

Abstract
Aims
To assess safety, pharmacokinetics (PK) and clinical efficacy of bimekizumab (formerly UCB4940), a novel humanized monoclonal antibody and dual inhibitor of interleukin (IL)‐17A and IL‐17F, in subjects with mild plaque psoriasis.
Methods
Randomized, double‐blind, first‐in‐human study of bimekizumab in 39 subjects who received single‐dose intravenous bimekizumab (8–640 mg) or placebo (NCT02529956).
Results
Bimekizumab demonstrated dose‐proportional linear PK and was tolerated across the dose range assessed. No subject discontinued due to treatment‐emergent adverse events and no severe adverse events were reported. Bimekizumab demonstrated fast onset of clinically‐meaningful effects on skin of patients with mild psoriasis as early as Week 2. Maximal improvements (100% or near 100% reductions from baseline) in all measures of disease activity were observed between Weeks 8–12 in subjects receiving 160–640 mg bimekizumab. The duration of effect at doses ≥160 mg was evident up to Weeks 12–20 after a single intravenous dose, dependent on endpoint.
Conclusions
This is the first study to demonstrate the safety, tolerability and clinical efficacy of a dual IL‐17A and IL‐17F inhibitor, in subjects with mild psoriasis. Bimekizumab showed fast onset of clinically‐meaningful efficacy by Week 2, with a maximal or near‐maximal magnitude of response that was maintained up to study Weeks 12–20. These findings support the continued clinical development of bimekizumab for diseases mediated by both IL‐17A and IL‐17F, including psoriasis.
Keywords: anti‐IL17A, anti‐IL17F, bimekizumab, interleukin‐17, psoriasis, UCB4940
What is Already Known about this Subject
Interleukin (IL)‐17A, and potentially IL‐17F, are believed to drive pathogenesis in psoriasis.
Targeting the IL‐17 pathway with anti‐IL‐17A antibodies has demonstrated clinical efficacy in subjects with moderate‐to‐severe psoriasis.
Inhibition of the IL‐17 pathway may leave subjects more vulnerable to infection; neutropenia is a potential anti‐IL‐17 class effect.
What this Study Adds
First demonstration of safety, tolerability and clinical efficacy of a dual IL‐17A and IL‐17F inhibitor in mild psoriasis.
Bimekizumab showed fast onset of clinically‐meaningful efficacy; maximal or near‐maximal magnitude of response was maintained up to Weeks 12–20.
Supports continued clinical development of bimekizumab for IL‐17A and IL‐17F‐mediated diseases.
Tables of Links
| LIGANDS | |
|---|---|
| Adalimumab | Infliximab |
| Brodalumab | Ixekizumab |
| Cyclophosphamide | Methotrexate |
| Etanercept | Psoralen |
| IL‐17A | Secukinumab |
| IL‐17F | Vitamin D3 |
| IL‐25 (IL‐17E) | |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2, 3.
Introduction
Psoriasis is a chronic and debilitating immune‐mediated inflammatory skin disease that affects 1–3% of the worldwide population 4. Approximately 80% of individuals with psoriasis have mild‐to‐moderate forms of the disease 5. People with psoriasis typically develop erythematosus and scaly plaques on their skin, and extent of skin surface involvement is a key determinant of disease severity 5. Beyond physical symptoms, psoriasis imposes a substantial psychosocial burden on individuals and many struggle to cope with the stigma associated with their disease 6. Moreover, it has been reported that psoriasis can impair quality of life to the same extent as clinically severe conditions such as cancer and diabetes 4. Psoriasis is also associated with significant comorbidities such as cardiovascular disease, diabetes and depression 7. Current treatment recommendations are based on psoriasis symptom severity: topical creams (corticosteroids and vitamin D3) for mild‐to‐moderate disease; and systemic or phototherapies for moderate‐to‐severe disease 5.
The aetiology of psoriasis is poorly understood; however, it is known to have an immune‐mediated pathogenesis 8. Tumour necrosis factor (TNF) plays a role in the pathogenesis of psoriasis 9 and biological treatments include TNF inhibitors (adalimumab, etanercept and infliximab) 10. Interleukin (IL)‐17 also plays a critical role in psoriasis 8. Release of IL‐17 triggers a proinflammatory signalling cascade in skin keratinocytes, which is believed to drive chronic inflammation in conditions such as psoriasis 11. IL‐17 family members share a structural similarity, which is particularly evident in the IL‐17A and IL‐17F isoforms 11. IL‐17A is pathogenic in mouse models of immune‐inflammatory disease, with IL‐17F considered less important 12. In humans, IL‐17A and IL‐17F are key proinflammatory cytokines that share similar and overlapping biological functions 13, 14. Both are drivers of chronic inflammation and coexpressed in immune‐inflammatory diseases 14, 15. Although there are limited studies in humans, there is evidence for a role for IL‐17F in the pathogenesis of psoriasis 16, 17.
The use of agents targeting the IL‐17 pathway has been evaluated extensively in clinical studies for psoriasis 8 with therapeutic interventions either approved or in clinical development. Notably, secukinumab (IL‐17A inhibitor) is now approved for the treatment of moderate‐to‐severe plaque psoriasis, psoriatic arthritis and ankylosing spondylitis in the USA and EU 18, 19. Ixekizumab (IL‐17A inhibitor) is also approved for the treatment of patients with moderate‐to‐severe plaque psoriasis in the USA and EU 20, 21. Brodalumab (IL‐17RA blocker) is under clinical development for moderate‐to‐severe psoriasis 8, 22, 23. Inhibitors of the IL‐17RA receptor, such as brodalumab, exert broad blockade, encompassing inhibition of not only IL‐17A and IL‐17F but also IL‐25 (IL‐17E); IL‐25 has anti‐inflammatory effects in human cells 24.
