

Abstract
Aim
The aim was to investigate the ability of a battery of pain models to detect analgesic properties of commonly used analgesics in healthy subjects.
Methods
The battery consisted of tests eliciting electrical, mechanical and thermal (contact heat and cold pressor)‐pain and included a UVB model, the thermal grill illusion and a paradigm of conditioned pain modulation. Subjects were administered fentanyl 3 μg kg–1, phenytoin 300 mg, (S)‐ketamine 10 mg and placebo (part I), or imipramine 100 mg, pregabalin 300 mg, ibuprofen 600 mg and placebo (part II). Pain measurements were performed at baseline and up to 10 h post‐dose. Endpoints were analysed using a mixed model analysis of variance.
Results
Sixteen subjects (8 female) completed each part. The pain tolerance threshold (PTT) for electrical stimulation was increased (all P < 0.05) compared to placebo for (S)‐ketamine (+10.1%), phenytoin (+8.5%) and pregabalin (+10.8%). The PTT for mechanical pain was increased by pregabalin (+14.1%). The cold pressor PTT was increased by fentanyl (+17.1%) and pregabalin (+46.4%). Normal skin heat pain detection threshold was increased by (S)‐ketamine (+3.3%), fentanyl (+2.8%) and pregabalin (+4.1%). UVB treated skin pain detection threshold was increased by fentanyl (+2.6%) and ibuprofen (+4.0%). No differences in conditioned pain modulation were observed.
Conclusion
This study shows that these pain models are able to detect changes in pain thresholds after administration of different classes of analgesics in healthy subjects. The analgesic compounds all showed a unique profile in their effects on the pain tasks administered.
Keywords: analgesics, human pain models, pharmacodynamics, pharmacokinetics
What is Already Known about this Subject
Human pain models can assist to bridge preclinical findings and those in the clinical situation. However, one human pain model cannot be used exclusively to screen the pharmacological mechanism of a compound.
What this Study Adds
This battery of pain models is able to detect changes in pain detection and pain tolerance thresholds after administration of different classes of analgesic compounds in healthy male and female subjects. Compounds with different mechanisms of action demonstrated a distinct response pattern on the different pain models.
This battery of pain models can be used to benchmark analgesic properties of new drugs against established analgesics in early phase clinical studies in healthy subjects.
Tables of Links
| TARGETS | |
|---|---|
| GPCRs 2 | Ligand‐gated ion channels 4 |
| Opioid receptor http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=249 | NMDA receptor |
| Enzymes 3 | Voltage‐gated ion channels 5 |
| Cyclooxygenase | Voltage‐gated calcium channel (α2d subunit) |
| Fatty acid amide hydrolase | Voltage‐gated sodium channels |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2, 3, 4, 5.
Introduction
Pharmaceutical science continues to search for suitable biomarkers that can assist in predicting the therapeutic potential of analgesic medication and, therefore, its efficacy in the target population. Data intensive, early‐phase studies provide a valuable opportunity that can offer this translational information 6. A series of nociceptive pain tests used early in drug development could bridge preclinical findings and those in the clinic to provide valuable information about the mechanism of action of a new drug and to benchmark new drugs to existing analgesics. The need to use a comprehensive battery of pain models is highlighted by studies in which only a single pain model, thought to relate to the clinical situation, demonstrates lack of efficacy 7, 8. A single evoked model cannot replicate the complex nature of clinical pain. Therefore, one evoked pain model cannot be used exclusively to screen the pharmacological mechanism of action of a new compound, for which this mechanism has not been demonstrated earlier. The aim of this study was to pharmacologically validate an integrated range of human pain models that can be used as a combined screening tool for early stage clinical drug development.
Each pain model in this battery has been used before 9, 10, 11, 12. However, the integrated execution of these tests has not yet been investigated, and it is mostly unclear how well‐known and frequently used analgesic compounds influence the pain tests when used in this integrated manner. Data obtained from early phase clinical studies may be used for the determination or confirmation of a drug's mechanism of action. Furthermore, results obtained from pain models could be useful for the prediction of the efficacy of the drug in future clinical populations or potential disease states 13. This battery of tests should be able to help establish whether a drug is acting centrally or peripherally, whether it is more suitable for a particular modality of pain (nociceptive, neuropathic or inflammatory), and which other effects contribute to its mode of action (sedation, tolerance etc.). Nociceptive tests, when used in combination with pharmacokinetic (PK) parameters, can be used to provide information regarding future dose selection of new drugs. Particularly if used in combination with pharmacokinetic–pharmacodynamic (PK/PD) modelling and simulation techniques, the establishment of a threshold of pharmacological activity may be determined and used for dose prediction 14.
The models in this study were chosen to represent a broad range of pain modalities and nociceptor function, combined with the possibility to perform these pain tests in a standardised setting in clinical studies. Regarding the choice of compounds, a selection was made of distinctly different, relevant, targets of analgesia. The analgesic mechanism of action of these compounds was compared using the existing literature 9, 10, 11, 15, 16, 17, 18. Specific compounds, representative of a range of mechanistic classes, were chosen if they showed analgesic efficacy in previous pain models in humans or if their efficacy in pain models was expected but yet unknown. It was hypothesised that the battery of pain models would show distinct response patterns for the different analgesic classes.
Methods
Subject and study design
The study was approved by the Medical Ethics Committee of the Leiden University Medical Center (Leiden, The Netherlands). The study was conducted according to the Dutch Act on Medical Research Involving Human Subjects (WMO) and in compliance with Good Clinical Practice (ICH‐GCP) and the Declaration of Helsinki.
Healthy male and female subjects between 18 and 45 years with a body mass index of 18–30 kg m–2 were enrolled. All subjects gave written informed consent. The subjects underwent a full medical screening, including taking medical history, a physical examination, blood chemistry and haematology, urinalysis, electrocardiogram, and assessment of the minimal erythema dose (MED) for UVB light to assess eligibility. Subjects with a clinically significant known medical condition, in particular any existing condition that would affect sensitivity to cold or pain were excluded. Subjects with Fitzpatrick skin type V or VI, wide‐spread acne, tattoos or scarring on the back were excluded due to the inability to assess MED accurately. Also, subjects who were regular users of any illicit drugs, had a history of drug abuse or a positive drug screen at screening were excluded. Smoking and the use of xanthine‐containing products was not allowed during dosing days. Alcohol was not allowed at least 24 h before each scheduled visit or during the stay in the research unit. Except for contraception, subjects were not allowed to use prescription medications within 7 days and over‐the‐counter analgesics within 3 days of nociceptive assessments. Female subjects were required to have an intrauterine device, a contraceptive implant or were willing to continuously use oral contraceptives (i.e. skip their menstruation) during the study period, to prevent influences of menstrual phase 19.
This was a two‐part, randomised, double‐blind, placebo‐controlled, four‐way crossover, single‐dose study. The total number of planned subjects was 16 in each part. In part I, subjects received the study drug or placebo intravenously over a 30‐min time period in the antecubital vein. Treatment consisted of fentanyl 3 μg kg–1 (Hameln Pharmaceuticals GmbH, Hameln, Germany), phenytoin 300 mg (Diphantoïne; Apotex Europe Ltd, Leiden, The Netherlands), (S)‐ketamine 10 mg (Ketanest‐S 5; Eurocept BV, Ankeveen, The Netherlands) and sodium chloride 0.9% (placebo). In part II, subjects received the over encapsulated study drug or placebo orally with 150 ml of still water. Treatment consisted of imipramine hydrochloride 100 mg (Centrafarm B.V.; Etten‐Leur, The Netherlands), pregabalin 300 mg (Lyrica, Pfizer Limited, Kent, UK), ibuprofen 600 mg (Nurofen oval tablet, Reckitt Benckiser Healthcare B.V., Hoofddorp, The Netherlands) and placebo tablets (lactose monohydrate with 1% magnesium stearate). Subjects participated in either part I or part II in which they received all four treatments. The study treatments were randomly allocated based on a 4 × 4 William's square. The randomisation code was generated by a study‐independent statistician using SAS version 9.1.3 (SAS Institute Inc., Cary, NC, USA).
Each treatment period consisted of two study visits to the clinical research unit. During the first visit, UVB erythema was induced. On the morning of the next day, subjects received the study treatment after which the PD and PK assessments were performed (Figure 1). Subjects were discharged at the end of the study day. There was a 1‐week washout period between treatment periods.
Figure 1.
