0 | 3 |
| CYP3A4 | 75.32 | UGT1A8 | 0 | 3 |
| CYP3A5 | ‐ | UGT1A6 | 0 | 3 |
| UGT1A9 | 0 | 208 | ||
| UGT2B4 | 4 | 0 | ||
In vitro incubation of 14C‐canagliflozin (2 μmol l–1) in Escherichia coli cells expressing human CYP isoforms (100 pmol) after 2 h
In vitro incubation of 14C‐canagliflozin (100 μmol l–1) in UGT Supersomes (1 mg ml−1) after 2 h
UGT enzyme phenotyping
Twelve recombinant human UGT isoforms (Supersomes UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B4, UGT2B7, UGT2B15, UGT2B17) were used to determine the UGTs involved in the formation of two major circulating glucuronide conjugates of canagliflozin (M5, M7). The calculated metabolism rate at 2 h by various isoforms is presented in Table 3. M7 was mainly formed with UGT1A9, and M5 with UGT2B4. The amount of M5 formed was small (0.51%). A significant amount of M7 was formed in UGT1A9, with ~16% and ~25% conversion of canagliflozin to M7 after incubation for 1 and 2 h, respectively. An insignificant amount (<0.5%) of canagliflozin was converted to M7 in UGT1A3, 1A6, 1A7, and 1A8 isoforms. These isoforms were considered to have a minimal role in the metabolism of canagliflozin.
In vitro CYP inhibition
Inhibition potential of CYP by canagliflozin for a panel of 10 CYP isoforms (1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4/5) was assessed in human liver microsomes, and the results are shown in Table 4. Canagliflozin showed a weak inhibition of CYP3A4 testosterone hydroxylation (IC50 = 27 μmol l–1) but not CYP3A4/5‐mediated ‐midazolam 1′‐hydroxylation (IC50 > 100 μmol l–1). CYP2C9, CYP2B6 and 2C8 were also weakly inhibited, with IC50 values of 80, 16 and 75 μmol l–1, respectively. In addition, canagliflozin did not exhibit TDI potential for any CYPs (1A2, 2C9, 2C19, 2D6 or 3A4). At the highest concentration tested (50 μmol l–1) in the TDI assay, residual percent CYP activity in NADPH preincubated samples was 93–118% of the corresponding incubations without NADPH for all CYPs examined. Standard positive control inhibitors for both reversible and TDI of CYPs showed expected inhibition in the respective assays.
Table 4.
In vitro evaluation of canagliflozin as an inhibitor of human CYP and UGT enzymes
| Enzyme | Enzyme reaction | Direct inhibition | |
|---|---|---|---|
| IC50 (SE) μmol l–1 | (%) Inhibition at 100 μmol l–1 | ||
| CYP1A2 | Phenacetin O‐deethylase | >100 | 25 |
| CYP2A6 | Coumarin 7‐hydroxylation | >100 | 41 |
| CYP2B6 | Bupropion 1‐hydroxylation | 16 (2.1) | 100 |
| CYP2C8 | Desethylation of amodiaquine | 75 (6.4) | 61 |
| CYP2C9 | Tolbutamide hydroxylation | 80 (8.1) | 56 |
| CYP2C19 | S‐Mephenytoin 4‐hydroxylation | >100 | 41 |
| CYP2D6 | Bufuralol 1′‐hydroxylation | >100 | 35 |
| CYP2E1 | 6‐Hydroxylation of chlorzoxazone | >100 | 4 |
| CYP3A4 | 6β‐Hydroxylation of testosterone | 27 (4.9) | 84 |
| CYP3A4/5 | Midazolam 1′‐hydroxylation | >100 | 21 |
| UGT1A1 | 17β‐Estradiol 3‐β‐D‐glucuronidation | 91 (53) | 45 |
| UGT1A4 | Trifluoperazine glucuronidation | >100 | 42 |
| UGT1A6 | 1‐Naphthol glucuronidation | 50 (4.0) | 78 |
| UGT1A9 | Propofol glucuronidation | > 100 | 40 |
| UGT2B7 | Morphine 3‐β‐D‐glucuronidation | > 100 | NA |
NA = Not applicable. Inhibition was not observed at the highest concentration tested.
IC50 = half maximal inhibitory concentration; SE = standard error. SE values presented for the IC50 estimates
Based on the above in vitro assessment, DDI potential for canagliflozin was evaluated by basic models suggested by FDA 10 and EMA 11 for four CYPs (3A4, 2B6, 2C9, and 2C8; Table 5). Based on the FDA approach, r values for all four CYPs were >1.1, indicating potential for CYP inhibition‐mediated DDIs for these CYPs. However, based on the EMA approach, r values indicated that interaction potential of canagliflozin with all four CYPs can be excluded (see PBPK section for further assessment).
Table 5.
Evaluation of the drug–drug interaction potential for canagliflozin using the r approach
| CYP/UGT Enzyme | Ki (μmol l–1)c | FDA | EMA | ||
|---|---|---|---|---|---|
| R = 1 + [Ia]/Ki | Clinical study warrantedd? | R = [Iu b]/Ki | Clinical study warrantede? | ||
| CYP3A4 | 13.5 | 1.78 | Yes | 0.008 | No |
| CYP2C9 | 40 | 1.26 | Yes | 0.003 | No |
| CYP2B6 | 8 | 2.31 | Yes | 0.014 | No |
| CYP2C8 | 37.5 | 1.28 | Yes | 0.003 | No |
| UGT1A1 | 45.5 | 1.23 | Yes | 0.002 | No |
| UGT1A6 | 25 | 1.42 | Yes | 0.004 | No |
[I] represents mean steady‐state total peak concentration in plasma following once daily administration of highest clinical dose 300 mg (10.5 μmol l–1)
[Iu] represents unbound mean steady‐state peak concentration in plasma following once daily administration of highest clinical dose 300 mg (0.11 μmol l–1)
Ki = half maximal inhibitory concentration/2, assumption of competitive inhibition
Interaction potential if R >1.1
Interaction potential if R ≥0.02
CYP, cytochrome P450; EMA, European Medicines Agency; FDA, US Food & Drug Administration; UGT, uridine 5′‐diphospho‐glucuronosyltransferase
CYP induction
Induction potential of canagliflozin (3, 15, 75 μmol l–1) was evaluated in human hepatocytes from three separate donors, and the results are shown in Table 6. Positive control inducers showed expected increases in CYP1A2, 2B6 and 3A4 activities. Due to observed cytotoxicity, CYP induction of canagliflozin at 75 μmol l–1 could not be assessed. At 3 and 15 μmol l–1, there was no significant increase in CYP activity higher than 20% of the respective positive control inducer or higher than two‐fold the activity of the solvent control, indicating that canagliflozin was not an inducer of these CYPs within the tested concentration range.
Table 6.