The currently approved agents have demonstrated clinical efficacy in subjects with moderate‐to‐severe plaque psoriasis 25, 26, 27, 28, and are considered generally well tolerated 29. However, IL‐17A and IL‐17F have a critical function in adaptive immune defence from bacterial and fungal infection, and inhibition of this action could potentially leave subjects more vulnerable to infection 30. In clinical studies involving anti‐IL‐17A or anti‐IL‐17 receptor inhibitors, the most common adverse events (AEs) reported by subjects included: worsening of disease, upper respiratory tract infections, headache, nausea and nasopharyngitis 29. Neutropenia is also a potential class effect, specifically reported in subjects who have received secukinumab, ixekizumab or brodalumab 26, 27, 28.
Bimekizumab (formerly UCB4940) is a novel humanized monoclonal antibody of the immunoglobulin G1 isotype, rationally designed to be able to bind at a similar site on both IL‐17A and IL‐17F, conveying dual inhibition of both isoforms. Comparing this unique mechanism of action with other agents, bimekizumab might improve therapeutic efficacy through its potent selectivity for the IL‐17A and IL‐17F isoforms. Bimekizumab is in early clinical development in several indications, including psoriasis. Here we report the safety, tolerability, pharmacokinetics (PK) and effect on clinical features of plaque psoriasis of single‐dose bimekizumab in a first‐in‐human study of subjects with mild psoriasis.
Methods
Study design
This was a Phase I, first‐in‐human, randomized, double‐blind, placebo‐controlled, single‐dose, dose‐escalating study (NCT02529956). It was conducted in accordance with the applicable regulatory and International Conference on Harmonisation – Good Clinical Practice requirements, the ethical principles originating in the Declaration of Helsinki, and local laws. The protocol was reviewed and approved by the National Research Ethics Service Committee South Central‐Berkshire (Research Ethics Committee reference: 12/SC/0564). All subjects provided documented consent prior to study entry. Treatment administration and data collection were carried out at a single clinical pharmacology site in the UK.
Each subject underwent a ≤19‐day screening period, followed by predosing assessments over a 2‐day period, prior to infusion of bimekizumab or placebo. Subjects were randomized to one of six treatment groups to receive: bimekizumab single intravenous infusion dose of 8, 40, 160, 480 or 640 mg, or placebo. A safety follow‐up (SFU) period was also included in the study; subjects were evaluated during a 20‐week period after dosing.
Study treatment
Dosing
Each subject received a single intravenous infusion of bimekizumab (at the dose specified for that cohort), or placebo over a duration of ≥60 min. Subjects were assigned sequentially to cohorts and were randomly assigned to study treatment. The first treatment group received the lowest dose, 8 mg, bimekizumab, or placebo; dose escalations were based on reviews of the safety, PK and laboratory data from the previous cohort.
Study population
The key inclusion criteria for this study were: male or female (age ≥18 to ≤70 years); subjects with a confirmed diagnosis of mild‐to‐moderate plaque‐type psoriasis for ≥6 months involving ≤5% of body surface area (excluding the scalp); and at least two psoriatic lesions with at least one plaque in suitable biopsy sites. Key exclusion criteria included: use of systemic nonbiological psoriasis therapy (methotrexate, steroids or cyclophosphamide) or psoralen plus UVA/UVB phototherapy within 4 weeks prior to screening; and treatment with biological agents ≤12 months before the study.
Objectives
The primary objective was to evaluate the safety of single‐dose bimekizumab. Secondary objectives were to characterize the PK profile of single‐dose bimekizumab and to assess its effects on clinical features of plaque psoriasis. An exploratory objective was to assess the immunogenicity of bimekizumab.
Study outcome variables
Safety
Safety variables were assessed during this study, e.g. AEs, vital signs, electrocardiogram (ECG) data, clinical chemistry, haematology (including coagulation/haemostasis tests) and urinalysis. All variables were assessed during the study predose and 0–48‐h postdrug time points, all safety variables except ECG data were assessed during the study screening period. Additionally, AEs were assessed at each time point up to Week 20 of the postinfusion SFU period; vital signs were monitored during Weeks 1–20 of the SFU. Clinical chemistry and haematology parameters were assessed at Weeks 1–4, and Weeks 8, 12 and 20. Urinalysis was measured at Weeks 1, 4, 12 and 20.
Pharmacokinetics
Bimekizumab concentrations were determined in plasma using a sandwich ligand‐binding assay and visualized using electrochemiluminescence. The assay had a range of quantitation between 150 and 18 000 ng ml–1. The inter‐ and intraday assay precision, as determined by coefficient of variation, was ≤20%. The impact of interference was not evaluated. Blood samples were taken predose, then at 0–48 h, 72 h and 96 h postdose, and during Weeks 1–20 of the SFU.
Immunogenicity
Plasma samples were also used to test for the detection of anti‐bimekizumab antibodies at predose, Week 4 and Week 20 of the SFU period.
Effects on clinical features of plaque psoriasis
Lesion severity score (LSS), Psoriasis Area and Severity Index (PASI) and hysician's Global Assessment (PGA) of disease severity were investigated. LSS was assessed using five‐point severity scales of lesion redness, induration and scaling (0 = none, 4 = severe), on separate psoriatic lesions to those taken for biopsy. PASI was assessed by the investigator, based on a five‐point scale, as for LSS. PASI scores ranged from 0 to 72 (higher scores indicating more severe disease 31). PGA was assessed by the investigator based on their impression of disease severity. A seven‐point scale was used to grade PGA (0 = clear, 6 = severe). These variables were measured during the predose and dosing periods, and during Weeks 1–20 of SFU.