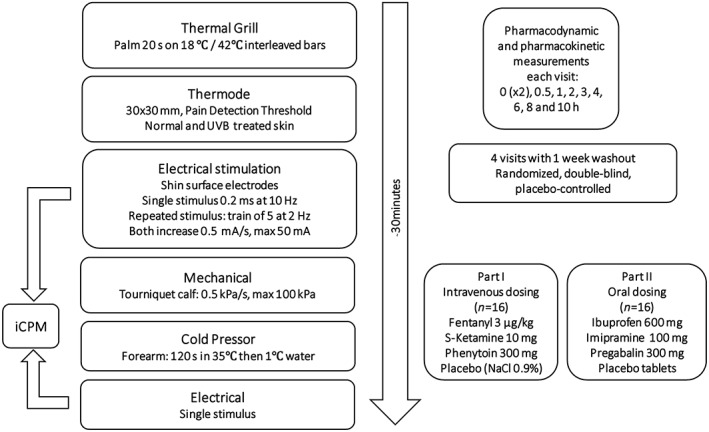
Overview of study design
PD assessments
Nociceptive (pain) detection and tolerance thresholds were measured using a battery of human pain models. The battery is an integrated range of tests for measuring different modalities of nociception and takes approximately 30 min to complete (Figure 1) 20. It aims to assess as objectively as possible the levels of pain induced by several noxious mechanisms in human subjects. A training session was included as part of the screening examination to reduce learning effects during the study. All tests have previously been shown to be sensitive to the effects of analgesics in healthy adults. All measurements were performed in a quiet room with ambient illumination. Per session, there was only one subject in the same room.
For the electrical stimulation tests, the pressure stimulation test and the cold pressor test, pain intensity was measured continuously (beginning from when the first stimulus was applied until the predetermined end of the test) using an electronic visual analogue scale (eVAS) scale ranging from 0 (no pain) to 100 (most intense pain tolerable). Equipment was programmed to cease giving stimuli if pain intensity reaches the maximum possible score. For each test the pain detection threshold (PDT), pain tolerance threshold (PTT) and area under the curve (AUC) were determined. The AUC was calculated as the surface under the pain intensity–stimulation (–time for cold pressor) curve.
Thermal grill
The thermal grill consisted of a set of eight juxtaposed bars of cold and warm innocuous temperatures (18°C and 42°C) on which the subject placed their dominant hand for 20 s. During this time, the subject rated unpleasantness, pain sensation and thermal sensation using the eVAS‐slider.
Thermode testing and UVB model
The method of UVB irradiation was based on methods previously described 21. UVB irradiation (TL01 [narrow‐band], Phillips, Amsterdam, The Netherlands) was applied at the screening visit in ascending doses to determine the individual UVB dose that produced the first clearly discernible erythema. The three‐fold individual MED of UVB was applied 24 h prior to dosing to the subject's back to produce local cutaneous inflammation, thereby inducing a homogeneous area of skin erythema and hyperalgesia. The area of skin irradiated was 3 x 3 cm. Subsequently, a 3 × 3 cm thermode (TSA‐II; Medoc Ltd., St Ramat Yishai, Israel) was used to measure pain detection thresholds (initially 34°C, ramp 0.5°C/s, average of three stimuli) on the normal skin contralateral to the site of UVB irradiation and on the UVB irradiated skin (cut‐off 50°C).
Electrical stimulation test
For cutaneous electrical pain, Ag–AgCl electrodes (3M Red‐Dot) were placed on cleaned, scrubbed, and if required, shaved skin, 10 cm distal from the patella overlying the tibia. Electrical resistance between electrodes was to be <2 kΩ. The electrical stimulus was delivered as two different paradigms by a computer‐controlled constant current stimulator (DS5; Digitimer, Cambridge, UK).
For the single stimulus, adapted from methods previously described 22, 23 (10 Hz tetanic pulse with a duration of 0.2 ms), current intensity increased from 0 mA in steps of 0.5 mA s–1 (cut‐off 50 mA).
For the repeated stimulus, adapted from methods previously described 24, each single stimulus (train of five 1‐ms square wave pulses repeated at 200 Hz) was repeated five times with a frequency of 2 Hz at the same current intensity with a random interval of 3–8 s between the repetitions. Current intensity increased from 0 mA in steps of 0.5 mA s–1 (cut‐off 50 mA). Pain detection threshold was taken as the value (mA) whereby a subject indicated either: all five stimuli were painful, or the train of five stimuli started feeling nonpainful but ended feeling painful (VAS > 0). The pain intensity for each stimulation was measured using the eVAS slider, until pain tolerance threshold or a maximum of 50 mA was reached.
Pressure stimulation test
The method of mechanical pressure pain induction was based on methods previously described, and was shown to primarily assess nociception generated from the muscle with minimal contribution by cutaneous nociceptors 25, 26. Briefly, an 11‐cm wide tourniquet cuff (VBM Medizintechnik GmbH, Sulz, Germany) was placed over the gastrocnemius muscle with a constant pressure rate increase of 0.5 kPa s–1. The pneumatic pressure was increased until the subject indicated maximum pain tolerance using the eVAS slider, or a maximum pressure of 100 kPa was achieved, at which point the device released pressure to the cuff.
Cold pressor test
The method of cold pressor pain was based on the methods previously described 27, 28 and is the most commonly used test to induce conditioned pain modulation (CPM, previously known as ‘diffuse noxious inhibitory control’) 29. Subjects placed their nondominant hand into a water bath at 35 ± 0.5°C for 2 min. At 1 min 45 s, a blood pressure cuff on the upper‐arm was inflated to 20 mmHg below resting diastolic pressure. At 2 min the subject then moved that hand from the warm water bath, directly into a similar sized water bath at 1.0 ± 0.5°C. The subjects were instructed to indicate when pain detection threshold was reached (first change in sensation from cold nonpainful to painful) as well as the pain intensity, by moving the eVAS slider. When pain tolerance or a time limit (120 s) was reached, subjects were instructed to remove their hand from the water, at which point the blood pressure cuff was deflated.
CPM
CPM is the activation of the pain‐modulatory mechanism, as part of the descending endogenous analgesia system 29. The degree of CPM was assessed by comparing the electrical pain thresholds for the single stimulus paradigm before and within 5 min after the cold pressor test.
Measurements of drug concentrations in plasma
Samples for determination of compounds in plasma were obtained at baseline, 0.5, 1, 2, 3, 4, 6, 8 and 10 h after the start of administration. Samples were collected in 6 ml K2EDTA tubes. Plasma was separated within 30 min of blood collection by centrifugation at 2000 g for 10 min. All samples were stored in an upright position at – 40°C. Drug concentrations in plasma were determined using Liquid Chromatography–Mass Spectrometry (LC–MS/MS). The analytical range was 0.200–50.0 ng ml–1 for fentanyl, 1.00–200 ng ml–1 for (S)‐ketamine, 0.500–100 ng ml–1 for norketamine, 20.0–10 000 ng ml–1 for phenytoin, 0.5–100 ng ml–1 for imipramine and desipramine, 20.0–20 000 ng ml–1 for pregabalin and 100–100 000 ng ml–1 for ibuprofen. Quality control for the analytical performance of the assays for all compounds showed acceptable performance (Table S3). Standard curves were linear for the ranges tested (r > 0.99 for all compounds). Control runs were performed in low, medium and high concentrations of each compound. Coefficients of variation varied from 1.5% to 7.9%.
Statistical analysis
The sample size calculation was based on previous studies performed in our centre. The detectable effect sizes using a paired t‐test with a 0.050 two‐sided significance level and 16 subjects were as follows (standard deviations [SDs] are rounded): electrical stimulation repeated stimulus AUC 225 (6%; assuming an SD of 300), electrical stimulation single stimulus AUC 450 (16%; assuming an SD of 600), pressure stimulation AUC 525 (9%; assuming an SD of 700), cold pressor area above the curve 337 (17%; assuming an SD of 450).
PK analysis was performed using noncompartmental analysis. The peak concentration and the time to the peak concentration were recorded as observed. In addition, the terminal half‐life, the area under the plasma concentration–time curve (AUC) from time zero to the time of the last sample (AUC0–last) and from time zero to infinity (AUC0–inf), the volume of distribution (Vd), and the clearance were determined for all compounds. AUC's were calculated using the linear trapezoidal method. Calculations were performed using R v2.12.0 (R Foundation for Statistical Computing, Vienna, Austria).
PDT and PTT variables follow a log‐normal distribution and were therefore log‐transformed before analysis. Transformed parameters were back‐transformed after analysis.