In vitro evaluation of cytochrome P450 (CYP) induction potential in human hepatocytes
| Treatment | (μmol l–1) | % CYP induction relative to positive control | Fold induction relative to solvent controlb | ||||
|---|---|---|---|---|---|---|---|
| CYP1A2 | CYP2B6 | CYP3A4 | CYP1A2 | CYP2B6 | CYP3A4 | ||
| Solvent control | – | – | – | 1.0 ± 0.0 | 1.0 ± 0.0 | 1.0 ± 0.0 | |
| Positive control a | 100 | 100 | 100 | 32.7 ± 18.1 | 17.1 ± 7.6 | 10.4 ± 3.4 | |
| Canagliflozin | 3 | –0.2 ± 1.2 | –0.3 ± 0.1 | 0.7 ± 0.7 | 1.0 ± 0.15 | 0.7 ± 0.1 | 0.7 ± 0.1 |
| 15 | 5.4 ± 4.1 | 0.2 ± 0.5 | 5.9 ± 1.7 | 1.9 ± 0.8 | 1.2 ± 0.6 | 0.50 ± 0.15 | |
Positive controls: CYP1A2 = 50 μmol l–1 β‐naphthoflavone; CYP2B6 = 1 mmol l–1 phenobarbital; CYP3A4 = 10 μmol l–1 rifampicin
Solvent control: 0.1% dimethyl sulfoxide
Data expressed as mean ± standard deviation, n = 3 donors.
UGT inhibition
UGT inhibition results are shown in Table 4. Under the experimental conditions examined, canagliflozin showed a weak inhibition of UGTs at high concentrations with IC50 values of >100 μmol l–1 for UGT1A4 and UGT1A9, 91 μmol l–1 for UGT1A1, and 50 μmol l–1 for UGT1A6. No inhibition was observed for UGT2B7 up to the highest concentration (100 μmol l–1) tested. Both positive controls (troglitazone and probenecid) showed expected inhibition of the UGT isoforms in vitro.
DDI potential for canagliflozin was evaluated by basic prediction models recommended by FDA 10 and EMA 11 for two UGT isoforms (1A1, 1A6), and the r and Ki values are presented in Table 5. The r values based on the FDA approach were higher than Ki values, indicating potential for UGT inhibition‐mediated DDI for both UGTs. However, based on the EMA approach, interaction potential for canagliflozin was excluded.
Substrate activity and inhibition potential of efflux transporters
The efflux ratios (B‐A/A‐B) were calculated for Pgp, MRP2 and BCRP, and the results are presented in Figure 1. Digoxin, etoposide and topotecan, which are prototypical substrates for human Pgp, MRP2 and BCRP, showed expected efflux ratios of 26, 7.9 and 5, respectively. The efflux ratios for canagliflozin were >2 for all three efflux transporters (Pgp, 11.6; MRP2, 6.5; BCRP, 12.5). In the presence of respective positive control inhibitors (cyclosporine, cisplatin, KO143), the efflux ratios for canagliflozin were <2, which indicates that canagliflozin is a substrate of all three efflux transporters in vitro.
Figure 1.
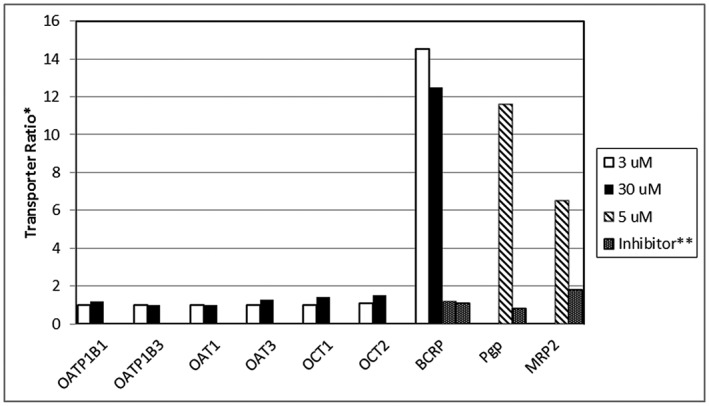
In vitro evaluation of canagliflozin as a substrate of human uptake and efflux transporters. *Transporter ratio for the efflux transporters (P‐glycoprotein [Pgp], multidrug resistance‐associated protein‐2 [MRP2], breast cancer resistant protein [BCRP]) is based on the permeability of canagliflozin from A‐B/B‐A. For the uptake transporters (organic anion transporter polypeptide isoforms OAT1B1, OATP1B3, organic anion transporters OAT1 and OAT3, and organic cationic transporters OCT1 and OCT2) transporter ratio is based on the uptake of canagliflozin in transporter containing system to control (water injected oocyte system or parental HEK293 line). **Ratio in presence of specific transport inhibitor (1 μmol l–1 KO143 for BCRP, 100 μmol l–1 cyclosporine for Pgp or 100 μmol l–1 cisplatin for MRP2)
Using prototypical substrates, the potential of canagliflozin to inhibit these efflux transporters was evaluated. Results are shown in Table 7. Canagliflozin showed a concentration dependent inhibition of Pgp and MRP2 substrate transport. IC50 values for Pgp and MRP2 were 19.3 and 21.5 μmol l–1, respectively. With respect to BCRP, toxicity was observed when canagliflozin was tested at >5 μmol l–1 in the presence of topotecan. Within the assay system, no BCRP inhibition was observed with canagliflozin up to 5 μmol l–1. To assess transporter inhibition potential of canagliflozin further, R values were calculated for intestinally and renally expressed efflux transporters according to the FDA 10 and EMA 11 guidelines, and the values are shown in Table 8. The results suggest that canagliflozin was likely an in vivo inhibitor of Pgp, MRP2, and BCRP.
Table 7.
In vitro evaluation of canagliflozin as an inhibitor of human efflux and uptake transporters
| Prototypical substrate | IC50 (SE) μmol l–1 | |
|---|---|---|
| Efflux transporters | ||
| Pgp | [3H]‐Digoxin | 19.3 (7.1) |
| MRP2 | [3H]‐Etoposide | 21.5 (3.2) |
| BCRP | [3H]‐Topotecan | NAa |
| Uptake transporters | % inhibition at 100 μmol l–1 | |
| OATIP1B1 | [3H]‐Estrone‐3‐sulfate | 24 |
| OATP1B3 | [3H]‐Estradiol‐17 β‐glucuronide | 32.6 |
| OAT1 | [3H]‐Para‐aminohippuric acid | NA |
| OAT3 | [3H]‐Estrone‐3‐sulfate | NA |
| OCT1 | [14C]‐Tetraethyl ammonium bromide | 5.2 |
| OCT2 | [14C]‐Tetraethyl ammonium bromide | 44 |
No inhibition at the highest feasible concentration (10.5 μmol l–1)
NA = Not applicable.
SE, standard error. SE values presented for the IC50 estimates
Table 8.