Statistical methods
This study was designed to have >80% power to detect a 60% improvement in LSS from placebo at Week 2. The power calculations were based on a two‐sided t test with a standard deviation (SD) of 0.84, placebo mean score of 6.3, statistical significance level of 5%, and six subjects on bimekizumab and 10 subjects on placebo.
Demographic, safety, PK and clinical features of plaque psoriasis were summarized descriptively by treatment group and visit. Descriptive statistics on continuous data included arithmetic means, geometric means (where appropriate), medians, SDs and ranges. Categorical data were summarized using frequency counts and percentages.
PK parameters were calculated via noncompartmental analysis methods from the concentration‐time data. Immunogenicity was assessed using the validated method QBR113786 (LGC Limited, UK) to quantify anti‐bimekizumab antibodies, with a screening cut‐off point of 290 ng ml–1.
The LSS at Week 2 was treated as a continuous variable and was analysed using an analysis of covariance (ANCOVA) with baseline value (predose) as a covariate and dose as a fixed effect. Estimated means, differences from placebo and 95% confidence intervals were presented by dose.
Additionally, a Bayesian analysis of LSS and PASI scores at Week 2 was performed to support internal decision making for bimekizumab and the model was similar to the ANCOVA described above. Vague normal prior distributions were used for these analyses. From the posterior distributions of the model parameters, the posterior mean, 95% credible interval and posterior probability of the treatment effect exceeding certain prespecified thresholds were reported. The results of the Bayesian analyses only are reported in the subsequent section, which were consistent with the ANCOVA results.
A decision was made a‐priori to not use P‐values and so they are not reported here. Statistical confidence in the treatment effect was based on confidence/credible intervals and posterior probabilities. There was no imputation of missing data, nor correction for multiple comparisons.
Results
Subject disposition is summarized in Figure 1. Of the 234 subjects screened for study inclusion, 195 (83.3%) failed screening; 39 (comprising the full analysis set [FAS] and PK per‐protocol set) were randomized to treatment (bimekizumab, n = 26; placebo, n = 13). A total of 37 subjects completed the study; two were withdrawn (one each from the bimekizumab 8 mg group and 480 mg group, neither because of an AE). No subjects received concomitant medication for psoriasis during the study.
Figure 1.
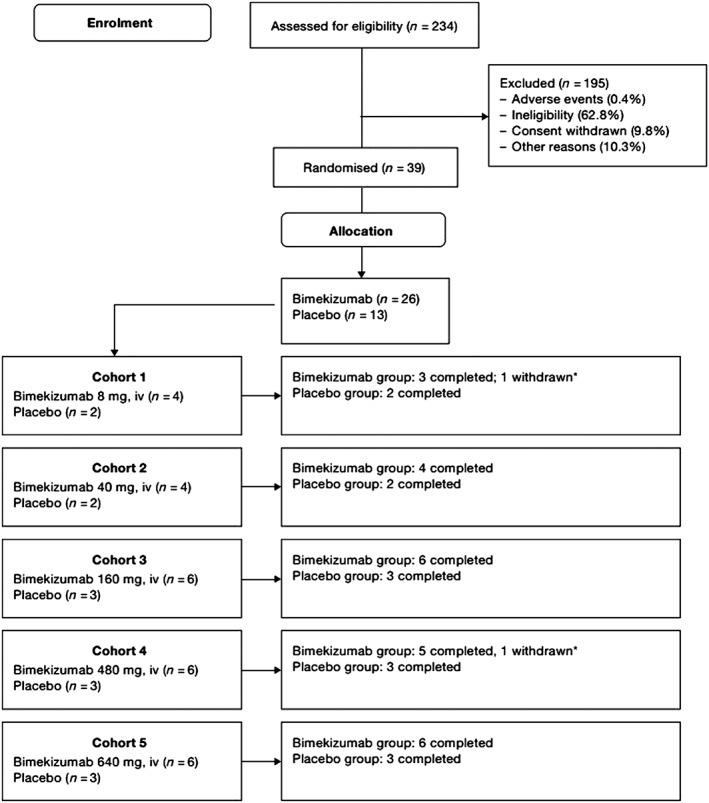
Subject disposition. iv, intravenous. * Subject 00019 (bimekizumab 8 mg group) and subject 00111 (bimekizumab 480 mg group) withdrew themselves from the study. For both subjects, the reason for discontinuation was listed as withdrawal by subject and was not the result of an adverse event
Subject demography and disease characteristics
A summary of subject demographics at baseline is shown in Table 1. The majority of subjects were male (76.9%) and Caucasian (94.9%), and mean age was 39.53 years (range: 21.4–65.0 years). The median PASI score for the study population at baseline was 3.50 (range 0.8–6.7), corresponding to PASI scores for a population with mild psoriasis. The first subject was enrolled on 22nd November 2012; the last subject completed the study protocol on 13th January 2014.
Table 1.
