

In this study we determined the major mechanotransduction pathway by which blood flow-driven shear stress activates cyclooxygenase-2 (COX-2) and prostaglandin I2 (PGI2) release in endothelial cells. Our work has demonstrated for the first time that COX-2/PGI2 mechanotransduction is mediated by the mechanosensor platelet endothelial cell adhesion molecule-1 (PECAM-1).
Keywords: prostaglandin I2, cyclooxygenase-2, PECAM-1, integrins, shear stress, glycocalyx, syndecan-4, primary cilia
Abstract
Vascular endothelial cells play an important role in the regulation of vascular function in response to mechanical stimuli in both healthy and diseased states. Prostaglandin I2 (PGI2) is an important antiatherogenic prostanoid and vasodilator produced in endothelial cells through the action of the cyclooxygenase (COX) isoenzymes COX-1 and COX-2. However, the mechanisms involved in sustained, shear-induced production of COX-2 and PGI2 have not been elucidated but are determined in the present study. We used cultured endothelial cells exposed to steady fluid shear stress (FSS) of 10 dyn/cm2 for 5 h to examine shear stress-induced induction of COX-2/PGI2. Our results demonstrate the relationship between the mechanosensor platelet endothelial cell adhesion molecule-1 (PECAM-1) and the intracellular mechanoresponsive molecules phosphatidylinositol 3-kinase (PI3K), focal adhesion kinase (FAK), and mitogen-activated protein kinase p38 in the FSS induction of COX-2 expression and PGI2 release. Knockdown of PECAM-1 (small interference RNA) expression inhibited FSS-induced activation of α5β1-integrin, upregulation of COX-2, and release of PGI2 in both bovine aortic endothelial cells (BAECs) and human umbilical vein endothelial cells (HUVECs). Furthermore, inhibition of the PI3K pathway (LY294002) substantially inhibited FSS activation of α5β1-integrin, upregulation of COX-2 gene and protein expression, and release of PGI2 in BAECs. Inhibition of integrin-associated FAK (PF573228) and MAPK p38 (SB203580) also inhibited the shear-induced upregulation of COX-2. Finally, a PECAM-1−/− mouse model was characterized by reduced COX-2 immunostaining in the aorta and reduced plasma PGI2 levels compared with wild-type mice, as well as complete inhibition of acute flow-induced PGI2 release compared with wild-type animals.
NEW & NOTEWORTHY In this study we determined the major mechanotransduction pathway by which blood flow-driven shear stress activates cyclooxygenase-2 (COX-2) and prostaglandin I2 (PGI2) release in endothelial cells. Our work has demonstrated for the first time that COX-2/PGI2 mechanotransduction is mediated by the mechanosensor platelet endothelial cell adhesion molecule-1 (PECAM-1).
vascular endothelial cells (ECs) are located at the interface between flowing blood and the vascular wall where they play a significant role in regulating circulatory functions. Blood flow-induced fluid shear stress (FSS) partially regulates EC function by stimulating mechanoreceptors that activate intracellular signaling pathways modulating the expression of genes that encode growth factors, adhesion molecules, chemoattractants, and the release of vasoactive agents such as nitric oxide (NO), and prostaglandin I2 (PGI2) (9, 52). PGI2 is an important antiatherogenic prostanoid, responsible, in part, for regulating the homeostasis of the cardiovascular system in response to FSS (20, 32). PGI2 is a potent vasodilator produced in ECs by the conversion of arachidonic acid to prostaglandin H2 (PGH2) through the action of the cyclooxygenase (COX) isoenzymes COX-1 and COX-2. PGH2 is then enzymatically converted by prostacyclin (PGI2) synthase in ECs to produce PGI2. In blood, PGI2 functions to counteract the effects of thromboxane A2 (TXA2), a known vasoconstrictor and a promoter of platelet aggregation (9, 15, 32). These two prostanoids provide an important balance in cardiovascular homeostasis. In addition, COX enzymes participate in acute inflammatory responses leading to PGI2 release, edema formation, and pain (33, 53). However, despite many studies of the mechanotransduction mechanisms involved in the transmission of FSS across the endothelium to regulate EC function, the detailed mechanisms involved in the upregulation of COX-2 and associated release of PGI2 in ECs remain unclear.
In this study we hypothesized that one or more primary mechanotransducer candidates, including the cell surface endothelial glycocalyx layer (EGL), the basal proteoglycan syndecan-4 (SDC4), primary cilia, basal integrins, and platelet endothelial adhesion molecule-1 (PECAM-1) sense FSS and activate intracellular signaling to upregulate COX-2 and PGI2. The rationale for considering these candidates is described below.