Evaluation of transporter inhibition potential for canagliflozin using basic approaches
| Transporter and location | In vitro Inhibition | FDAb | EMAc | ||
|---|---|---|---|---|---|
| Intestinal | Ki a | I d /Ki < 10 | Interaction warranted | R = 0.1x I d | Interaction warranted |
| Pgp | 9.65 | 125.6 | Yes | 268 | Yes |
| MRP2 | 10.76 | 139.5 | Yes | 268 | Yes |
| Hepatic | Ki a | 1 + ([I uinlet,max ) < 1.25 | R = 25x [I u, inlet max ] | ||
| OATP1B3 | >50 | 1.174 | No | 4.35 | No |
| Renal | Ki a | I u, /IC50 < 0.1 | R = 50x [I u, ] | ||
| OCT2 | >50 | 0.0011 | No | 5.5 | No |
| Pgp | 9.65 | 0.0057 | No | 5.5 | No |
[I] represents mean steady‐state total Cmax in plasma following once daily administration of highest clinical dose 300 mg (10.5 μmol l–1)
[Iu] represents unbound mean steady‐state Cmax in plasma following once daily administration of highest clinical dose 300 mg (0.11 μmol l–1)
Ki = IC50/2, assumption of competitive inhibition
Id = Dose of canagliflozin/250 ml
Iu, inlet max =0.174 μmol l–1, unbound hepatic inlet concentration after oral administration at 300 mg canagliflozin
If R < the given factor, no in vivo DDI study is needed.
If Ki > R, interaction potential is excluded
Substrate activity and inhibition potential of uptake transporters
Uptake transport of 14C‐canagliflozin was measured in solvent injected oocytes and specific human transporter injected oocytes (OATP1B1, OAT1, OAT3, OCT1 and OCT2) at 2 concentrations (3 and 30 μmol l–1) in vitro. The uptake ratios (transporter‐injected oocyte/solvent‐injected oocyte) were calculated in each of these systems and the results are presented in Figure 1. The prototypical tritiated substrates of OATP1B1 and OAT3 (estrone 3‐sulfate), OAT1 (para‐aminohippuric acid), and OCT1 and OCT2 (tetraethylammonium bromide) showed uptake ratios of 20, 18, 59, 5 and 44, respectively. In the presence of transporter specific positive control inhibitors, uptake ratios for all prototypical substrates were <2, confirming active transport occurred in the system. In contrast, uptake ratios for canagliflozin were <2 for all uptake transporters tested in the oocyte injected models, indicating that canagliflozin is not a substrate of these uptake transporters. Due to unavailability of oocyte models for OATP1B3, canagliflozin was tested in HEK293 cells stably transfected with OATP1B3. The uptake ratio (transfected cells versus non‐transfected control cells) of [3H]estradiol‐17‐β‐glucuronide, a prototypical substrate of OATP1B3, was 3.2, and uptake was inhibited by rifampicin (positive control inhibitor). For canagliflozin, the uptake ratio was <2 and unaffected by rifampicin, indicating that canagliflozin is not a substrate of OATP1B3.
Using prototypical substrates, the potential of canagliflozin to inhibit the above six human uptake transporters was evaluated. The inhibition of uptake for OATP1B1, OATP1B3, OAT1, OAT3, OCT1 and OCT2 at 100 μmol l–1 canagliflozin concentration is shown in Table 7. Although no transporter reached 50% inhibition in vitro, one hepatic (OATP1B3) and one renal (OCT2) transporter that showed highest inhibition was selected to assess in vivo inhibition potential of canagliflozin. The R values calculated for OCT2 and OATP1B3 10, 11 shown in Table 8 indicate that an in vivo uptake transporter inhibition was not expected with canagliflozin.
PBPK simulations: canagliflozin PK model performance verification
PK parameters and concentration–time plots of the observed and simulated data for canagliflozin 300 mg are presented in Table 9 and Figure 2A. Visual predictive checks as well as the AUC simulated/AUC observed and Cmax simulated/Cmax observed ratios suggest that, in general, the models effectively simulated the observed canagliflozin PK parameters within 33%. The average (± SD) simulated oral plasma clearance for canagliflozin was 15.0 ± 5.7 l h–1 and the observed average oral clearance based on nine subjects was 17.3 ± 3.54 l h–1. The simulated canagliflozin PK were dose linear between 100 and 300 mg QD dosing after single and repeated oral administration (Figure 2B), and the results are consistent with the observed data 4. There were no meaningful differences in the model performance and outcomes of canagliflozin PBPK DDI simulations between Simcyp versions 10, 11 and 15.
Table 9.
Simulated and observed canagliflozin oral pharmacokinetics parameters after a single 300 mg dose in healthy human subjects
| Simulated (90 subjects) (9 subjects/trial, 10 trials) | Observed (9 subjects) 10 | |||||||
|---|---|---|---|---|---|---|---|---|
| AUC0‐48h (ng h ml–1) | Tmax (h) | Cmax (ng ml−1) | CL/F (l h–1) | AUC0‐48h (ng h ml−1) | Tmax (h) | Cmax (ng ml−1) | CL/F (l h–1) | |
| Mean | 23 015 | 1.3 | 2968 | 15.0 | 17 375 | 1.89 | 2504 | 17.3 |
| Median | 22 525 | 1.25 | 2919 | 13.3 | 17 455 | 1.50 | 2504 | 17.0 |
| 5th percentile | 12 282 | 0.95 | 2234 | 8.10 | ||||
| 95th percentile | 37 002 | 1.80 | 3695 | 24.5 | ||||
| CV (%) | 41.0 | 19.0 | 17.0 | 38.0 | 20.0 | 41.0 | 19.0 | 26.0 |
| Min Val | 8857 | 0.90 | 1880 | 5.10 | 13 009 | 1.00 | 1820 | 12.6 |
| Max Val | 59 113 | 2.10 | 4889 | 33.9 | 23 547 | 3.00 | 3450 | 22.7 |
| S.D | 9327 | 0.25 | 501 | 5.70 | 3555 | 0.78 | 482 | 3.54 |
Figure 2.