Subject demography, baseline characteristics and disease characteristics (FAS)
| Bimekizumab | |||||||
|---|---|---|---|---|---|---|---|
| Variable | Placebo | 8 mg | 40 mg | 160 mg | 480 mg | 640 mg | Total |
| n = 13 | n = 4 | n = 4 | n = 6 | n = 6 | n = 6 | n = 39 | |
| Age, years | |||||||
| Mean (SD) | 38.22 (13.31) | 34.70 (9.73) | 44.80 (10.46) | 43.53 (8.43) | 39.52 (9.10) | 38.05 (7.19) | 39.53 (10.39) |
| Sex, n (%) | |||||||
| Male | 12 (92.3) | 4 (100) | 2 (50.0) | 4 (66.7) | 3 (50.0) | 5 (83.3) | 30 (76.9) |
| Racial cohort, n (%) | |||||||
| Asian | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 1 (2.6) |
| Other/mixed | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 1 (2.6) |
| Caucasian | 13 (100) | 4 (100) | 4 (100) | 6 (100) | 6 (100) | 4 (66.7) | 37 (94.9) |
| Ethnicity, n (%) | |||||||
| Not Hispanic or Latino | 13 (100) | 4 (100) | 4 (100) | 6 (100) | 6 (100) | 6 (100) | 39 (100) |
| BMI, kg m–2 | |||||||
| Mean (SD) | 26.92 (3.49) | 27.18 (2.67) | 28.35 (1.13) | 24.68 (2.94) | 25.05 (3.47) | 27.33 (5.13) | 26.53 (3.48) |
| LSS | |||||||
| Mean (SD) | 5.0 (1.4) | 4.5 (1.3) | 4.5 (1.7) | 5.0 (0.6) | 4.2 (1.5) | 4.3 (1.4) | 4.5 (1.2) |
| PASI | |||||||
| Median | 3.00 | 2.60 | 3.65 | 3.30 | 3.40 | 3.75 | 3.50 |
| Min, max | 1.8, 6.1 | 1.2, 6.7 | 2.6, 4.4 | 1.0, 6.2 | 0.8, 6.2 | 2.4, 5.4 | 0.8, 6.7 |
BMI, body mass index; FAS, full analysis set; LSS, lesion severity score; max, maximum; min, minimum; PASI, Psoriasis Area and Severity Index; SD, standard deviationPercentages are based on the numbers of subjects per treatment group (in the FAS)
Safety
Bimekizumab was tolerated across the dose range assessed. Treatment‐emergent AEs (TEAEs) are reported in Table 2. Subjects in both bimekizumab and placebo groups experienced TEAEs (84.6% vs. 76.9%, respectively). The majority of TEAEs were of mild intensity (bimekizumab, 61.5%; placebo, 53.8%). Commonly reported TEAEs occurring in ≥10% of all subjects receiving bimekizumab were headache (n = 6 subjects; 23.1%), oropharyngeal pain (n = 5 subjects; 19.2%), nasopharyngitis (n = 4 subjects; 15.4%) and medical device (ECG) site reaction (n = 3 subjects; 11.5%; data not shown).
Table 2.
Overview of treatment‐emergent adverse events (TEAEs; FAS)
| Placebo | Bimekizumab | ||||||
|---|---|---|---|---|---|---|---|
| Category | 8 mg | 40 mg | 160 mg | 480 mg | 640 mg | Total | |
| n = 13 | n = 4 | n = 4 | n = 6 | n = 6 | n = 6 | n = 26 | |
| n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | |
| Any TEAEs | 10 (76.9) | 1 (25.0) | 4 (100) | 5 (83.3) | 6 (100) | 6 (100) | 22 (84.6) |
| Serious TEAEs | 0 | 0 | 1 (25.0) | 0 | 0 | 0 | 1 (3.8) |
| Discontinuations due to TEAEs | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Treatment‐related TEAEs | 4 (30.8) | 1 (25.0) | 3 (75.0) | 3 (50.0) | 3 (50.0) | 2 (33.3) | 12 (46.2) |
| Severe TEAEs | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Deaths | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
FAS, full analysis set; TEAE, treatment‐emergent adverse event
Treatment‐related TEAEs were reported in 12 (46.2%) and 4 (30.8%) subjects in the bimekizumab and placebo groups, respectively. One serious AE of vomiting (moderate intensity), occurring 37 days after dosing, was experienced by a subject receiving 40 mg bimekizumab; this was considered not related to the study medication. No study discontinuations due to TEAEs, severe TEAEs or deaths were reported during this study. No infusion‐site reactions were reported. Mean neutrophil counts were similar between treatment groups (Figure 2), and no subject experienced clinically meaningful changes in neutrophil count.
Figure 2.
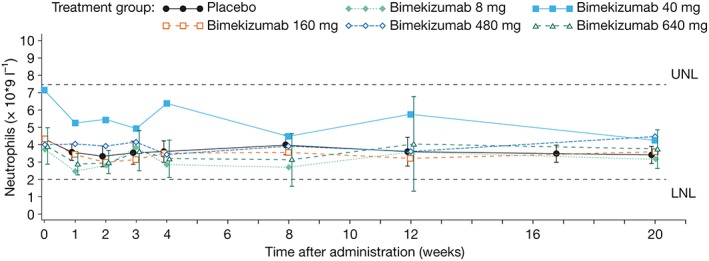
Mean neutrophil count by treatment group (full analysis set); LNL, lower normal limit; UNL, upper normal limit. Note: 95% confidence intervals are included for the placebo and bimekizumab highest treatment groups only. The figure was based on protocol‐specified time after administration time points for the x‐axis; however, the time points presented in this figure are offset for each treatment group to avoid overlap of symbols and confidence intervals
No notable differences in mean values, or consistent or clinically meaningful changes from baseline, for any laboratory parameters were reported. There were no reports of clinically meaningful ECG changes or physical examination abnormalities during the study. Overall, there were no consistent or clinically meaningful changes from baseline in vital signs observed during the study.
PK
The PK of bimekizumab increased with dose in a linear fashion (Figure 3). Following the end of drug infusion, bimekizumab exhibited a biphasic disposition and a linear elimination across the dose range evaluated.
Figure 3.