The apical surfaces of ECs are covered with a layer that includes proteoglycans with extended covalently bound sulfated glycosaminoglycan (GAG) side chains [heparan sulfate (HS) and chondroitin sulfate (CS)], the nonsulfated GAG hyaluronic acid (HA) bound primarily to its surface receptor CD44, as well as sialic acids, glycocproteins, and plasma proteins that constitute the endothelial glycocalyx layer (EGL). Earlier studies from our laboratory showed that removal of ~50% of the surface HS from bovine aortic endothelial cells (BAECs) with the specific enzyme heparinase III completely blocked the shear-induced upregulation of NO production (19). A subsequent study in BAECs (45) showed that cells treated with similar doses of additional EGL-degrading enzymes (hyaluronidase for HA and neuraminidase for sialic acid) also blocked shear-induced NO production. Surprisingly, the same enzyme depletions that blocked NO production had no significant effect on shear-induced PGI2 upregulation in response FSS for 3 h. However, a recent study in rat fat pad ECs (66) showed that the same enzyme treatments at much higher doses could almost completely and specifically remove HS, HA, or CS from the EC surface. This leaves open the possibility that complete removal of an individual GAG component might inhibit shear-induced production of COX-2/PGI2.
EGL transmembrane proteoglycans, syndecan (SDC)1, 2, and 4, are associated with mechanotransduction models (28). Among the syndecan family, SDC4 is a transmembrane proteoglycan that colocalizes with integrin receptors on the basal aspect of EC to modulate the formation of focal adhesion complexes in epithelial cells (24, 36, 41, 55). Due to its cell surface localization and interactions with integrins, cytoskeletal elements, and focal adhesion complex proteins, SDC4 has been implicated as a cell surface mechanosensor that transmits extracellular signals into intracellular signaling pathways (2, 60).
Primary cilia have been identified recently as a potential mechanosensor responsible for the amplification and transduction of FSS-induced activation of COX-2 mRNA in bone (25, 31, 34). Primary cilia are nonmotile microtubule-based organelles found on most mammalian cells including ECs, where they develop from a mother centriole of the centrosome. The cilium is an isolated structure, usually distributed one per cell on the surface of nonproliferating cells, where it is localized with many cell surface signaling receptors. Anchored to the basal body it extends its antenna-like protrusion into the lumen where it is directly in contact with the circulating blood possibly mediating signal transduction through cell-cell junctions and basal focal adhesions.
PECAM-1, is a 130-KDa transmembrane glycoprotein that belongs to the immunoglobulin (Ig) superfamily, primarily expressed in ECs, platelets, leukocytes, and neutrophils (38). In confluent ECs PECAM-1 localizes at intercellular junctions, where it regulates homophilic binding between ECs. By virtue of its cellular location it has been implicated in a mechanosensory complex leading to the activation of several signaling pathways involved in “inside out” integrin activation upon phosphorylation (61). Forces on PECAM-1 result in its rapid tyrosine phosphorylation and have been shown to induce phosphatidylinositol 3-kinase (PI3K) activation and downstream activation of integrins, FAK (Tyr 397), and ERK in ECs (10–12, 44, 58). Furthermore, treatment of ECs with the PI3K inhibitor LY294002 (LY29) completely blocked activation of the major fibronectin integrin α5β1 after exposure to FSS for short times (43, 61). Taken together these studies provide evidence for the role of PECAM-1 in initiating the transduction of mechanical signals across the membrane to regulate PI3K-mediated integrin activation and subsequent activation of the downstream signaling molecule FAK and MAPKs, which may be involved in the shear-induced regulation of COX-2 and PGI2 in ECs.
To test our hypothesis: 1) EGL components CS, HA, and HS were selectively depleted using EGL-degrading enzymes heprinase (HepIII), hyaluronidase (Hyal.), and chondroitinase (Chond.) at high doses; 2) FSS activation of α5β1-integrin was assessed using a specialized fusion protein GST-FNIII9–11; 3) SDC4, primary cilia and PECAM-1 were depleted by siRNA; 4) signaling molecules PI3K, FAK, and MAPKs were suppressed by specific inhibitors; and 5) a PECAM-1−/− mouse model was used to confirm in vitro findings.