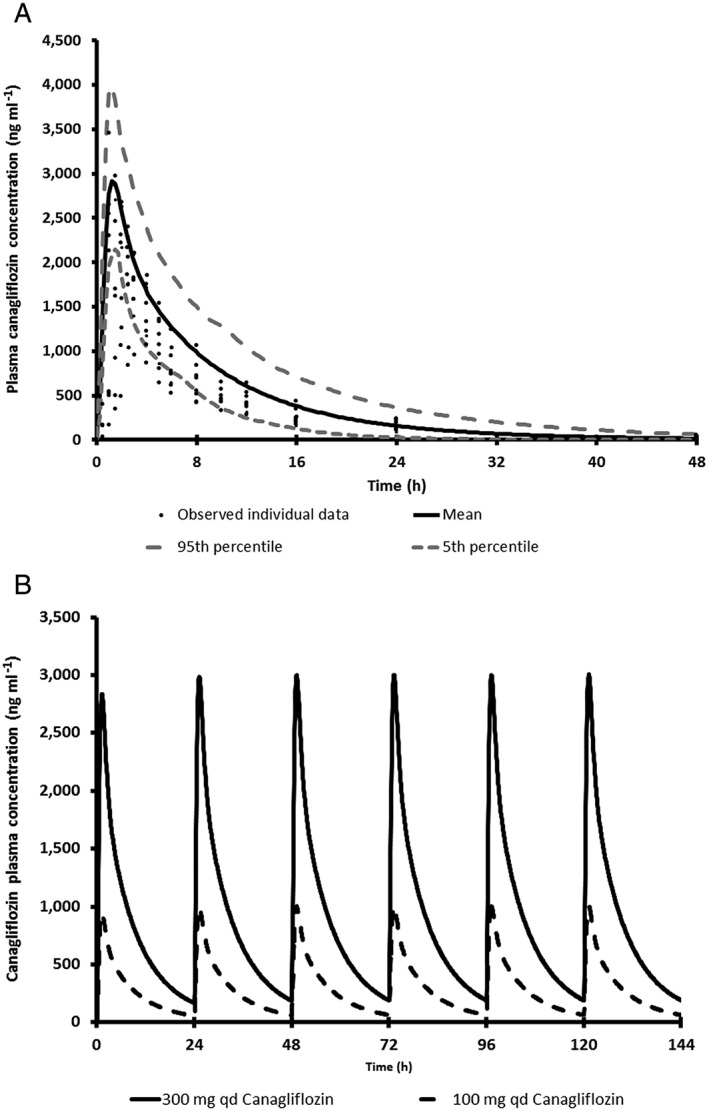
(A) Overlay plot of simulated mean (and 5th and 95th percentile) and observed canagliflozin pharmacokinetics following a single oral dose of 300 mg to nine healthy subjects. (B) Simulated pharmacokinetic profiles of canagliflozin following repeated oral administration of 100 and 300 mg to healthy subjects for 6 days
DDI simulations between canagliflozin and clinical CYP probe substrates
The simulated mean plasma concentration profiles of simvastatin (CYP3A probe), S‐warfarin (CYP2C9 probe), bupropion (CYP2B6 probe) and repaglinide (CYP2C8 probe) in virtual Caucasian healthy subjects administered each clinical probe alone or with repeated‐dose administration of canagliflozin are shown in Figures 3, 4, 5, 6. The simulated PK parameters of each clinical probe substrate with and without canagliflozin are shown in Table 10. The ratios of simulated average Cmax and AUC for all clinical probe substrates (simvastatin, S‐warfarin, bupropion or repaglinide) with and without canagliflozin administration were 1.00–1.13, suggesting that canagliflozin would not be a clinically relevant inhibitor of CYP3A, 2B6, 2C8, or 2C9.
Figure 3.
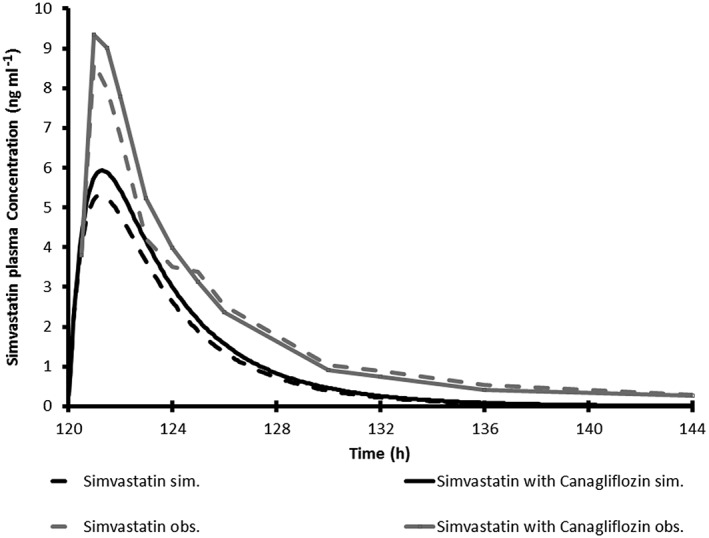
Simulated and observed average plasma concentration time profile for a single 40 mg oral dose of simvastatin with and without 300 mg canagliflozin once daily in healthy subjects
Figure 4.
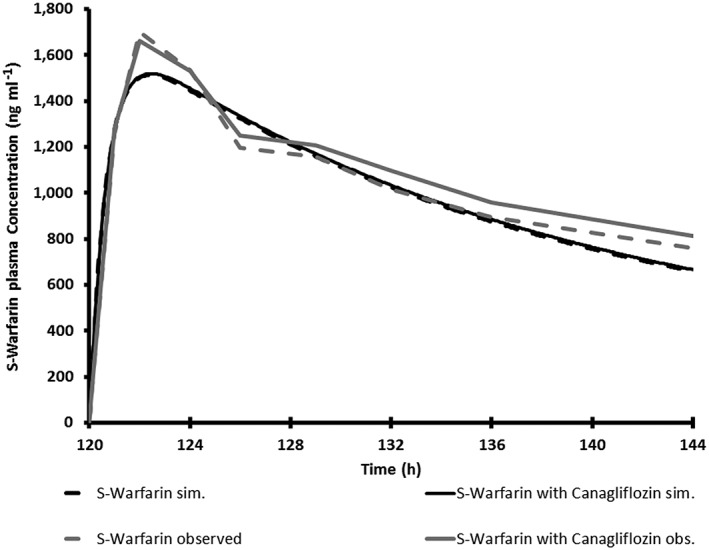
Simulated average plasma concentration time profile for a single 15 mg oral dose of S‐warfarin with and without 300 mg once daily canagliflozin in healthy subjects compared with observed plasma concentration time profile for a single 30 mg oral dose of warfarin (which contained 50% S‐warfarin) with and without 300 mg once daily canagliflozin in healthy subjects
Figure 5.
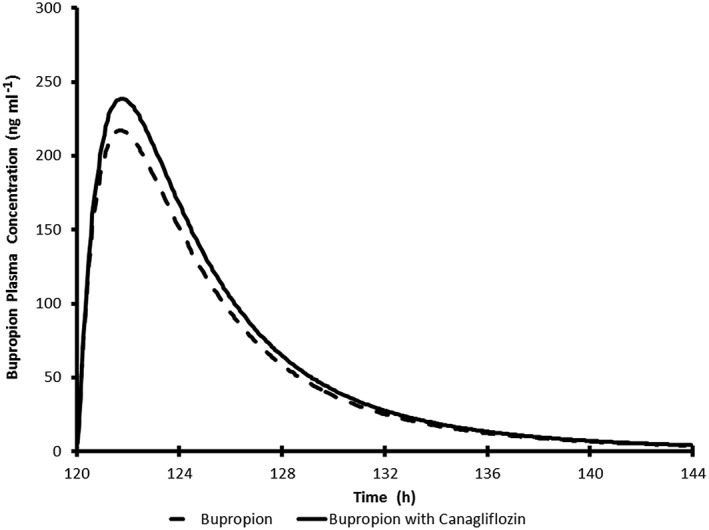
Simulated average plasma concentration time profile for a single 150 mg oral dose of buproprion with and without 300 mg once daily canagliflozin in healthy subjects
Figure 6.