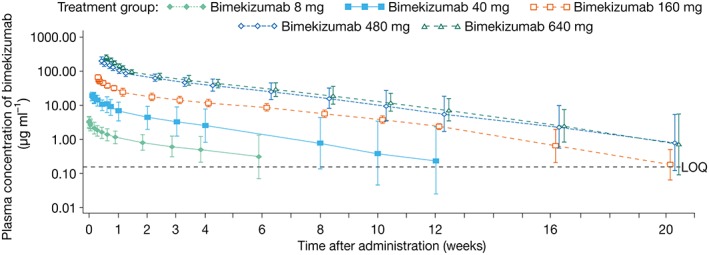
Geometric mean (with 95% confidence intervals) plasma concentration‐time profile of bimekizumab by dose (pharmacokinetic per‐protocol set). LOQ, limit of quantitation; Note: the figure was based on protocol‐specified time after administration time points for the x‐axis; however, the time points presented in this figure are offset for each treatment group to avoid overlap of symbols and confidence intervals
PK parameters are summarized in Table 3. Moderate‐to‐high interindividual variability for the area under the curve (AUC; geometric coefficient of variation [%] range: 22.0–64.1%) was observed in the bimekizumab dose groups. Geometric mean (GeoMean) half‐life ranged from 17 days (bimekizumab 40 mg) to 22 days (bimekizumab 160 mg) across the treatment groups. GeoMean volume of distribution in terminal phase ranged from 4.25 l (480 mg group) to 5.82 l (8 mg group). GeoMean of total body clearance ranged from 0.15 l day–1 (480 mg group) to 0.19 l day–1 (8 mg group). GeoMean AUC0–∞ increased in proportion with increasing doses of bimekizumab (Figure 4), as did Cmax (data not shown).
Table 3.
PK parameters of bimekizumab (PK‐PPS)
| Bimekizumab | |||||
|---|---|---|---|---|---|
| 8 mg n = 4 | 40 mg n = 4 | 160 mg n = 6 | 480 mg n = 6 | 640 mg n = 6 | |
| Cmax (ng ml–1) | |||||
| GeoMean | 2.9 | 17.5 | 60.9 | 211.1 | 260.0 |
| GeoCV (%) | 27.1 | 16.4 | 30.4 | 11.7 | 21.4 |
| AUC0–∞ (day × μg ml–1) | |||||
| GeoMean | 42.8 | 222.7 | 1019.0 | 3298.0 | 3787.0 |
| GeoCV (%) | 58.2 | 64.1 | 22.0 | 36.3 | 39.0 |
| AUC0–t (day × μg ml–1) | |||||
| GeoMean | 35.0 | 216.0 | 988.6 | 3251.0 | 3709.0 |
| GeoCV (%) | 61.2 | 64.3 | 21.6 | 35.1 | 36.6 |
| tmax (days) | |||||
| Median | 0.06 | 0.05 | 0.06 | 0.09 | 0.06 |
| Min, max | 0.04 | 0.04 | 0.06 | 0.06 | 0.06 |
| 0.13 | 0.13 | 0.13 | 0.21 | 0.13 | |
| t1/2 (days) | |||||
| GeoMean | 21.6 | 17.1 | 22.0 | 20.2 | 20.3 |
| GeoCV (%) | 47.0 | 40.2 | 14.9 | 32.4 | 39.3 |
| CL (l day–1) | |||||
| GeoMean | 0.19 | 0.18 | 0.16 | 0.15 | 0.17 |
| GeoCV (%) | 58.28 | 64.12 | 22.08 | 36.21 | 38.97 |
| Vz (l) | |||||
| GeoMean | 5.82 | 4.45 | 4.98 | 4.25 | 4.94 |
| GeoCV (%) | 11.12 | 29.92 | 23.11 | 26.95 | 25.93 |
AUC0–∞, area under the plasma concentration–time curve from time zero to infinity; AUC0–t, area under the plasma concentration–time curve from zero to the time of last quantifiable concentration; CL, total body clearance; Cmax, maximum plasma concentration; GeoCV, geometric coefficient of variation; GeoMean, geometric mean; max, maximum; min, minimum; PK, pharmacokinetic; PK‐PPS, pharmacokinetic‐per‐protocol set; t1/2, terminal elimination half‐life; tmax, time to reach Cmax; Vz, volume of distribution in terminal phase
GeoMeans and GeoCV(%)s were only calculated if less than one‐third of the parameters were not calculated
For Subject 00 111, only Cmax and tmax were reported. Other parameters were not calculated because sufficient data points were not available in the elimination phase of the PK profile
Figure 4.
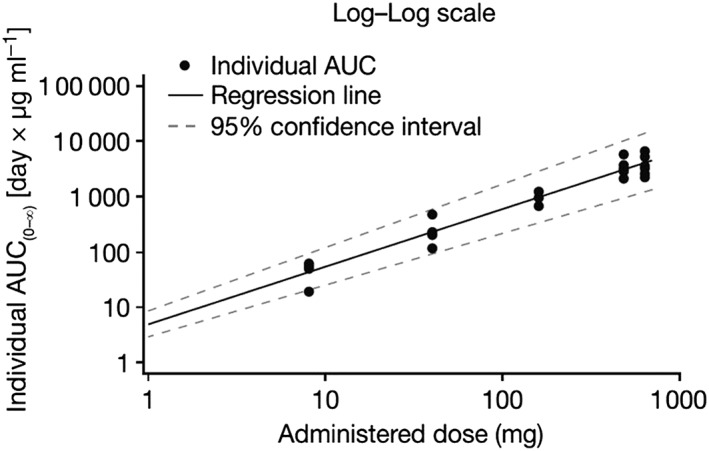
Dose proportionality (pharmacokinetic per‐protocol set): individual area under the curve (AUC) vs. bimekizumab dose. Note: at bimekizumab 480 mg, the AUC was reported for five subjects. The AUC was not calculated for one subject because insufficient data points were available in the elimination phase of the pharmacokinetic profile
Immunogenicity
Anti‐bimekizumab antibodies were detected and confirmed in one subject predose (160 mg treatment group), and five subjects at the SFU visit (Week 20; two subjects in the 8 mg group, two subjects in the 40 mg group, and one subject in the 160 mg group). There was no effect of anti‐bimekizumab antibodies on PK parameters (data not shown).