Our results demonstrate that complete removal of EGL components, knockdown of SDC4 or primary cilia, and inhibition of ERK phosphorylation had no significant effect on FSS induction of COX-2. However, inhibition of the PI3K pathway completely blocked the FSS-induced activation of fibronectin integrin α5β1, upregulation of COX-2 protein and mRNA expression, and PGI2 release in BAECs. In addition, inhibition of FAK and MAPK p38, but not ERK, blocked the shear-induced upregulation of COX-2 protein expression in BAECs. Knockdown of PECAM-1 expression in both BAECs and HUVECs substantially inhibited COX-2 protein expression, activation of the major fibronectin integrin α5β1, and PGI2 release. Finally, the PECAM-1−/− mouse model was characterized by reduced COX-2 immunostaining in the aorta and reduced plasma PGI2 levels compared with wild-type (WT) mice, as well as complete inhibition of acute flow-induced PGI2 release compared with the WT animals.
MATERIALS AND METHODS
Cell culture.
Passage 4–7 BAECs were purchased from VEC Technologies. BAECs were cultured in MCDB131 containing 10% FBS, 0.05 mg/ml gentamycin. Human umbilical vein endothelial cells (HUVECs) (Lonza) were used between passages 1–3. BAECs and HUVECs are the most widely studied ECs in the context of mechanotransduction. HUVECs were cultured according to Lonza instructions in EGM-MW growth medium supplemented with 5% fetal bovine serum (FBS), 0.04% hydrocortisone, 0.4% human fibroblast growth factor, 0.1% vascular endothelial growth factor, 0.1% R3-insulin-like growth factor-1, 0.1% ascorbic acid, 0.1% human recombinant epidermal growth factor, and 0.1% gentamicin and amphotericin-B (GA-1000) (all from Lonza) at 37°C and 5% CO2. The cells were first subclutured in T-75 tissue culture flasks until they reached confluency within 3–5 days and then passaged to fibronectin coated six-well plates and allowed to reach confluency 3–5 days before experiments. Confluent monolayers were washed twice with experimental media containing 1% BSA and 1% penicillin and streptomycin or gentamycin before each treatment.
Fluid shear stress.
BAECs were subjected to fluid shear stress (10 dyn/cm2), using a previously described six-well parallel-disk viscometer designed in our laboratory (16, 19). A time course of EC exposure to FSS was conducted to determine the length of time required to significantly induce COX-2 expression (see Fig. 15). Subsequently, all fluid shear stress experiments were performed for 5 h in an incubator at 37°C with a supply of 5% CO2, with each well containing 3 ml of experimental media. Static controls were also incubated under the same conditions.
Fig. 15.
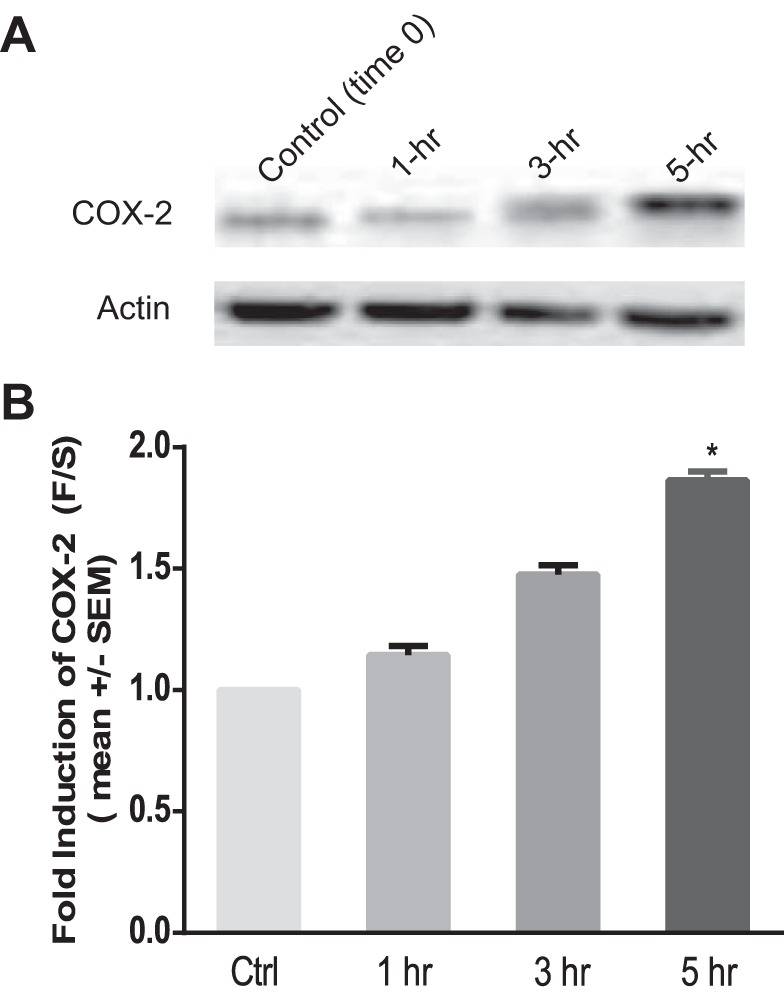
FSS-induced upregulation of COX-2 in BAECs over time. A: Western blot showing shear-induced upregulation of COX-2 protein in BAECs exposed to a time course of FSS. B: densitometric analysis of COX-2 expression under shear stress over time. Densitometric analysis was accomplished by dividing the target protein densitometric units by β-actin (loading control) densitometric units to normalize for loading and then normalizing by the control (time 0) (n = 4). Values are means ± SE. *P < 0.05 vs. static (time 0) controls.