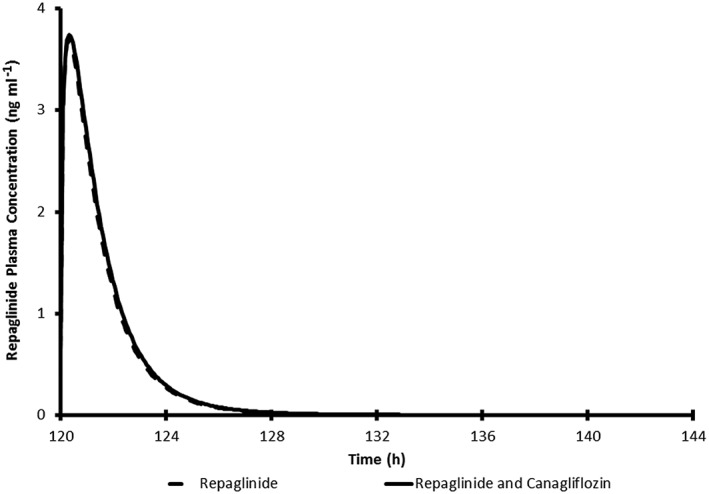
Simulated average plasma concentration time profile for a single 0.25 mg oral dose of repaglinide with and without 300 mg canagliflozin once daily in healthy subjects
Table 10.
Simulated pharmacokinetic parameters for simvastatin, s‐warfarin, bupropion or repaglinide with and without canagliflozin (300 mg) in healthy human subjects
| Simulated pharmacokinetic parameters of simvastatin (40 mg) in virtual subjects (n = 100) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Simvastatin without canagliflozin | Simvastatin with canagliflozin | Ratio with canagliflozin over without canagliflozin | ||||||
| AUC0‐48h (ng h ml−1) | Tmax (h) | Cmax (ng ml−1) | AUC0‐48h (ng h ml−1) | Tmax (h) | Cmax (ng ml−1) | AUC ratio | Cmax ratio | |
| Mean | 24.41 | 1.35 | 5.40 | 27.58 | 1.39 | 6.03 | 1.13 | 1.12 |
| 5th percentile | 5.26 | 0.90 | 1.07 | 6.11 | 0.95 | 1.19 | 1.09 | 1.07 |
| 95th percentile | 67.01 | 1.85 | 15.22 | 78.58 | 1.86 | 16.70 | 1.20 | 1.19 |
| CV (%) | 86 | 23 | 85 | 85 | 21 | 84 | 3.0 | 3.0 |
| SD | 20.92 | 0.31 | 4.59 | 23.42 | 0.29 | 5.07 | 0.03 | 0.04 |
| Simulated pharmacokinetic parameters of s‐warfarin (15 mg) in virtual subjects (n = 20) | ||||||||
|---|---|---|---|---|---|---|---|---|
| S‐warfarin without canagliflozin | S‐warfarin with canagliflozin | ratio with canagliflozin over without canagliflozin | ||||||
| AUC (ng h ml−1) | Tmax (h) | Cmax (ng ml−1) | AUC (ng h ml−1) | Tmax (h) | Cmax (ng ml−1) | AUC ratio | Cmax ratio | |
| Mean | 45 662.23 | 2.57 | 1522.60 | 46 270.15 | 2.60 | 1528.59 | 1.01 | 1.00 |
| 5th percentile | 14 608.29 | 1.99 | 731.39 | 14 790.17 | 1.99 | 734.10 | 1.01 | 1.00 |
| 95th percentile | 90 662.77 | 3.61 | 2606.95 | 92 839.30 | 3.61 | 2618.17 | 1.02 | 1.00 |
| CV (%) | 63.00 | 22.00 | 42.00 | 63.00 | 22.00 | 42.00 | ||
| SD | 28590.11 | 0.57 | 640.55 | 28984.54 | 0.57 | 643.18 | ||
| Simulated pharmacokinetic parameters of bupropion (150 mg) in virtual subjects (n = 100) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Bupropion without canagliflozin | Bupropion with canagliflozin | Ratio with canagliflozin over without canagliflozin | ||||||
| AUC0‐24h (ng h ml−1) | Tmax (h) | Cmax (ng ml−1) | AUC0‐24h (ng h ml−1) | Tmax (h) | Cmax (ng ml−1) | AUC ratio | Cmax ratio | |
| Mean | 1374.90 | 1.76 | 223.77 | 1503.70 | 1.81 | 244.66 | 1.13 | 1.12 |
| 5th percentile | 296.88 | 1.18 | 69.51 | 349.13 | 1.25 | 81.49 | 1.03 | 1.03 |
| 95th percentile | 4248.25 | 2.74 | 501.93 | 4393.12 | 2.75 | 561.87 | 1.22 | 1.24 |
| CV (%) | 0.0 | 27 | 0.0 | 0.0 | 26 | 0.0 | 5.0 | 6.0 |
| SD | 1292.81 | 0.48 | 150.55 | 1331.18 | 0.46 | 155.56 | 0.06 | 0.06 |
| Simulated pharmacokinetic parameters of repaglinide (0.25 mg) in virtual subjects (n = 100) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Repaglinide alone | Repaglinide + canagliflozin | Ratio with canagliflozin over without canagliflozin | ||||||
| AUC0‐24h (ng h ml−1) | Tmax (h) | Cmax (ng ml−1) | AUC0‐24h (ng h ml−1) | Tmax (h) | Cmax (ng ml−1) | AUC0‐24h | Cmax | |
| Mean | 6.66 | 0.34 | 3.82 | 6.98 | 0.36 | 3.88 | 1.04 | 1.02 |
| 5th percentile | 1.92 | 0.19 | 1.63 | 1.97 | 0.20 | 1.65 | 1.02 | 1.00 |
| 95th percentile | 13.2 | 0.55 | 6.36 | 13.9 | 0.60 | 6.41 | 1.09 | 1.04 |
| CV (%) | 59 | 47 | 41 | 60 | 47 | 41 | 2.0 | 1.0 |
| SD | 3.94 | 0.16 | 1.56 | 4.18 | 0.17 | 1.59 | 0.02 | 0.01 |
AUC, area under the concentration–time curve; [Cmax, peak plasma concentration; CV, coefficient of variance; SD, standard deviation; Tmax, time to reach peak plasma concentration
Discussion
The evaluation of drug interaction potential of a new drug is an integral part of risk assessment during drug development and regulatory review, and provides guidance to clinicians. In general, the main mechanisms of drug interactions involve DME (CYP, UGT enzymes), as well as efflux and uptake drug transporters. It is important to understand the interaction potential of a new drug with medications that are likely to be coadministered to patients. Here we present an integrated assessment of DDI potential of canagliflozin, a drug recently approved for the treatment of type 2 diabetes.