To establish whether significant treatment effects could be detected on the PD outcome variables, variables were analysed with a mixed model analysis of variance with treatment, time, sex, treatment by time and treatment by sex as fixed factors and subject, subject by treatment and subject by time as random factors and the average baseline measurement as covariate. The Kenward–Roger approximation was used to estimate denominator degrees of freedom and model parameters were estimated using the restricted maximum likelihood method. The general treatment effect and specific contrasts were reported with the estimated difference and the 95% confidence interval, the least squares mean estimates and the P‐value. Graphs of the least squares means estimates over time by treatment were presented with 95% confidence intervals as error bars. The contrasts for the relevant time periods based on the PK profiles of the compounds (0–1 h for (S)‐ketamine, 0–5 h for fentanyl ibuprofen and pregabalin, 0–10 h for phenytoin and imipramine) are presented. All calculations of the pharmacodynamic parameters were performed using SAS for Windows version 9.1.3 (SAS Institute Inc., Cary, NC, USA). The main SAS procedure that was used in the analysis was PROC MIXED. No adjustments for multiple comparisons were employed.
Results
A total of 39 subjects, of whom 18 were female, were randomised by treatment (Figure 2); subjects had a mean age of 22.5 ± 2.8 years and had a mean body mass index of 21.8 ± 1.7 kg m–2. In part I, where we studied the effects of intravenous analgesics, 18 subjects received placebo treatment, 17 fentanyl, 17 (S)‐ketamine and 20 phenytoin. In one subject the dose administration was prematurely stopped due to an adverse event (syncope) during phenytoin administration. In the oral part II, 16 subjects received placebo, 17 ibuprofen, 17 imipramine and 16 pregabalin. In both parts, 16 subjects completed all four study periods.
Figure 2.
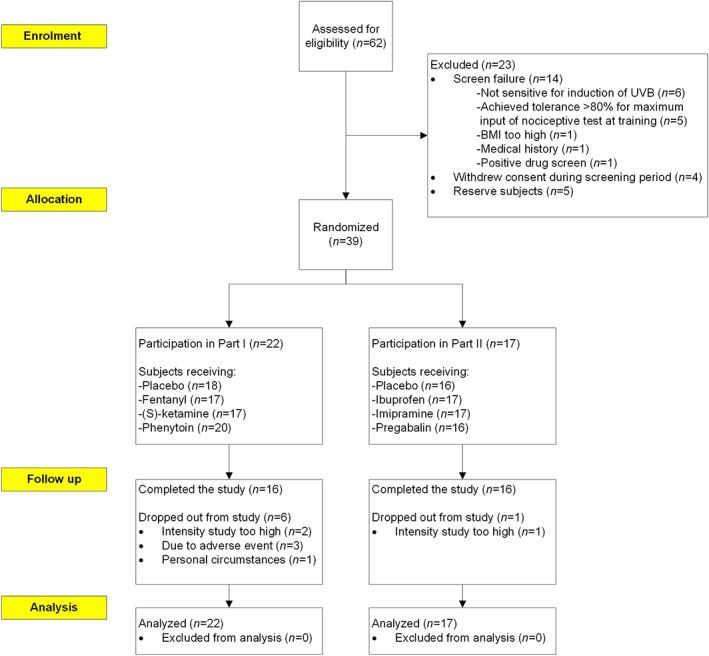
Disposition of subjects
An overview of the pharmacodynamic output variables is provided in Table 1 (part I), Table 2 (part II), Table S1, Table S2, Figure 3 and Figure 4. Differences compared to placebo for the cold pressor test were observed after administration of fentanyl (pain tolerance threshold, PTT; estimate of difference [95% confidence interval]: 17.1% [2.3%–33.9%]) and pregabalin (pain detection threshold, PDT and PTT; 36.8% [5.9%–76.8%] / 46.4% [27.1%–68.6%]). Electrical stimulation single stimulus parameters changed after administration of (S)‐ketamine (PTT; 10.1% [0.2%–20.9%]), phenytoin (PDT and PTT; 31.5% [10.3%–56.8%] / 8.5% [1.4%–16.1%]), and pregabalin (PTT; 10.8% [2.4%–19.9%]). The PTT for pressure pain was only increased by pregabalin (14.1% [4.3%–24.9%]). The normal skin heat PDT increased after administration of (S)‐ketamine (3.3% [1.1%–5.6%]), fentanyl (2.8% [1.1%–4.5%]) and pregabalin (4.1% [1.3%–7.0%]). UVB‐treated skin PDT increased after administration of fentanyl (2.6% [1.2%–4.1%]) and ibuprofen (4.0% [1.8%–6.3%]). Thermal grill maximum unpleasantness was not influenced by any of the compounds administered. After administration of ibuprofen, an increase was observed for the thermal grill pain intensity (1.25 [0.25–2.25]). Inhibitory conditioned pain modulation was influenced by administration of imipramine and pregabalin. These compounds also caused an increase in the difference between pre‐ and postcold pressor electrical stimulation PDT (0.88 [0.06–1.70] / 1.95 [0.84–3.06]). The effect sizes for the compounds during the relevant analysis period compared to placebo for the different pain models are shown in Figure 5.
Table 1.
Least squares means for pharmacodynamic outcome measures and estimates of difference, 95% confidence intervals and P‐values for main contrasts for part I
| LS Means | Contrasts | ||||||
|---|---|---|---|---|---|---|---|
| Parameter | Placebo (0–1 h/ 0–5 h/ 0–10 h) | Fentanyl 3 μg kg–1 (0–5 h) | (S)‐ketamine 10 mg (0–1 h) | Phenytoin 300 mg (0–10 h) | Fentanyl 3 μg kg–1 Placebo up to 5 h | (S)‐ketamine 10 mg Placebo up to 1 h | Phenytoin 300 mg Placebo up to 10 h |
| Cold PDT (s) | 4.3 / 4.5 / 4.4 | 4.4 | 6.1 | 5.2 | –3.6% (–34.2%, 41.0%) P = 0.8435 | 39.7% (–14.1%, 127.1%) P = 0.1758 | 18.8% (–16.0%, 67.9%) P = 0.3143 |
| Cold PTT (s) | 22.0 / 21.6 / 20.7 | 25.3 | 24.8 | 21.4 | 17.1% (2.3%, 33.9%) P = 0.0230 | 12.7% (–5.5%, 34.4%) P = 0.1820 | 3.6% (–8.2%, 17.0%) P = 0.5562 |
| Electrical Repeat PDT (mA) | 2.7 / 2.7 / 2.7 | 2.8 | 2.6 | 2.9 | 5.0% (–15.4%, 30.2%) P = 0.6523 | –1.9% (–27.0%, 31.7%) P = 0.8957 | 4.3% (–14.4%, 27.2%) P = 0.6692 |
| Electrical Repeat PTT (mA) | 10.3 / 10.4 / 10.5 | 11.2 | 11.6 | 10.8 | 8.3% (–1.2%, 18.8%) P = 0.0871 | 12.7% (–0.2%, 27.3%) P = 0.0533 | 3.7% (–4.8%, 12.9%) P = 0.3934 |
| Electrical Single PDT (mA) | 6.7 / 7.2 / 7.3 | 7.9 | 8.0 | 9.6 | 9.8% (–9.4%, 33.0%) P = 0.3324 | 18.3% (–11.4%, 57.9%) P = 0.2525 | 31.5% (10.3%, 56.8%) P = 0.0032 |
| Electrical Single PTT (mA) | 21.4 / 22.1 / 22.3 | 23.5 | 23.6 | 24.2 | 6.7% (–0.7%, 14.7%) P = 0.0770 | 10.1% (0.2%, 20.9%) P = 0.0447 | 8.5% (1.4%, 16.1%) P = 0.0193 |
| CPM PDT (mA) | 0.50 / 0.37 / 0.68 | 1.18 | –0.11 | 0.37 | 0.811 (–0.319, 1.941) P = 0.1576 | –0.611 (–2.913, 1.692) P = 0.6022 | –0.305 (–1.196, 0.585) P = 0.4925 |
| CPM PTT (mA) | 0.84 / 0.69 / 0.74 | 0.62 | 0.62 | 1.13 | –0.074 (–0.938, 0.789) P = 0.8648 | –0.215 (–1.880, 1.449) P = 0.7994 | 0.394 (–0.306, 1.095) P = 0.2622 |
| Pressure PDT (kPa) | 15.0 / 12.8 / 12.7 | 13.0 | 14.9 | 14.9 | 1.6% (–15.2%, 21.6%) P = 0.8636 | –0.8% (–24.2%, 29.7%) P = 0.9527 | 16.9% (–0.5%, 37.4%) P = 0.0572 |
| Pressure PTT (kPa) | 42.2 / 42.7 / 41.8 | 45.8 | 45.5 | 42.3 | 7.2% (–0.2%, 15.1%) P = 0.0571 | 7.9% (–3.4%, 20.5%) P = 0.1765 | 1.1% (–5.0%, 7.6%) P = 0.7291 |
| Normal skin‐heat PDT (°C) | 43.88 / 44.18 / 44.13 | 45.41 | 45.33 | 44.61 | 2.8% (1.1%, 4.5%) P = 0.0018 | 3.3% (1.1%, 5.6%) P = 0.0034 | 1.1% (–0.4%, 2.6%) P = 0.1508 |
| UVB skin‐heat PDT (°C) | 39.11 / 38.94 / 38.93 | 39.95 | 39.44 | 38.93 | 2.6% (1.2%, 4.1%) P = 0.0006 | 0.8% (–1.1%, 2.9%) P = 0.4075 | –0.0% (–1.2%, 1.2%) P = 0.9813 |
| Grill Unpleasantness VAS Max (mm) | 9.0 / 8.5 / 8.2 | 6.2 | 7.8 | 9.3 | –2.33 (–5.06, 0.40) P = 0.0923 | –1.22 (–4.92, 2.47) P = 0.5140 | 1.13 (–1.40, 3.65) P = 0.3709 |
| Grill Pain intensity VAS (mm) | 5.6 / 4.4 / 4.5 | 4.4 | 7.3 | 5.6 | –0.03 (–1.51, 1.44) P = 0.9661 | 1.77 (–0.85, 4.38) P = 0.1844 | 1.11 (–0.15, 2.36) P = 0.0824 |
CPM, conditioned pain modulation; LS indicates least squares; PDT, pain detection threshold; PTT, pain tolerance threshold; VAS, visual analogue scale
Table 2.