Effects on clinical features of plaque psoriasis
After administration, bimekizumab produced dose‐dependent improvements across the evaluated clinical features of plaque psoriasis, LSS, PASI and PGA (Figure 5). The 95% confidence intervals (CIs) for the placebo and bimekizumab 640 mg groups did not overlap at any time point postbaseline for all of these endpoints, indicating a difference between the two treatment groups.
Figure 5.
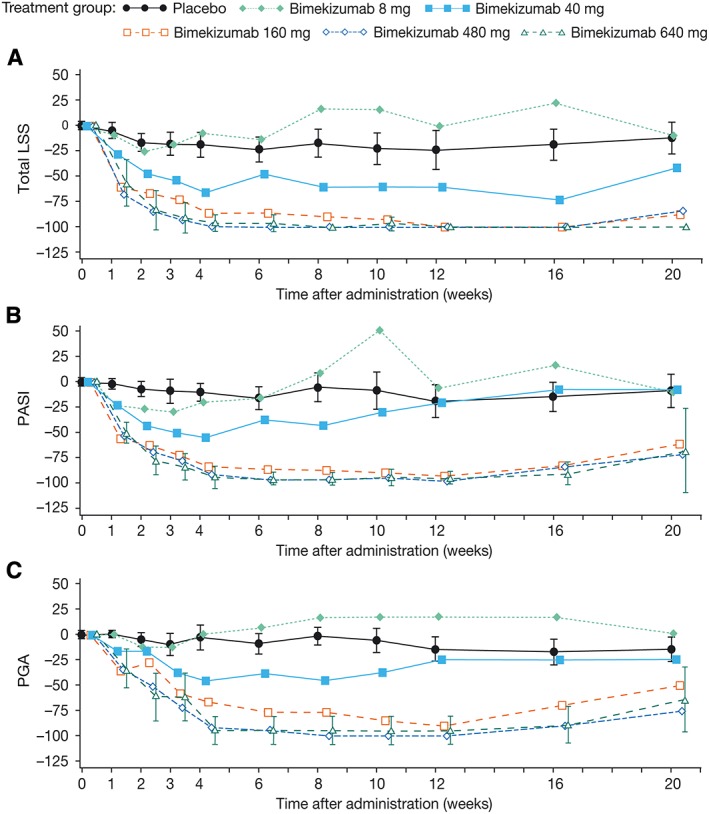
Mean percentage change from baseline in (A) LSS, (B) PASI and (C) PGA in the placebo and bimekizumab treatment groups (full analysis set). LSS, lesion severity score; PASI, Psoriasis Area and Severity Index; PGA, physician's global assessment. Note: 95% confidence intervals are included for the placebo and bimekizumab highest treatment groups only, to enable clarity of the plots. Note: figure was based on protocol‐specified time after administration time points for the x‐axis; however, the time points presented in this figure are offset for each treatment group to avoid overlap of symbols and confidence intervals
LSS
A fast onset of response was observed with a mean reduction of >80% from baseline LSS in the top two dose groups at study Week 2. At this time point, in the 40–640 mg treatment groups, it was estimated from the Bayesian analysis that the probability of achieving >0% improvement in LSS over placebo was >99%; in the top two treatment groups, probability of ≥60% improvement versus placebo was >90%. The magnitude of response was reflected by a maximal reduction in mean change from baseline in LSS of >90% in the 160 mg group. This was achieved by Week 8 and was maintained to Week 16. In the 480 mg group a maximal magnitude of response, 100%, was achieved as early as Week 4. The response was also maintained to Week 16. In the 640 mg group, the maximal reduction (100%) was achieved by Week 8 and was maintained between Weeks 12 and 20 (Figure 5A). In addition to the 640 mg dose group, no overlap in CIs between the other bimekizumab treatment groups (40–480 mg) at Week 2 vs. placebo was observed, indicating a difference between the two groups. Similar results were obtained for the area under the effect curve (Weeks 0–4, [AUEC0‐4w]) variable (data not shown).
PASI score
A fast onset of response was observed with a mean reduction of >65% from baseline PASI scores in the top two treatment groups at Week 2. Bayesian analysis revealed that the posterior probability of ≥60% improvement over placebo at this time point was >80%. For the bimekizumab 160 mg treatment group, the magnitude of response was reflected by a maximal reduction in the mean change from baseline in PASI score of >85% achieved by Week 6. This response was maintained to study Week 12. In the 480 mg and 640 mg treatment groups, a maximal magnitude of response of ≥94% was observed. This was achieved by Week 6 in the 480 mg group and Week 4 in the 640 mg group. In both groups the response was maintained to Week 12 (Figure 5B). Additionally, for bimekizumab 40–640 mg treatment groups at Week 2, the posterior probability of a >0% improvement in PASI score over placebo was >99%.
PGA
A mean reduction of >50% from baseline in PGA was observed at the highest doses of bimekizumab (480 mg and 640 mg) assessed. Maximal reductions in mean change from baseline in PGA scores of >75% were observed in the bimekizumab 160 mg group. This reduction was achieved by Week 6 and was maintained to Week 12. Maximal reductions in mean change from baseline in PGA scores were 100% and 94%, respectively for bimekizumab 480 mg and 640 mg treatment groups (Figure 5C). These reductions were achieved by Week 8 and were maintained to Week 12 in the 480 mg group, and achieved by Week 4 and maintained to Week 12 in the 640 mg group.
Discussion
This is the first time that safety, tolerability and clinical efficacy of a dual IL‐17A and IL‐17F inhibitor have been demonstrated in subjects with mild psoriasis.