A three‐tier approach (i.e., in vitro studies coupled with basic models and PBPK simulations) was adopted in the DDI assessment of canagliflozin as a perpetrator of inhibition based interactions. Initially, standardized in vitro assays were used to examine whether canagliflozin was a substrate or an inhibitor of a given DME or transporter. If identified as an inhibitor in the in vitro assay, basic predictive models that are recommended by health authorities (FDA, EMA) were used to further assess the likelihood of interaction. If basic model(s) suggested drug interaction potential, then where applicable, either PBPK DDI models or clinical DDI studies were used to confirm the likelihood of a clinically relevant drug interaction.
In the radiolabelled human mass balance–metabolism study, a single oxidative metabolite (M9) was identified as present in circulation at <10% of the drug‐related species, suggesting that it is a minor circulating metabolite 8. In the CYP phenotyping study, the relative rate of M9 formation by the 3A4 isoform was ~29 times higher than 2C9 and ~14 times higher than 2D6, suggesting that 3A4 may be primarily involved in the formation of M9 from canagliflozin. However, the role of CYP3A4 metabolism was considered minimal based on limited contribution in overall disposition of canagliflozin (~7.6% of the administered oral dose).
CYP inhibition studies in human liver microsomes revealed a weak to moderate inhibition of CYP2B6, 2C8, 2C9 and 3A4. Further assessment of DDI potential using two basic models 10, 11 gave conflicting results on likelihood of in vivo interaction. Hence, DDI risk was further assessed by PBPK simulations.
To enable PBPK DDI simulation, first a PBPK model was developed for canagliflozin in virtual healthy human subjects. Model performance was verified by comparing the simulated PK profile of canagliflozin at 300 mg QD (maximum clinical dose) with observed clinical PK data 4. Overall, the simulated Cmax, AUC, oral clearance (CL/F), and Tmax and interindividual variability in PK were comparable to the observed values (Table 9).
Using the verified canagliflozin PBPK model, DDI CYP inhibition simulation trials were conducted with model substrates of CYP3A4 (simvastatin), CYP2C9 (S‐warfarin), CYP2B6 (bupropion) and CYP2C8 (repaglinide) that are available within the SIMCYP compound library (v11.1). Simulated profiles for all four probe substrates were comparable to the profiles obtained in the presence of canagliflozin (300 mg QD), suggesting that clinical interaction with CYP3A4, CYP2C9, CYP2B6 or CYP2C8 substrates was excluded. Later, lack of clinical interaction was confirmed in clinical DDI studies with simvastatin and S‐warfarin 15, 16. Since there was good agreement of results from DDI simulations to clinical DDI studies for CYP3A4 (simvastatin) and CYP2C9 (S‐warfarin), DDI simulation results for CYP2B6 and CYP2C8 were considered sufficient to confirm the lack of interaction and these results were accepted by health authorities to register the product. No further clinical DDI studies were conducted for CYP2B6 and 2C8.
As shown in the simulated PK of canagliflozin, the predicted mean Tmax was achieved at 1.30 h, slightly earlier than the observed mean Tmax (1.89 h). To investigate the impact of the predicted Tmax on DDI simulations, simulations were repeated with a modified dosing regimen where canagliflozin was dosed 0.6 h later than the comedication (substrate) to mimic a scenario of achieving a canagliflozin Tmax at 1.89 h after dosing of the comedication. DDI simulation outcomes under the modified scenario were similar to predicted Tmax (1.30 h) for the clinical probe substrates (bupropion, S‐warfarin, repaglinide or simvastatin) vs canagliflozin. Therefore simulations under this modified scenario were not presented in the manuscript. Based on this work it can be concluded that the identified differences between predicted Tmax versus observed Tmax did not have an impact on the predicted extent of DDI with bupropion and repaglinide as substrates.
Canagliflozin did not show induction of CYP3A4, 2B6 or 1A2 up to maximum feasible concentrations (15 μmol l–1) in human hepatocytes in vitro. Further, there was no evidence of CYP induction in clinical DDI studies on simvastatin 15, glyburide 15, S‐warfarin 16, ethinylestradiol 16 and paracetamol 17. Based on the body of evidence from the in vitro human hepatocyte induction study and several clinical DDI studies, clinically meaningful induction‐related DDI by canagliflozin was excluded.
Based on a human mass balance–metabolism study 8 and canagliflozin metabolism in recombinant UGT systems, it appears that UGTs 1A9 and 2B6 are involved in the metabolism of canagliflozin. In an in vitro UGT inhibition study in human liver microsomes, canagliflozin showed a weak inhibition of UGT1A1 (IC50 = 91 μmol l–1) and UGT1A6 (IC50 = 50 μmol l–1). Similar to CYPs, two basic predictive approaches 10, 11 (Table 5) gave conflicting results regarding likelihood of in vivo interaction. Review of existing clinical pharmacology studies performed during development indicated that there was no effect of canagliflozin (300 mg dose) on the PK of paracetamol (~1000 mg dose), a substrate of UGT1A1, UGT1A6 and UGT1A9 enzymes in healthy subjects 17. It is noteworthy that glucuronidation by these enzymes is the primary route of paracetamol clearance, accounting for ~57% of the administered dose 18, 19. Although paracetamol is not an ideal clinical probe substrate for UGT1A6, no interaction of canagliflozin on the PK of paracetamol provides general evidence for lack of significant inhibition of the UGTs. Overall, in vitro and in vivo DDI results suggest that canagliflozin has a low potential to cause drug interactions via inhibition of UGTs.
No in vitro studies were conducted to test the effect of other drugs on canagliflozin metabolism by UGTs; however, clinical DDI studies with probenecid (clinical probe inhibitor for multiple UGTs) and rifampicin (clinical probe inducer of DMEs) were conducted later in the development program. In the probenecid‐canagliflozin clinical DDI study 20, canagliflozin Cmax and AUC were marginally increased (↑13%, ↑21%) in the presence of probenecid. The metabolite (M5 or M7) to parent ratios for Cmax and AUC in canagliflozin plus probenecid group were 10–20% higher than in the canagliflozin alone group, indicating that there was no clinically meaningful interaction observed with probenecid. In the rifampicin–canagliflozin clinical DDI study 20, canagliflozin Cmax and AUC were significantly decreased (↓28%, ↓51%) in the presence of rifampicin. The glucuronide metabolite (M5 or M7) to parent ratios for Cmax and AUC in canagliflozin plus rifampicin group were 1.4× to 2.2× higher than in the canagliflozin alone group, indicating that there was a clinically significant interaction with rifampicin. Based on the clinical DDI study, coadministration of canagliflozin with broad inducers such as rifampicin (e.g., phenytoin, barbiturates, phenobarbital, ritonavir, carbamazepine, efavirenz and St John's wort [Hypericum perforatum]) may warrant a dose increase adjustment. It is recommended that, for patients who are receiving canagliflozin 100 mg QD, glycosylated haemoglobin should be monitored and, if additional glycaemic control is needed, an increase in the canagliflozin dose to 300 mg QD should be considered 20. For patients receiving the 300 mg QD dose, a 50% reduction in canagliflozin exposure is still predicted to provide plasma exposures and glycosylated haemoglobin‐lowering efficacy that is slightly greater than those in patients receiving 100 mg QD canagliflozin 7, 20, 21.