Least squares means for pharmacodynamic outcome measures and estimates of difference, 95% confidence intervals and P‐values for main contrasts for part II
| LS means | Contrasts | ||||||
|---|---|---|---|---|---|---|---|
| Parameter | Placebo (0–5 h / 0–10 h) | Ibuprofen 600 mg (0–5 h) | Imipramine 100 mg (0–10 h) | Pregabalin 300 mg (0–5 h) | Ibuprofen 600 mg Placebo up to 5 h | Imipramine 100 mg Placebo up to 10 h | Pregabalin 300 mg Placebo up to 5 h |
| Cold PDT (s) | 4.1 / 3.7 | 3.8 | 3.6 | 5.6 | –8.3% (–29.5%, 19.3%) P = 0.5109 | –4.7% (–24.3%, 19.9%) P = 0.6698 | 36.8% (5.9%, 76.8%) P = 0.0174 |
| Cold PTT (s) | 17.2 / 16.0 | 17.7 | 18.1 | 25.1 | 2.9% (–10.7%, 18.7%) P = 0.6850 | 12.6% (–1.3%, 28.6%) P = 0.0769 | 46.4% (27.1%, 68.6%) P = <.0001 |
| Electrical Repeat PDT (mA) | 2.1 / 2.0 | 2.3 | 2.3 | 2.0 | 13.1% (–20.0%, 60.1%) P = 0.4779 | 16.6% (–16.0%, 61.8%) P = 0.3497 | –0.6% (–30.5%, 42.0%) P = 0.9719 |
| Electrical Repeat PTT (mA) | 8.9 / 8.8 | 9.9 | 9.3 | 10.1 | 11.3% (–1.1%, 25.4%) P = 0.0749 | 5.1% (–6.2%, 17.7%) P = 0.3808 | 13.1% (–0.3%, 28.4%) P = 0.0561 |
| Electrical Single PDT (mA) | 7.5 / 7.3 | 7.6 | 8.0 | 7.3 | 0.6% (–19.4%, 25.6%) P = 0.9565 | 9.4% (–10.4%, 33.5%) P = 0.3673 | ‐2.7% (–22.3%, 22.0%) P = 0.8112 |
| Electrical Single PTT (mA) | 19.9 / 19.7 | 20.1 | 20.5 | 22.0 | 1.2% (–6.2%, 9.2%) P = 0.7525 | 3.9% (–3.3%, 11.6%) P = 0.2887 | 10.8% (2.4%, 19.9%) P = 0.0121 |
| CPM PDT (mA) | ‐0.19 / 0.31 | 0.70 | 1.19 | 1.76 | 0.895 (–0.213, 2.003) P = 0.1122 | 0.879 (0.060, 1.699) P = 0.0364 | 1.950 (0.840, 3.061) P = 0.0007 |
| CPM PTT (mA) | 0.56 / 0.76 | 1.08 | 0.88 | 1.21 | 0.519 (–0.160, 1.198) P = 0.1319 | 0.117 (–0.452, 0.685) P = 0.6795 | 0.644 (–0.036, 1.323) P = 0.0630 |
| Pressure PDT (kPa) | 14.5 / 14.2 | 14.4 | 13.4 | 13.9 | –0.7% (–22.2%, 26.7%) P = 0.9528 | ‐5.6% (–24.8%, 18.5%) P = 0.6062 | ‐4.6% (–25.3%, 21.9%) P = 0.6998 |
| Pressure PTT (kPa) | 41.1 / 41.7 | 44.0 | 44.3 | 47.6 | 5.3% (–3.9%, 15.4%) P = 0.2576 | 7.7% (–0.9%, 17.0%) P = 0.0773 | 14.1% (4.3%, 24.9%) P = 0.0052 |
| Normal skin‐heat PDT (°C) | 43.32 / 43.25 | 44.08 | 43.62 | 45.09 | 1.7% (–1.0%, 4.5%) P = 0.2080 | 0.9% (–1.7%, 3.5%) P = 0.5108 | 4.1% (1.3%, 7.0%) P = 0.0049 |
| UVB skin‐heat PDT (°C) | 38.63 / 38.49 | 40.17 | 38.92 | 39.08 | 4.0% (1.8%, 6.3%) P = 0.0006 | 1.1% (–0.9%, 3.1%) P = 0.2589 | 1.2% (–0.9%, 3.3%) P = 0.2671 |
| Grill Unpleasantness VAS Max (mm) | 1.4 / 1.7 | 3.0 | 2.6 | 2.3 | 1.57 (–0.41, 3.55) P = 0.1177 | 0.90 (–0.97, 2.78) P = 0.3357 | 0.89 (–1.09, 2.86) P = 0.3698 |
| Grill Pain intensity VAS (mm) | 1.0 / 1.2 | 2.3 | 1.0 | 1.7 | 1.25 (0.25, 2.25) P = 0.0151 | ‐0.16 (–1.10, 0.77) P = 0.7250 | 0.73 (–0.25, 1.71) P = 0.1425 |
CPM, conditioned pain modulation; LS, least squares; PDT, pain detection threshold; PTT, pain tolerance threshold; VAS, visual analogue scale
Figure 3.
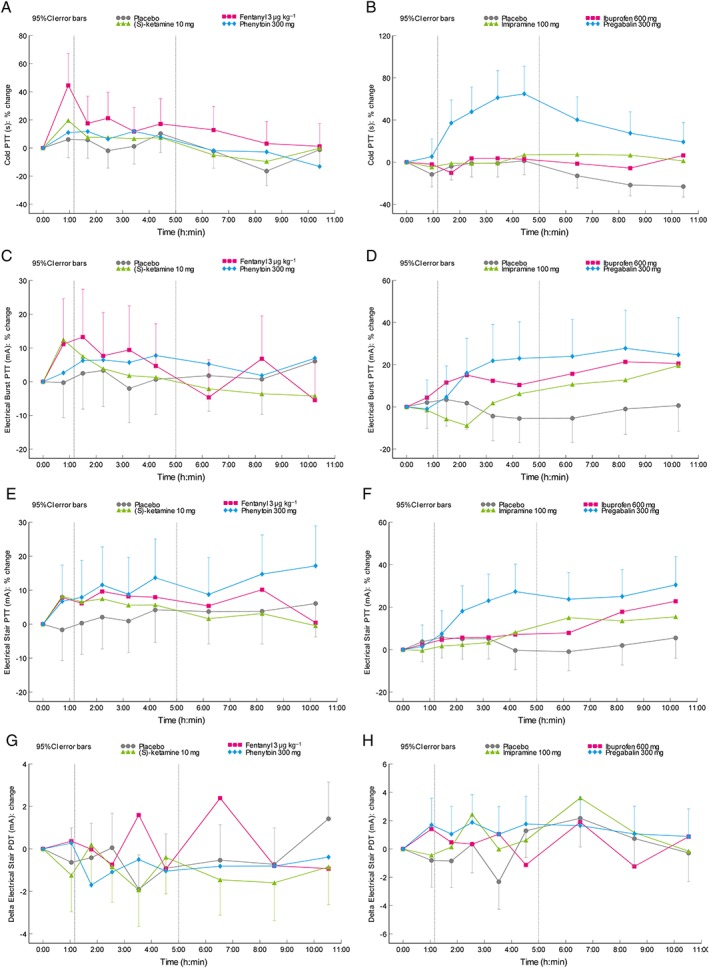
Time course of the mean change from baseline profile in least squares means for (A,B) the pain tolerance threshold for cold pressor, electrical stimulation (C,D: repeated stimulus) and (E,F: single stimulus), and the (G,H) electrical stimulation (single stimulus) delta pain detection threshold after administration of the different compounds in (A,C,E,G) part I and (B,D,F,H) part II
Figure 4.