Bimekizumab, administered by intravenous infusion in a single ascending dose, was tolerated across the dose range assessed (≤640 mg); no safety concerns were identified in this study. The most frequently reported TEAE was headache and only one serious AE (vomiting of moderate intensity) was reported, but it was not considered related to the study treatment. Furthermore, no subject experienced clinically meaningful changes in neutrophil count and no infusion‐site reactions occurred during the study. Importantly, the specific dual inhibition of IL‐17A and IL‐17F by bimekizumab is achieved without interacting with the IL‐17RA receptor, which also binds IL‐25. As such, by sparing IL‐25 in tissues, bimekizumab may reduce the potential for proinflammatory consequences that could result from interfering with the IL‐25 signalling pathway.
Bimekizumab demonstrated linear and dose‐proportional PK and a typical half‐life ranging from 17–26 days. The moderate‐to‐high interindividual variability for AUC in the bimekizumab treatment groups is likely to be due to the small number of subjects in the study. Estimated values for volume of distribution in terminal phase were similar to predicted total blood volume distribution, suggesting that distribution of bimekizumab was restricted to the extravascular compartment with limited distribution to peripheral tissues, as expected for a biological agent. The presence of anti‐bimekizumab antibodies in six subjects with mild psoriasis did not have a negative impact on PK or clinical outcomes. The single‐dose nature of this study should be considered when discussing development of anti‐bimekizumab antibodies and any potential effect on clinical outcomes; this topic may be further explored in multiple‐dose studies.
All indicators of disease activity measured at baseline were consistent with those expected in subjects with mild psoriasis. Bimekizumab‐treated subjects achieved a fast onset response, observed from study Week 2. Responses reached a maximal magnitude (75–100% reduction in disease activity scores) as early as Week 4. Responses were also maintained up to Weeks 12–20 for 160–640 mg groups, in all measures of disease activity compared with the placebo group, as demonstrated by reductions in LSS, PASI scores and PGA. Observed clinical efficacy appeared to increase with increasing doses of bimekizumab. These data are consistent with the hypothesis that inhibition of both IL‐17A and IL‐17F will lead to significant clinical outcomes in moderate to severe psoriasis and requires larger clinical studies in moderate to severe psoriasis patients to confirm this hypothesis and to explore further dosage regimes.
Conclusion
This is the first demonstration of safety, tolerability and clinical efficacy using a dual inhibitor of IL‐17A and IL‐17F in subjects with mild psoriasis. Subjects receiving bimekizumab achieved fast onset of clinically‐meaningful efficacy from Week 2. A maximal magnitude of response was observed as early as Week 4 that was maintained up to Weeks 12–20, in all clinical features of plaque psoriasis. These findings support the continued clinical development of bimekizumab for diseases mediated by both IL‐17A and IL‐17F, including psoriasis.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (http://www.icmje.org/coi_disclosure.pdf) (available on request from the corresponding author) and declare: S.G., E.H., B.H., F.S., G.P., P.V., O.H. and S.S. had support from UCB Pharma for the submitted work; S.G., E.H., B.H., F.S., G.P., P.V., O.H. and S.S. are/have been employees of UCB Pharma in the previous 3 years; BH, PV, OH and SS receive UCB Pharma stocks and/or stock options. JL declares no relevant conflicts of interest.
The study was funded by UCB Pharma. Additional statistical support was provided by Emma Jones of Veramed Ltd and Ros Walley of UCB. The authors acknowledge Dr Penny Ward's contribution to the study protocol while at UCB. The authors would like to acknowledge Ailsa Dermody, PhD, of iMed Comms, an Ashfield Company, part of UDG Healthcare plc for medical writing support that was funded by UCB Pharma.
Contributors
Statistical oversight and interpretation: F.S. Protocol development: G.P., E.H., P.V., F.S. Conducting the study: E.H., J.L. Medical monitoring: G.P. Analysis and interpretation of data: G.P., E.H., P.V., J.L. Manuscript development and review: S.G., E.H., B.H., F.S., G.P., P.V., O.H., J.L., S.S.
Glatt, S. , Helmer, E. , Haier, B. , Strimenopoulou, F. , Price, G. , Vajjah, P. , Harari, O. A. , Lambert, J. , and Shaw, S. (2017) First‐in‐human randomized study of bimekizumab, a humanized monoclonal antibody and selective dual inhibitor of IL‐17A and IL‐17F, in mild psoriasis. Br J Clin Pharmacol, 83: 991–1001. doi: 10.1111/bcp.13185.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 2016; 44: D1054–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, et al. The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 2015; 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 2015; 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Raho G, Koleva DM, Garattini L, Naldi L. The burden of moderate to severe psoriasis: an overview. Pharmacoeconomics 2012; 30: 1005–1013. [DOI] [PubMed] [Google Scholar]
- 5. Kivelevitch DN, Hebeler KR, Patel M, Menter A. Emerging topical treatments for psoriasis. Expert Opin Emerg Drugs 2013; 18: 523–532. [DOI] [PubMed] [Google Scholar]
- 6. Augustin M, Radtke MA. Quality of life in psoriasis patients. Expert Rev Pharmacoecon Outcomes Res 2014; 14: 559–568. [DOI] [PubMed] [Google Scholar]
- 7. National Psoriasis Foundation . Facts about psoriasis. Available at www.psoriasis.org/document.doc/?id=1492. (last accessed 21 July 2015).
- 8. Gooderham M, Posso‐De Los Rios CJ, Rubio‐Gomez GA, Papp K. Interleukin‐17 (IL‐17) inhibitors in the treatment of plaque psoriasis: a review. Skin Ther Lett 2015; 20: 1–5. [PubMed] [Google Scholar]
- 9. Krueger J, Bowcock A. Psoriasis pathophysiology: current concepts of pathogenesis. Ann Rheum Dis 2005; 64: ii30–ii36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. American Academy of Dermatology (AAD) . Clinical guidelines. Available at www.aad.org/education/clinical‐guidelines. (last accessed 21 July 2015).