In studies with uptake transporters, a maximum % inhibition (44%) was observed with OCT2 at 100 μmol l–1 of canagliflozin. Based on basic predictive models (Table 8), in vivo inhibition of OCT2 at the renal level was excluded. Further, in a clinical DDI study with metformin 2000 mg QD dose (a substrate of OCT2, mainly cleared by the renal route in humans), metformin renal clearance was similar for metformin alone and in combination with canagliflozin 300 mg QD, indicating no clinically relevant inhibition of OCT2 or multidrug and toxin extrusion protein 1 transporters 15. For OATP1B3, 32.6% inhibition was observed at 100 μmol l–1; however, the relevance to potential inhibition in vivo was excluded based on the basic predictive models (Table 8). No meaningful inhibition was observed for the other uptake transporters (i.e., OATP1B1, OCT1, OAT1 and OAT3) at 100 μmol l–1; therefore, the potential for in vivo inhibition of these transporters by canagliflozin was excluded. Canagliflozin was not a substrate of any uptake transporters investigated in vitro, suggesting that victim interactions based on uptake transporters can be excluded for canagliflozin.
Canagliflozin was found to be a substrate of Pgp, MRP2 and BCRP transporters in vitro. Based on limited excretion of unchanged drug in urine and bile in preclinical species and humans 8, 13, it appears that efflux transporters are less likely to play a significant role in the disposition of canagliflozin. Further, a lack of clinically meaningful interaction with cyclosporine indicated that canagliflozin is less likely to be a candidate for victim‐based DDIs with efflux transporter inhibitors 21. In published DDI studies, cyclosporine was shown to effectively inhibit biliary excretion of irinotecan (a substrate of MRP2, Pgp and BCRP) and increase systemic concentrations of irinotecan 22, 23, suggesting that cyclosporine was a suitable clinical probe inhibitor to study efflux transporter DDIs.
In inhibition assays, canagliflozin was shown to be an in vitro inhibitor of Pgp and MRP2 but not BCRP. Based on recommended basic predictive models (Table 8), a theoretical possibility exists for canagliflozin to inhibit MRP2 and Pgp at the intestinal level in vivo at the highest recommended clinical dose (300 mg). However, a lack of interaction in the clinical Pgp DDI study with digoxin 16 indicates that canagliflozin is not likely to cause clinically relevant drug interactions for Pgp substrates. Recently, Lin et al. 24 summarized the efflux transporter properties of 63 marketed drugs in the Caco‐2 cell system. It was noted that most of these drugs are substrates of Pgp and have overlapping substrate specificity for Pgp and at least one or two other efflux transporters. In addition, there were no published reports of well characterized MRP2 intestinal inhibition in clinical DDI studies. Furthermore, studies in knock‐out animals showed a limited role of MRP2 in intestinal absorption of different drugs compared to Pgp [25–27]. For paclitaxel (substrate of mouse Pgp and mrp2 and human Pgp and MRP2), oral bioavailability was 8.5‐fold higher in Pgp knock‐out (mdr1 a/b (−/−)) mice compared to the wild‐type mice, whereas there was no change observed in bioavailability of paclitaxel between mrp2(−/−) and wild‐type mice 25, suggesting a limited role of MRP2 at the intestinal level. Thus, clinically relevant interaction due to MRP2 inhibition at the intestinal level was considered unlikely. The potential for in vivo inhibition at the systemic level (kidney or liver) was evaluated based on basic predictive regulatory models (Table 8), and the results indicated that the inhibition of efflux transporters at systemic level by canagliflozin can be excluded in vivo.
From the perspective of DDI assessment, in vitro studies followed by application of basic predictive model was useful in new drug development. There were some differences in predictive approaches recommended by FDA vs. EMA. For canagliflozin, the EMA basic model predicted less potential for an in vivo interaction with CYPs and UGTs, whereas the FDA approach predicted an interaction potential for both enzyme systems. However, there was good agreement in the transporter DDI assessment results between EMA and FDA predictive basic models for canagliflozin. Internationally harmonized basic predictive models for both enzyme‐ and transporter‐based interactions might be beneficial to the industry for DDI assessment before proceeding to mechanistic static or advanced PBPK simulations. Lack of relevant in vivo inhibition of CYPs by canagliflozin was confirmed by PBPK simulations.
In conclusion, an integrated assessment of drug interaction potential of canagliflozin with metabolic enzymes and transporters was successfully performed using in vitro DDI studies, scientific criteria provided by major regulatory authorities, and PBPK simulations together with available clinical DDI studies that were done during development. In vitro predictive models and PBPK DDI simulations were useful tools in drug development and most of the positive DDI in vitro results for canagliflozin were addressed by in vitro predictive models or PBPK DDI simulations without conducting clinical DDI studies. Based on overall assessment, it was concluded that canagliflozin is neither a perpetrator nor victim of clinically meaningful drug interactions, except when coadministered with UGT inducers such as rifampicin.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: the submitted work was supported by Janssen Pharmaceutical Companies of Johnson & Johnson. All authors have been employees and shareholders of Johnson & Johnson. No financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work.
The authors are thankful to Verna Hillsamer (Janssen Research & Development, LLC, Employee and shareholder of Johnson & Johnson USA) for writing and editorial assistance in the development of this manuscript.
Supporting information
Table S1 Cytochrome P450 and uridine 5′‐diphospho‐glucuronosyltransferase identification (ID), induction, and inhibition studies with canagliflozin
Table S2 Positive controls, probe substrates and standards used in cytochrome P450 and uridine 5′‐diphospho‐glucuronosyltransferase studies
Table S3 Transporter studies with canagliflozin
Table S4 Positive controls, probe substrates, and standards used in transporter studies
Supporting info item
Mamidi, R. N. V. S. , Dallas, S. , Sensenhauser, C. , Lim, H. K. , Scheers, E. , Verboven, P. , Cuyckens, F. , Leclercq, L. , Evans, D. C. , Kelley, M. F. , Johnson, M. D. , and Snoeys, J. (2017) In vitro and physiologically‐based pharmacokinetic based assessment of drug–drug interaction potential of canagliflozin. Br J Clin Pharmacol, 83: 1082–1096. doi: 10.1111/bcp.13186.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 2015; 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Devineni D, Curtin CR, Polidori D, Gutierrez MJ, Murphy J, Rusch S, et al. Pharmacokinetics and pharmacodynamics of canagliflozin, a sodium glucose co‐transporter 2 inhibitor, in subjects with type 2 diabetes mellitus. J Clin Pharmacol 2013; 53: 601–610. [DOI] [PubMed] [Google Scholar]
- 5. Liang Y, Arakawa K, Ueta K, Matsushita Y, Kuriyama C, Martin T, et al. Effect of canagliflozin on renal threshold for glucose, glycemia, and body weight in normal and diabetic animal models. PLoS One 2012; 7: e30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rosenstock J, Aggarwal N, Polidori D, Zhao Y, Arbit D, Usiskin K, et al. Dose‐ranging effects of canagliflozin, a sodium‐glucose cotransporter 2 inhibitor, as add‐on to metformin in subjects with type 2 diabetes. Diabetes Care 2012; 35: 1232–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sha S, Devineni D, Ghosh A, Polidori D, Chien S, Wexler D, et al. Canagliflozin, a novel inhibitor of sodium glucose co‐transporter 2, dose dependently reduces calculated renal threshold for glucose excretion and increases urinary glucose excretion in healthy subjects. Diabetes Obes Metab 2011; 13: 669–672. [DOI] [PubMed] [Google Scholar]
- 8. Mamidi RN, Cuyckens F, Chen J, Scheers E, Kalamaridis D, Lin R, et al. Metabolism and excretion of canagliflozin in mice, rats, dogs, and humans. Drug Metab Dispos 2014; 42: 903–916. [DOI] [PubMed] [Google Scholar]
- 9. Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, et al. Drug–drug interactions for UDP‐glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos 2004; 32: 1201–1208. [DOI] [PubMed] [Google Scholar]
- 10. FDA . Draft Guidance for Industry: Drug Interaction Studies – Study Design, Data Analysis, and Implications for Dosing and Labeling Recommendations. Silver Spring, MD2012.