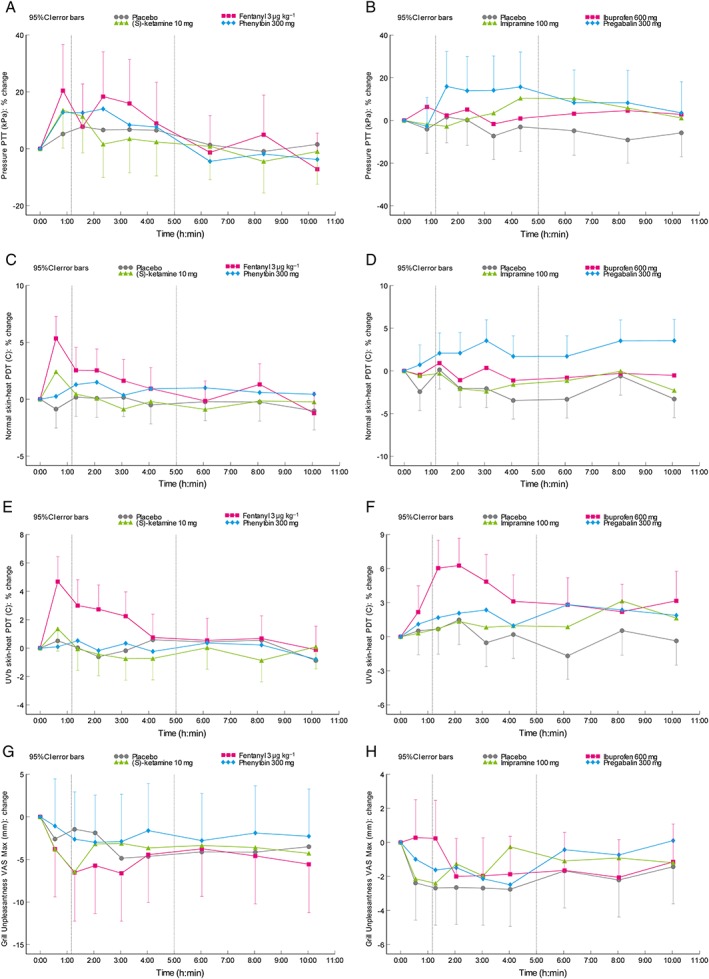
Time course of the mean change from baseline profile in least squares means for the pain tolerance threshold for (A,B) pressure stimulation, (C,D) the heat pain detection threshold for thermal testing on normal skin, and (E,F) UVB‐irradiated skin and (G,H) the thermal grill maximum unpleasantness VAS after administration of the different compounds in (A,C,E,G) part I and (B,D,F,H) part II
Figure 5.
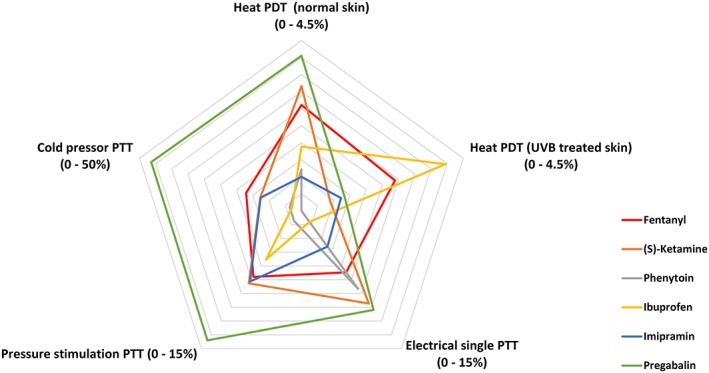
Radar chart of effect sizes of the compounds used. Effect sizes are given as the contrast between the different compounds and placebo
The observed PK parameters for the compounds and their active metabolites are listed in Table 3.
Table 3.
Pharmacokinetic parameters for administered compounds
| Fentanyl | (S)‐ketamine | Norketamine | Phenytoin | Ibuprofen | Imipramine | Desipramine | Pregabalin | |
|---|---|---|---|---|---|---|---|---|
| C max (ng ml –1 ) | 1.23 ± 0.66 | 52.4 ± 15.7 | 23.2 ± 5.7 | 8280 ± 1740 | 53 300 ± 9360 | 77.2 ± 21.1 | 25.4 ± 14.6 | 9650 ± 1880 |
| T max (h) | 0.50 (0.50–1.22) | 0.50 (0.50–2.33) | 1.10 (1.05–3.05) | 0.55 (0.50–2.13) | 2.02 (1.08–3.00) | 3.12 (1.08–6.00) | 6.00 (2.07–10.00) | 1.56 (1.08–5.00) |
| T 1/2 (h) | 2.52 ± 1.08 | 3.12 ± 1.30 | 4.89 ± 1.14 | 12.50 ± 2.86 | 1.80 ± 0.29 | 6.54 ± 1.96 | 56.2 ± 8.7 | 5.30 ± 0.58 |
| AUC 0–last (ng hr ml –1 ) | 2.21 ± 0.72 | 102 ± 18.6 | 129 ± 35.7 | 54 500 ± 7980 | 174 000 ± 30 200 | 441 ± 128 | 163 ± 99.2 | 49 700 ± 7340 |
| AUC 0–inf (ng hr ml –1 ) | 3.10 ± 0.84 | 111 ± 22.2 | 180 ± 54.6 | 135 000 ± 34 800 | 182 000 ± 35 600 | 782 ± 193 | 517 ± 100 | 71 200 ± 9860 |
| Clearance (l hr –1 ) | 1.05 ± 0.32 | 93.6 ± 20.1 | 60.2 ± 17.7 | 2.36 ± 0.54 | NC | NC | NC | NC |
| V d (l) | 3.56 ± 1.29 | 410 ± 153 | 411 ± 106 | 40.60 ± 4.97 | NC | NC | NC | NC |
Data are means ± standard deviation and median (range) for Tmax.
AUC0–inf, estimated area under the plasma concentration‐time curve from time of dosing to infinity; AUC0–last, area under the plasma concentration‐time curve from time of dosing to the last observation; Cmax, maximum concentration; Tmax, time at which Cmax was observed; T1/2, apparent terminal half‐life; Vd, apparent volume of distribution of the drug; NC, not calculated.
All subjects experienced at least one adverse event (AE) during their participation. In part I, the incidence of AEs was 100% in the active treatment groups (fentanyl, (S)‐ketamine and phenytoin) compared to 33% in the placebo group. In part II, 100% of the subjects receiving imipramine, 87.5% of the subject receiving pregabalin, 41.2% of the subjects receiving ibuprofen and 50% of the subjects receiving placebo tablets reported AEs. In part I, the most reported AEs were: dizziness (82%), nausea (65%) and feeling hot (53%) for fentanyl; dizziness (82%), nausea (35%) and feeling abnormal (29%) for (S)‐ketamine; and pain in extremity at administration site (60%), dizziness (55%) and nausea (30%) for phenytoin. In part II, the most reported AEs were: nausea (12%), fatigue (12%) and dizziness (12%) for ibuprofen; somnolence (65%), nausea (59%) and dizziness (29%) for imipramine; and dizziness (56%), somnolence (31%) and nausea (31%) for pregabalin. All AEs were mild or moderate in severity.