- 11. Lynde CW, Poulin Y, Vender R, Bourcier M, Khalil S. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol 2014; 71: 141–150. [DOI] [PubMed] [Google Scholar]
- 12. Sarkar S, Justa S, Brucks M, Endres J, Fox DA, Zhou X, et al. Interleukin (IL)‐17A, F and AF in inflammation: a study in collagen‐induced arthritis and rheumatoid arthritis. Clin Exp Immunol 2014; 177: 652–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hot A, Miossec P. Effects of interleukin (IL)‐17A and IL‐17F in human rheumatoid arthritis synoviocytes. Ann Rheum Dis 2011; 70: 727–732. [DOI] [PubMed] [Google Scholar]
- 14. Johansen C, Usher PA, Kjellerup RB, Lundsgaard D, Iversen L, Kragballe K. Characterization of the interleukin‐17 isoforms and receptors in lesional psoriatic skin. Br J Dermatol 2009; 160: 319–324. [DOI] [PubMed] [Google Scholar]
- 15. van Baarsen LG, Lebre MC, van der Coelen D, Aarrass S, Tang MW, Ramwadhdoebe TH, et al. Heterogeneous expression pattern of interleukin 17A (IL‐17A), IL‐17F and their receptors in synovium of rheumatoid arthritis, psoriatic arthritis and osteoarthritis: possible explanation for nonresponse to anti‐IL‐17 therapy? Arthritis Res Ther 2014; 16: 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fujishima S, Watanabe H, Kawaguchi M, Suzuki T, Matsukura S, Homma T, et al. Involvement of IL‐17F via the induction of IL‐6 in psoriasis. Arch Dermatol Res 2010; 302: 499–505. [DOI] [PubMed] [Google Scholar]
- 17. Watanabe H, Kawaguchi M, Fujishima S, Ogura M, Matsukura S, Takeuchi H, et al. Functional characterization of IL‐17F as a selective neutrophil attractant in psoriasis. J Invest Dermatol 2009; 129: 650–656. [DOI] [PubMed] [Google Scholar]
- 18. European Medicines Agency E . Cosentyx®. Available at http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/003729/human_med_001832.jsp&mid=WC0b01ac058001d124. (last accessed 27 April 2016).
- 19. Food and Drug Administration (FDA) . Highlights of Prescribing Information Cosentyx®. 2016.
- 20. Food and Drug Administration (FDA) . FDA approves new psoriasis drug Taltz. Available at http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm491872.htm. (last accessed 27 April 2016).
- 21. GlobalData . Eli Lilly's Taltz Headed for Blockbuster Status with European Approval for Moderate‐to‐Severe Plaque Psoriasis. Available at https://healthcare.globaldata.com/resources/expert‐insights/pharmaceuticals/eli‐lillys‐taltz‐headed‐for‐blockbuster‐status‐with‐european‐approval‐for‐moderatetosevere‐plaque‐psoriasis. (last accessed 4 April 2016).
- 22. Bartlett HS, Million RP. Targeting the IL‐17‐T(H)17 pathway. Nat Rev Drug Discov 2015; 14: 11–12. [DOI] [PubMed] [Google Scholar]
- 23. PR Newswire . Valeant Announces FDA Acceptance of BLA Submission for Brodalumab in Moderate‐to‐Severe Plaque Psoriasis. Available at http://www.prnewswire.com/news‐releases/valeant‐announces‐fda‐acceptance‐of‐bla‐submission‐for‐brodalumab‐in‐moderate‐to‐severe‐plaque‐psoriasis‐300208845.html (last accessed 27 April 2016).
- 24. Caruso R, Stolfi C, Sarra M, Rizzo A, Fantini MC, Pallone F, et al. Inhibition of monocyte‐derived inflammatory cytokines by IL‐25 occurs via p38 Map kinase‐dependent induction of Socs‐3. Blood 2009; 113: 3512–3519. [DOI] [PubMed] [Google Scholar]
- 25. Hueber W, Patel DD, Dryja T, Wright AM, Koroleva I, Bruin G, et al. Effects of AIN457, a fully human antibody to interleukin‐17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med 2010; 2: 52ra72. [DOI] [PubMed] [Google Scholar]
- 26. Leonardi C, Matheson R, Zachariae C, Cameron G, Li L, Edson‐Heredia E, et al. Anti‐interleukin‐17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med 2012; 366: 1190–1199. [DOI] [PubMed] [Google Scholar]
- 27. Papp KA, Langley RG, Sigurgeirsson B, Abe M, Baker DR, Konno P, et al. Efficacy and safety of secukinumab in the treatment of moderate‐to‐severe plaque psoriasis: a randomized, double‐blind, placebo‐controlled phase II dose‐ranging study. Br J Dermatol 2013; 168: 412–421. [DOI] [PubMed] [Google Scholar]
- 28. Papp KA, Leonardi C, Menter A, Ortonne JP, Krueger JG, Kricorian G, et al. Brodalumab, an anti‐interleukin‐17‐receptor antibody for psoriasis. N Engl J Med 2012; 366: 1181–1189. [DOI] [PubMed] [Google Scholar]
- 29. Lonnberg AS, Zachariae C, Skov L. Targeting of interleukin‐17 in the treatment of psoriasis. Clin Cosmet Investig Dermatol 2014; 7: 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jin W, Dong C. IL‐17 cytokines in immunity and inflammation. Emerg Microbes Infect 2013; 2: e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Feldman SR, Krueger GG. Psoriasis assessment tools in clinical trials. Ann Rheum Dis 2005; 64 (Suppl 2): ii65–68 ; discussion ii69‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]