- 11. EMA . Guideline on the Investigation of Drug Interactions. Committee for Human Medicinal Products (CHMP), London, UK, 2012. [Google Scholar]
- 12. Bjornsson TD, Callaghan JT, Einolf HJ, Fischer V, Gan L, Grimm S, et al. The conduct of in vitro and in vivo drug–drug interaction studies: a Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab Dispos 2003; 31: 815–832. [DOI] [PubMed] [Google Scholar]
- 13. Devineni D, Murphy J, Wang S‐S, Stieltjes H, Rothenberg P, Scheers E, et al. Absolute oral bioavailability and pharacokinetics of canagliflozin: A microdose study in healthy participants. Clin Pharmacol Drug Dev 2015; 4: 295–304. [DOI] [PubMed] [Google Scholar]
- 14. Turner DB, Rostami‐Hodjegan A, Tucker GT, Yeo KR. Prediction of non‐specific hepatic microsomal binding from readily specific hepatic microsomal binding from readily available physicochemical properties. (2006) ISSX meeting Manchester, poster 1812. Available at https://www.certara.com/wpcontent/uploads/Resources/Posters/Turner_2006_ISSX‐E9_NSPB.pdf
- 15. Devineni D, Manitpisitkul P, Murphy J, Skee D, Wajs E, Mamidi RN, et al. Effect of canagliflozin on the pharmacokinetics of glyburide, metformin and simvastatin in healthy participants. Clin Pharmacol Drug Develop 2015; 4: 226–236. [DOI] [PubMed] [Google Scholar]
- 16. Devineni D, Manitpisitkul P, Vaccaro N, Bernard A, Skee D, Mamidi RN, et al. Effect of canagliflozin, a sodium glucose co‐transporter 2 inhibitor, on the pharmacokinetics of oral contraceptives, warfarin, and digoxin in healthy participants. Int J Clin Pharmacol Ther 2015; 53: 41–53. [DOI] [PubMed] [Google Scholar]
- 17. Polidori D, Sha S, Mudaliar S, Ciaraldi TP, Ghosh A, Vaccaro N, et al. Canagliflozin lowers postprandial glucose and insulin by delaying intestinal glucose absorption in addition to increasing urinary glucose excretion: results of a randomized, placebo‐controlled study. Diabetes Care 2013; 36: 2154–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Patel M, Tang BK, Kalow W. Variability of acetaminophen metabolism in Caucasians and Orientals. Pharmacogenetics 1992; 2: 38–45. [DOI] [PubMed] [Google Scholar]
- 19. Prescott LF. Paracetamol overdosage. Pharmacological considerations and clinical management. Drugs 1983; 25: 290–314. [DOI] [PubMed] [Google Scholar]
- 20. Devineni D, Vaccaro N, Murphy J, Curtin C, Mamidi RN, Weiner S, et al. Effects of rifampin, cyclosporine A, and probenecid on the pharmacokinetic profile of canagliflozin, a sodium glucose co‐transporter 2 inhibitor, in healthy participants. Int J Clin Pharmacol Ther 2015; 53: 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wilding JP, Charpentier G, Hollander P, Gonzalez‐Galvez G, Mathieu C, Vercruysse F, et al. Efficacy and safety of canagliflozin in patients with type 2 diabetes mellitus inadequately controlled with metformin and sulphonylurea: a randomised trial. Int J Clin Pract 2013; 67: 1267–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chester JD, Joel SP, Cheeseman SL, Hall GD, Braun MS, Perry J, et al. Phase I and pharmacokinetic study of intravenous irinotecan plus oral ciclosporin in patients with fuorouracil‐refractory metastatic colon cancer. J Clin Oncol 2003; 21: 1125–1132. [DOI] [PubMed] [Google Scholar]
- 23. Innocenti F, Undevia SD, Ramirez J, Mani S, Schilsky RL, Vogelzang NJ, et al. A phase I trial of pharmacologic modulation of irinotecan with cyclosporine and phenobarbital. Clin Pharmacol Ther 2004; 76: 490–502. [DOI] [PubMed] [Google Scholar]
- 24. Lin X, Skolnik S, Chen X, Wang J. Attenuation of intestinal absorption by major efflux transporters: quantitative tools and strategies using a Caco‐2 model. Drug Metab Dispos 2011; 39: 265–274. [DOI] [PubMed] [Google Scholar]
- 25. Lagas JS, Vlaming ML, Tellingen O, Wagenaar E, Jansen RS, Rosing H, et al. Multidrug resistance protein 2 is an important determinant of paclitaxel pharmacokinetics. Clin Cancer Res 2006; 12 (20 Pt 1): 6125–6132. [DOI] [PubMed] [Google Scholar]
- 26. van Waterschoot RA, Lagas JS, Wagenaar E, Rosing H, Beijnen JH, Schinkel AH. Individual and combined roles of CYP3A, P‐glycoprotein (MDR1/ABCB1) and MRP2 (ABCC2) in the pharmacokinetics of docetaxel. Int J Cancer 2010; 127: 2959–2964. [DOI] [PubMed] [Google Scholar]
- 27. van Waterschoot RA, Heine R, Wagenaar E, Kruijssen CM, Rooswinkel RW, Huitema AD, et al. Effects of cytochrome P450 3A (CYP3A) and the drug transporters P‐glycoprotein (MDR1/ABCB1) and MRP2 (ABCC2) on the pharmacokinetics of lopinavir. Br J Pharmacol 2010; 160: 1224–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Cytochrome P450 and uridine 5′‐diphospho‐glucuronosyltransferase identification (ID), induction, and inhibition studies with canagliflozin
Table S2 Positive controls, probe substrates and standards used in cytochrome P450 and uridine 5′‐diphospho‐glucuronosyltransferase studies
Table S3 Transporter studies with canagliflozin
Table S4 Positive controls, probe substrates, and standards used in transporter studies
Supporting info item