Discussion
The main objective of this study was to investigate the ability of a battery of pain models to detect analgesic properties of commonly used analgesics in healthy subjects. A biomarker can be defined as “A characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention.” 30. This battery of different pain models was able to detect differences in pharmacological and analgesic properties, consistent with the PK properties of each individual compound. Each compound tested in this study demonstrated its own profile of effects on evoked pain in the different models included in the pain test battery. Most of these effects were in line with earlier described literature and with the expected PD and PK profile of the drugs. The drugs and doses used had already proven to be efficacious analgesics in clinical practice in either acute pain or in neuropathic pain. This battery of pain models can be used as a biomarker to assess the PD responses of analgesic drugs.
Strong opioids previously showed effects on electrical pain, cold pressor, thermal pain and the thermal grill 9, 16. In this study fentanyl affected pain thresholds in the cold pressor test and thermal testing. No effects were observed on the electrical pain tests, or the pressure pain paradigm. The effect of fentanyl on a broad range of pain tests corresponds with the many types of clinical pain that respond to strong opioids. Previous reports have shown decreases in pain intensity and unpleasantness after morphine administration on the thermal grill 16. Here, maximum unpleasantness and pain intensity did not change after fentanyl administration.
(S)‐ketamine, an N‐methyl‐D‐aspartate (NMDA) receptor antagonist, showed effects on the cold pressor test, electrical stimulation (both single and repeated stimulus) and thermal heat pain. The effects of (S)‐ketamine on the cold pressor test have not been reported before. In a study previously performed 21, the cold pressor test was used in combination with (S)‐ketamine, but only in order to induce a conditioned pain modulation (CPM) response. In a previous review 10, no differences were observed in PDT during heat skin stimulation. In the current study, we found an increase in heat PDT on the normal skin in the 1st h after dosing. Heat PDT in the UVB‐treated skin did not differ compared to placebo.
An effect of (S)‐ketamine on the thermal grill pain and unpleasantness was expected, as these effects were shown previously 15. Here, however, (S)‐ketamine did not result in a decrease in unpleasantness or pain sensation in the thermal grill paradigm. An explanation for these differences could be our method of dosing, where the bolus administration of (S)‐ketamine was not followed by a continuous infusion as described in other studies 15.
There is limited literature available about the effect of sodium channel blockers on human pain models. One study has been published in which the effects of phenytoin and lamotrigine on cold pressor pain were investigated 17; both phenytoin and lamotrigine reduced pain scores in healthy subjects. In the current study, we only observed an increase in PDT and PTT in the electrical stimulation single stimulus paradigm. The therapeutic range for phenytoin in epilepsy is between 8 and 25 μg ml–1 in plasma 32. The observed Cmax in the study was 8.3 μg ml–1, which is at the lower end of the therapeutic range. Higher doses or repeated dosing may lead to a more pronounced effect on the pain models.
In part II, pregabalin showed positive effects on cold pressor (PDT and PTT), electrical single stimulus (PTT), CPM (PDT) and thermal heat pain in normal skin. Alpha‐2δ ligands have previously been shown to affect pain in human pain models; gabapentin showed positive effects on pain in an electrical hyperalgesia model in healthy subjects 33. Conversely, gabapentin failed to show effects on heat PDT in healthy subjects 34. Pregabalin has not been investigated in pain models in healthy subjects but in in patients with painful chronic pancreatitis, pregabalin attenuated visceral pain 35. Of all compounds administered, pregabalin showed the largest effect on most of the pain paradigms (heat, cold, pressure and electrical pain). This might be due to the relatively large dose of pregabalin that was used. However, the same single dose of 300 mg was also used in other studies in which dosages of pregabalin of ≥300 mg showed a significant opioid sparing effect 36. Further studies are needed to show if these large effects can be replicated. Currently, pregabalin is mainly used in the treatment of neuropathic pain 37 and its use in acute postoperative pain is under investigation 36. The positive effects of pregabalin on several (acute) nociceptive pain models in this study may be an argument for its potential use also in the treatment of acute nociceptive pain.
Ibuprofen increased the heat PDT in UVB‐treated skin. These effects were also previously shown by others 9. Ibuprofen was the only compound administered that only increased heat PDT in UVB‐treated skin but not in normal skin. This in contrast with (S)‐ketamine and pregabalin (increased heat PDT in normal skin, but not in UVB treated skin) and fentanyl (increased heat PDT in normal and UVB‐treated skin). These effects of ibuprofen were expected and reflect its inhibition of cyclooxygenase by this nonsteroidal anti‐inflammatory drug, given the inflammatory type of pain that is caused in the UVB hyperalgesia model.
Imipramine only increased CPM, but did not affect other outcome measures. In previous research, imipramine increased acute pain tolerance after electrical stimulation, pressure pain and visceral pain 10, 18. Compared to the other compounds in this study, imipramine and its active metabolite desipramine have a relatively long half‐life of 6.54 h and 56.2 h, respectively. We only performed measurements up to 10 h after dose administration, which may partially explain the negative findings in this study. In favour of this argument is that an increasing trend could still be observed at the last measurements in the electrical repeat stimulation paradigm PTT. Furthermore, in the clinical setting a titration period of several weeks is needed for imipramine before its efficacy can be assessed 37. Here, we only administered a single dose. Imipramine was used as the tricyclic antidepressant of choice in this study because of previous positive results in human pain models. However, a recent meta‐analysis showed that there is only limited evidence for the use of imipramine in neuropathic pain 38.
In part I of the study, no effect was observed on CPM by either (S)‐ketamine, fentanyl or phenytoin. High variability in CPM measurements was observed throughout the study, for all delta electrical stimulation parameters (PDT, PTT and AUC). Previous research conducted has shown a potentiation of CPM after administration of strong opioids 39. Others observed no CPM response after ketamine treatment in healthy volunteers 31.
In part II of the study, both imipramine and pregabalin increased the difference in pain detection threshold after vs. before the cold pressor (delta PDT), which may be indicative for an increase in CPM. A study performed in patients with pancreatitis did not show changes in CPM responses after administration of pregabalin. To our knowledge no studies are published in which the CPM responses in healthy subjects after administration of α2δ ligands or tricyclic antidepressants were measured. The noradrenergic system plays an important role in central pain modulation 40; so the increase in delta PDT observed after administration of imipramine is likely to be explained by the enhancement of the inhibitory effect on noradrenaline reuptake.
No decrease on thermal grill maximum unpleasantness or maximum pain ratings could be observed in this study. However, overall, most subjects did not experience the thermal grill as unpleasant or painful, as reflected by the low scores on the eVAS for pain and unpleasantness, which resulted in a non‐normal distribution of the data, making them difficult to analyse.
Previous studies in which the thermal grill was used applied a range of combinations of warm and cold stimuli to assess relationships between painful and nonpainful sensations 16, 41. In the current study, a fixed temperature of the warm and cold bars was used. Furthermore, the occurrence of paradoxical pain elicited by the thermal grill illusion can be variable. A study by Bouhassiara and colleagues 42 reported a large subpopulation of subjects who only reported paradoxical pain when large cold‐warm differentials were applied. Due to the apparent necessity to tailor this method to each individual subject, it is difficult to standardize this method and incorporate it in a battery of pain models.
Multimodal testing with different pain models has been performed previously; with and without the administration of analgesic compounds 8, 18, 43. Here we combined both the execution of a broad range of human pain models and the administration of analgesic compounds with different mechanisms of action. An advantage of the battery of pain models was that the tests could be executed repeatedly in a relatively short time (~30 min) in a standardized fashion.
By repeatedly administering these pain tests in 1 day, this battery was able to determine time‐effect profiles of the drugs. Small individual differences between different compounds could be assessed. Although PK/PD modelling was not performed in this study, study designs using repeated application of this battery of pain models can be used to assess PK/PD relationships.
Overall, PK parameters measured in this study were reasonably consistent with the known PK data for these analgesics. Fentanyl's terminal half‐life and volume of distribution were somewhat lower compared to values reported in literature 44. Phenytoin, (S)‐ketamine and its active metabolite norketamine showed kinetics that were consistent with the literature 32, 45. The tmax of imipramine was as expected. The terminal half‐life was shorter, but this could have been related to the relatively short sampling period; the half‐life of its active metabolite desipramine was longer than expected 46. Ibuprofen and pregabalin showed PK that were consistent with the literature 47, 48.