Western blot analysis and deglycosylation.
After exposure to FSS, BAECs were quickly rinsed twice with ice cold PBS then harvested in ice-cold 1× RIPA buffer (Millipore). Protease and phosphotase inhibitors PMSF, NaSO4, leupeptin, aprotinin, and pepstain and denaturant DTT were added at a final concentration of 1 µg/ml. The whole cell lysates were centrifuged (13,000 g for 15 min at 4°C), and the resulting supernatant was collected for protein determination. Equal amounts of protein (30 µg) as determined by the Bradford method (Cayman Chemical) were separated on a 10% SDS-PAGE gel and transferred onto a nitrocellulose membrane. The membranes were washed in Tris saline buffer containing 0.5% Tween (TBS-T) and blocked for 30 min with TBS-T containing 5% milk and then incubated at 4°C overnight with one of the following antibodies: rabbit anti-COX-2 (1:200; Abcam); mouse anti-GST (1:1,000; Santa Cruz Biotechnology); human goat anti-PECAM-1 (1:200; C-20, Santa Cruz Biotechnology); bovine goat anti-PECAM-1 (1:200; M20, Santa Cruz Biotechnology); rabbit anti-phospho-FAK (1:1,000; Cell Signaling); rabbit anti-phospho-ERK (1:1,000; Cell Signaling); rabbit anti-phospho-p38(1:1,000; Cell Signaling); total rabbit anti-FAK, anti-ERK, anti-p38 (1:1,000; Cell Signaling); rabbit anti-IFT88 (1:1,000; Santa Cruz Biotechnology); and mouse anti-β-actin (1:3,000; Sigma). Following overnight primary incubation the membranes were washed four times (each wash 10 min) before being incubated for 1 h at room temperature with the goat anti-rabbit IgG, donkey anti-goat IgG, or goat anti-mouse IgG horseradish peroxidase-conjugated (1:10,000; GE) secondary antibodies. Their signals were detected using a chemiluminescent method and visualized by exposure to X-ray film or ChemiDoc XRS system with the Quantity One software (Bio-Rad). Specific bands were quantified by densitometry. For glycosylated samples (BAECs), deglycosylation was accomplished by treating 1–20 μg of whole cell lysate with PNGase F enzyme (NEB) according to the manufacturer's instructions. Degylcosylated samples were then separated as described above.
In vitro PGI2 AssayMedia samples were collected after 5 h of static or shear and assayed for 6-keto-prostaglandin F1α, a spontaneous hydrolysis product of the unstable PGI2, using an enzyme immunoassay kit from Abcam following the manufacturer’s instructions.
EGL-degrading enzyme treatments.
BAECs were grown to confluency in 3–5 days on fibronectin-coated six-well plates. The monolayers were then briefly washed twice with experimental media, and pretreated with 3 ml of experimental media containing Proteus vulgaris chondroitinase ABC (15 or 135 mU/ml; Sigma), Staphylococcus hyalurolyticus hyaluronidase (1.5 or 4.5 U/ml; Sigma), or Flavobacterium heparinum heparinase III (15 or 1,215 mU/ml; IBEX) for 2 h to degrade EGL components and then subjected to a 5-h static culture or fluid shear stress in the continued presence of each enzyme (66).
Kinase inhibition assays.
Confluent BAECs were pretreated with FAK, ERK, p38, and PI3K signaling pathway inhibitors PF-228 (10 µM; Cell Signaling), PD98059 (10 µM; Cell Signaling), SB203580 (10 µM; Cell Signaling), or LY294002 (30 uM; Cell Signaling), respectively, in MCDB131 media containing 1% BSA 1 h before static or flow conditions. The inhibitors were retained in the experimental media during the 5-h experiments.
Integrin activation assay.
After exposure to 5 h of FSS, BAECs or HUVECs were incubated with PBS containing magnesium, calcium, and 20 µg/ml of GST-FNIII9–11 (from A. W. Orr, Louisiana State University) for 30 min at 37°C. After incubation, cells were briefly washed and lysed in sample buffer and visualized via Western blotting using GST antibody (Santa Cruz Biotechnology).
siRNA transfection.