A large number of pain models were used in this study. This yielded an even greater number of outcome variables. No correction for multiple testing was applied. Therefore, this multimodal test battery should be considered as a screening tool for analgesic properties of compounds in development for the treatment of pain, and not as a way to definitively prove effects on a specific evoked pain model with statistical significance. When the analgesic effect of a new drug on a certain pain mechanism has already been established, predefining a primary outcome measure would prevent the need to correct for multiple testing. Maximum effect sizes differed for the pain models used. For instance, after pregabalin administration the contrast compared to placebo for heat PDT was 4.1%, while the contrast for cold PTT was 46.4%. Variability for these tests was also markedly different, with the coefficient of variation for the heat PTT being much lower than for the cold PTT (Table S2). To account for this variation, studies using this battery of pain models need to be adequately powered. Only one (expected analgesic) dose of each compound was used in our study. Therefore, one‐to‐one comparisons between different compounds cannot be made on individual pain models. However, the pharmacodynamic profiles of these single doses matched the plasma profile of the compounds used. Reproducibility of the pharmacological effects of the compounds on the pain models was not directly assessed in this study. We were able to replicate effects of different analgesics on individual pain tests as described before 9, 10, 11; however, future studies are needed to investigate the reproducibility of the effect profiles that we observed. One session of the battery of pain models lasted approximately 30 min. During one study period, 10 sessions were performed. This might have led to fatigue and diminishing concentration during the tests. This is also shown in Figures 3 and 4, where variation in the placebo group is observed between measurements during the day. In order to correct for these unavoidable effects, a crossover design with a placebo arm included was used. Somnolence was observed by 31% and dizziness by 56% of the subjects receiving pregabalin. Oral doses of imipramine also caused similar AEs, however imipramine did not show effects on the pain tasks administered. Other substances that are known to have strong sedating effects on the central nervous system also do not influence evoked pain tests. For instance, cannabinoids and benzodiazepines have limited effects on pain thresholds 10, 49. Therefore, we believe that the somnolence and the dizziness caused by the pregabalin is not responsible for the effects on the pain tasks administered.
Several drugs acting at different targets are currently under clinical development for the treatment of acute and neuropathic pain. These drugs are in different stages of the clinical development. Examples are selective sodium channel blockers, nerve growth factor antagonists and fatty acid amide hydrolase inhibitors 50, 51, 52. A recent review suggested that a limited set of human pain models could be sufficient to predict analgesic efficacy 7. With our integrated battery of pain models, it is possible to profile new compounds against currently existing analgesic compounds to predict their potential clinical use.
In conclusion, it was shown that this battery of pain models is able to detect changes in pain detection and pain tolerance thresholds after administration of different classes of analgesic compounds in healthy male and female subjects. The analgesic compounds all showed a unique profile in their effects on the pain tests administered. These profiles were in most cases compatible with the expected pharmacology. The knowledge of these profiles can be used to benchmark analgesic properties of these new drugs against established analgesics in early phase clinical studies in healthy subjects.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and P.O., G.A., M.K., J.S., J.G., A.D., J.H. and G.G. declare: no support from any organisation for the submitted work; no financial relationships with any organisations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work. R.B. and R.G. were employees of Pfizer Ltd. during study execution; the Centre for Human Drug Research received partial funding from Pfizer Ltd. for execution of the study. The funder reviewed and provided feedback on the paper.
Contributors
P.O., G.A., J.G., J.H. and G.G. wrote the manuscript, M.K., J.S., R.B., R.G. and A.D. reviewed and commented on the manuscript. P.O., J.G., R.B., R.G., A.D., J.H. and G.G. designed the research. P.O., J.H., G.A. and G.G. performed the research. M.K. and J.S. analysed the data.
Trial registration
The trial was registered in the trial register of the Committee on Research Involving Human Subjects (CCMO, https://www.toetsingonline.nl, NL46000.058.13).
Supporting information
Table S1 Summary data of the pharmacodynamic parameters for part I
Table S2 Summary data of the pharmacodynamic parameters for part II
Table S3 Analytical performance of the LC–MS/MS assays
Supporting info item
Supporting info item
Supporting info item
Okkerse, P. , van Amerongen, G. , de Kam, M. L. , Stevens, J. , Butt, R. P. , Gurrell, R. , Dahan, A. , van Gerven, J. M. , Hay, J. L. , and Groeneveld, G. J. (2017) The use of a battery of pain models to detect analgesic properties of compounds: a two‐part four‐way crossover study. Br J Clin Pharmacol, 83: 976–990. doi: 10.1111/bcp.13183.
Principal Investigator: G.J. Groeneveld, MD, PhD
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Fabbro D, Davenport AP, Kelly E, Marrion N, Peters JA, et al. The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 2015; 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E, et al. The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 2015; 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 2015; 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cohen AF, Burggraaf J, Gerven JM, Moerland M, Groeneveld GJ. The use of biomarkers in human pharmacology (Phase I) studies. Annu Rev Pharmacol Toxicol 2015; 55: 55–74. [DOI] [PubMed] [Google Scholar]
- 7. Lotsch J, Oertel BG, Ultsch A. Human models of pain for the prediction of clinical analgesia. Pain 2014; 155: 2014–2021. [DOI] [PubMed] [Google Scholar]
- 8. Staahl C, Reddy H, Andersen SD, Arendt‐Nielsen L, Drewes AM. Multi‐modal and tissue‐differentiated experimental pain assessment: reproducibility of a new concept for assessment of analgesics. Basic Clin Pharmacol Toxicol 2006; 98: 201–211. [DOI] [PubMed] [Google Scholar]
- 9. Oertel BG, Lotsch J. Clinical pharmacology of analgesics assessed with human experimental pain models: bridging basic and clinical research. Br J Pharmacol 2013; 168: 534–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Staahl C, Olesen AE, Andresen T, Arendt‐Nielsen L, Drewes AM. Assessing efficacy of non‐opioid analgesics in experimental pain models in healthy volunteers: an updated review. Br J Clin Pharmacol 2009; 68: 322–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Staahl C, Olesen AE, Andresen T, Arendt‐Nielsen L, Drewes AM. Assessing analgesic actions of opioids by experimental pain models in healthy volunteers – an updated review. Br J Clin Pharmacol 2009; 68: 149–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Olesen AE, Andresen T, Staahl C, Drewes AM. Human experimental pain models for assessing the therapeutic efficacy of analgesic drugs. Pharmacol Rev 2012; 64: 722–779. [DOI] [PubMed] [Google Scholar]
- 13. Arendt‐Nielsen L, Curatolo M, Drewes A. Human experimental pain models in drug development: translational pain research. Curr Opin Investig Drugs 2007; 8: 41–53. [PubMed] [Google Scholar]
- 14. Martini C, Olofsen E, Yassen A, Aarts L, Dahan A. Pharmacokinetic–pharmacodynamic modeling in acute and chronic pain: an overview of the recent literature. Expert Rev Clin Pharmacol 2011; 4: 719–728. [DOI] [PubMed] [Google Scholar]
- 15. Kern D, Pelle‐Lancien E, Luce V, Bouhassira D. Pharmacological dissection of the paradoxical pain induced by a thermal grill. Pain 2008; 135: 291–299. [DOI] [PubMed] [Google Scholar]
- 16. Kern D, Plantevin F, Bouhassira D. Effects of morphine on the experimental illusion of pain produced by a thermal grill. Pain 2008; 139: 653–659. [DOI] [PubMed] [Google Scholar]
- 17. Webb J, Kamali F. Analgesic effects of lamotrigine and phenytoin on cold‐induced pain: a crossover placebo‐controlled study in healthy volunteers. Pain 1998; 76: 357–363. [DOI] [PubMed] [Google Scholar]
- 18. Enggaard TP, Poulsen L, Arendt‐Nielsen L, Hansen SH, Bjornsdottir I, Gram LF, et al. The analgesic effect of codeine as compared to imipramine in different human experimental pain models. Pain 2001; 92: 277–282. [DOI] [PubMed] [Google Scholar]
- 19. Stening K, Eriksson O, Wahren L, Berg G, Hammar M, Blomqvist A. Pain sensations to the cold pressor test in normally menstruating women: comparison with men and relation to menstrual phase and serum sex steroid levels. Am J Physiol Regul Integr Comp Physiol 2007; 293: R1711–R1716. [DOI] [PubMed] [Google Scholar]
- 20. Hay JL, Okkerse P, Van Amerongen G, Groeneveld GJ. Determining pain detection and tolerance thresholds using an integrated, multi‐modal pain task battery. J Vis Exp 2016; 14: 53800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bishop T, Ballard A, Holmes H, Young AR, McMahon SB. Ultraviolet‐B induced inflammation of human skin: characterisation and comparison with traditional models of hyperalgesia. Eur J Pain 2009; 13: 524–532. [DOI] [PubMed] [Google Scholar]
- 22. Dahan A, Romberg R, Teppema L, Sarton E, Bijl H, Olofsen E. Simultaneous measurement and integrated analysis of analgesia and respiration after an intravenous morphine infusion. Anesthesiology 2004; 101: 1201–1209. [DOI] [PubMed] [Google Scholar]
- 23. Olofsen E, Romberg R, Bijl H, Mooren R, Engbers F, Kest B, et al. Alfentanil and placebo analgesia: no sex differences detected in models of experimental pain. Anesthesiology 2005; 103: 130–139. [DOI] [PubMed] [Google Scholar]
- 24. Arendt‐Nielsen L, Frokjaer JB, Staahl C, Graven‐Nielsen T, Huggins JP, Smart TS, et al. Effects of gabapentin on experimental somatic pain and temporal summation. Reg Anesth Pain Med 2007; 32: 382–388. [DOI] [PubMed] [Google Scholar]
- 25. Polianskis R, Graven‐Nielsen T, Arendt‐Nielsen L. Computer‐controlled pneumatic pressure algometry‐‐a new technique for quantitative sensory testing. Eur J Pain 2001; 5: 267–277. [DOI] [PubMed] [Google Scholar]
- 26. Polianskis R, Graven‐Nielsen T, Arendt‐Nielsen L. Pressure‐pain function in desensitized and hypersensitized muscle and skin assessed by cuff algometry. J Pain 2002; 3: 28–37. [DOI] [PubMed] [Google Scholar]
- 27. Eckhardt K, Li S, Ammon S, Schanzle G, Mikus G, Eichelbaum M. Same incidence of adverse drug events after codeine administration irrespective of the genetically determined differences in morphine formation. Pain 1998; 76: 27–33. [DOI] [PubMed] [Google Scholar]
- 28. Jones SF, McQuay HJ, Moore RA, Hand CW. Morphine and ibuprofen compared using the cold pressor test. Pain 1988; 34: 117–122. [DOI] [PubMed] [Google Scholar]
- 29. Pud D, Granovsky Y, Yarnitsky D. The methodology of experimentally induced diffuse noxious inhibitory control (DNIC)‐like effect in humans. Pain 2009; 144: 16–19. [DOI] [PubMed] [Google Scholar]
- 30. Atkinson AJJ, Colburn WA, DeGruttola VG, DeMets DL, Downing GJ, Hoth DF, et al. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 2001; 69: 89–95. [DOI] [PubMed] [Google Scholar]
- 31. Niesters M, Dahan A, Swartjes M, Noppers I, Fillingim RB, Aarts L, et al. Effect of ketamine on endogenous pain modulation in healthy volunteers. Pain 2011; 152: 656–663. [DOI] [PubMed] [Google Scholar]
- 32. Morselli PL, Franco‐Morselli R. Clinical pharmacokinetics of antiepileptic drugs in adults. Pharmacol Ther 1980; 10: 65–101. [DOI] [PubMed] [Google Scholar]
- 33. Boyle Y, Fernando D, Kurz H, Miller SR, Zucchetto M, Storey J. The effect of a combination of gabapentin and donepezil in an experimental pain model in healthy volunteers: results of a randomized controlled trial. Pain 2014; 155: 2510–2516. [DOI] [PubMed] [Google Scholar]
- 34. Gustorff B, Hoechtl K, Sycha T, Felouzis E, Lehr S, Kress HG. The effects of remifentanil and gabapentin on hyperalgesia in a new extended inflammatory skin pain model in healthy volunteers. Anesth Analg 2004; 98: 401–407 , table. [DOI] [PubMed] [Google Scholar]
- 35. Olesen SS, Graversen C, Olesen AE, Frokjaer JB, Wilder‐Smith O, van Goor H, et al. Randomised clinical trial: pregabalin attenuates experimental visceral pain through sub‐cortical mechanisms in patients with painful chronic pancreatitis. Aliment Pharmacol Ther 2011; 34: 878–887. [DOI] [PubMed] [Google Scholar]
- 36. Zhang J, Ho KY, Wang Y. Efficacy of pregabalin in acute postoperative pain: a meta‐analysis. Br J Anaesth 2011; 106: 454–462. [DOI] [PubMed] [Google Scholar]
- 37. Dworkin RH, O'Connor AB, Backonja M, Farrar JT, Finnerup NB, Jensen TS, et al. Pharmacologic management of neuropathic pain: evidence‐based recommendations. Pain 2007; 132: 237–251. [DOI] [PubMed] [Google Scholar]
- 38. Hearn L, Derry S, Phillips T, Moore RA, Wiffen PJ. Imipramine for neuropathic pain in adults. Cochrane Database Syst Rev 2014; (5): CD010769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Arendt‐Nielsen L, Andresen T, Malver LP, Oksche A, Mansikka H, Drewes AM. A double‐blind, placebo‐controlled study on the effect of buprenorphine and fentanyl on descending pain modulation: a human experimental study. Clin J Pain 2012; 28: 623–627. [DOI] [PubMed] [Google Scholar]
- 40. Ossipov MH, Dussor GO, Porreca F. Central modulation of pain. J Clin Invest 2010; 120: 3779–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Adam F, Alfonsi P, Kern D, Bouhassira D. Relationships between the paradoxical painful and nonpainful sensations induced by a thermal grill. Pain 2014; 155: 2612–2617. [DOI] [PubMed] [Google Scholar]
- 42. Bouhassira D, Kern D, Rouaud J, Pelle‐Lancien E, Morain F. Investigation of the paradoxical painful sensation (‘illusion of pain’) produced by a thermal grill. Pain 2005; 114: 160–167. [DOI] [PubMed] [Google Scholar]
- 43. Olesen AE, Brock C, Sverrisdottir E, Larsen IM, Drewes AM. Sensitivity of quantitative sensory models to morphine analgesia in humans. J Pain Res 2014; 7: 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McClain DA, Hug CC Jr. Intravenous fentanyl kinetics. Clin Pharmacol Ther 1980; 28: 106–114. [DOI] [PubMed] [Google Scholar]
- 45. Peltoniemi MA, Saari TI, Hagelberg NM, Laine K, Kurkinen KJ, Neuvonen PJ, et al. Rifampicin has a profound effect on the pharmacokinetics of oral S‐ketamine and less on intravenous S‐ketamine. Basic Clin Pharmacol Toxicol 2012; 111: 325–332. [DOI] [PubMed] [Google Scholar]
- 46. Ciraulo DA, Barnhill JG, Jaffe JH. Clinical pharmacokinetics of imipramine and desipramine in alcoholics and normal volunteers. Clin Pharmacol Ther 1988; 43: 509–518. [DOI] [PubMed] [Google Scholar]
- 47. Kapil R, Nolting A, Roy P, Fiske W, Benedek I, Abramowitz W. Pharmacokinetic properties of combination oxycodone plus racemic ibuprofen: two randomized, open‐label, crossover studies in healthy adult volunteers. Clin Ther 2004; 26: 2015–2025. [DOI] [PubMed] [Google Scholar]
- 48. Bockbrader HN, Radulovic LL, Posvar EL, Strand JC, Alvey CW, Busch JA, et al. Clinical pharmacokinetics of pregabalin in healthy volunteers. J Clin Pharmacol 2010; 50: 941–950. [DOI] [PubMed] [Google Scholar]
- 49. Vuilleumier PH, Besson M, Desmeules J, Arendt‐Nielsen L, Curatolo M. Evaluation of anti‐hyperalgesic and analgesic effects of two benzodiazepines in human experimental pain: a randomized placebo‐controlled study. PLoS One 2013; 8: e43896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Leite VF, Buehler AM, El AO, Benyamin RM, Pimentel DC, Chen J, et al. Anti‐nerve growth factor in the treatment of low back pain and radiculopathy: a systematic review and a meta‐analysis. Pain Physician 2014; 17: E45–E60. [PubMed] [Google Scholar]
- 51. Theile JW, Cummins TR. Recent developments regarding voltage‐gated sodium channel blockers for the treatment of inherited and acquired neuropathic pain syndromes. Front Pharmacol 2011; 2: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bisogno T, Maccarrone M. Latest advances in the discovery of fatty acid amide hydrolase inhibitors. Expert Opin Drug Discov 2013; 8: 509–522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Summary data of the pharmacodynamic parameters for part I
Table S2 Summary data of the pharmacodynamic parameters for part II
Table S3 Analytical performance of the LC–MS/MS assays
Supporting info item
Supporting info item
Supporting info item