The genes for core protein SDC4, intraflagellar transport protein (IFT88/Polaris) required for functional ciliogensis, or PECAM-1 were silenced using siRNA synthesized by IDT DNA Technologies. BAECs and HUVECs were seeded 24 h before transfection on fibronectin coated six-well plates at a cell confluency of ~60–80% in antibiotic free media. Lipofectamine RNAiMAX and siRNA sequences were diluted in Opti-MEM and added to cells following the manufacturer’s instruction. Controls were treated with certified negative control siRNA sequences (IDT). After 72 h of transfection, confluent monolayers were briefly rinsed and exposed 10 dyn/cm2 FSS for 5 h in experimental media containing antibiotics. Following exposure to FSS, we confirmed that expression of each target protein remained knocked down after 5 h of FSS by RT-PCR or Western blotting. siRNA sequences used for transfection were as follows: bovine SDC4, 5′-AGCAGAUCCCAAACUAUAACGUAGG-3′; bovine IFT88/Polaris, 5′- AGAAGUCACCUUACAAGAUUUA-3′; bovine PECAM-1, 5′-AUGCAACGUGAUCCUGAACAA- 3′ (10, 58); and human PECAM-1, 5′-GAAUUCUCGAGACCAGAAU-3′ (58).
RT-PCR gene expression.
RNA from sheared and static, treated and untreated, cells were harvested using Trizol reagent following the manufacturer’s instructions. ECs were lysed by incubation with Trizol LS Reagent (Invitrogen) for 3–5 min. Insoluble materials were removed by centrifuging the cell suspension for 15 min at 13,000 g and 4°C. Chloroform was added for phase separation followed by RNA isolation using the Purelink RNA Mini Kit (Invitrogen). RNA samples were then converted to cDNA by RT reaction. Reverse transcription was performed in a total volume of 28 μl containing Absolute Blue QPCR SYBR Green ROX Mix (Thermo Scientific), cDNA, and specific primer pairs. Target cDNA was amplified as previously described (39). Primer sequences were as follows: bovine PTGS-2 gene, forward 5′-TGCGACTGGAAGATTCCTAC-3′, reverse 5′-CTGGAACATGGTCTCACTCA-3′; bovine SDC-4, forward 5′-GTGAGTGAATGTTTGTGGGG-3′, reverse 5′-GTTTCTAGCCTAGTCGGGAG-3′; bovine housekeeping gene HPRT 5′-GAC CAGTCAACAGGCGACAT-3′, reverse 5′-GCTCGCAACCTTGACCATCT-3′; and β-actin, forward 5′-CATGTACGTGGCCATCCAGG-3′, reverse 5′-CCGTGGTGGTGAAGCTGTAG-3′. Gene expression was calculated using the DDCt method.
Immunofluorescence and confocal microscopy.
For primary cilia staining, confluent BAECs were briefly washed with PBS and fixed in 4% paraformaldehyde for 10 min, permeablized in 0.1% Triton-X in PBS for 5 min, blocked with1% BSA for 1 h and incubated overnight with monoclonal acetylated α-tubulin antibody (1:1,000; Sigma). On the following day, cells were washed three times with blocking solution and incubated with appropriate secondary antibody for 1 h and mounted for imaging. For FITC-GRGDSP experiments, confluent BAECs were briefly washed with experimental media and incubated with 400 µg/ml of FITC-GRGDSP for 2 h. After incubation the cells were washed twice with PBS to remove any excess peptide, after which they were fixed in 2% fixative for 10 min and mounted with glass coverslips for imaging. Confocal z-stacks of the cells were obtained on a LSM 510 confocal laser scanning system, using a ×40 and ×63/1.4 Oil DIC objective and analyzed using the Zeiss LSM software.
PECAM-1 knockout mice.
PECAM-1 knockout (KO) mice [B6.129S-Pecam1Gt(VICTR20)12Lex] and their WT littermates were generated by crossing PECAM-1 heterozygotes. Genotypes were confirmed by PCR. A total of 20 mice (females, 3 mo old) were used for the study. The animal use protocols were pre-approved by the Institutional Animal Care and Use Committee (IACUC) at the La Jolla Bioengineering Institute.
Immunohistochemistry and confocal microscopy.
For en face immunostaining, mice were anesthetized by intraperitoneal injection with ketamine (90 mg/kg)/xylazine (10 mg/kg) and monitored until unresponsive to toe pinch. Mice were then placed in a tray on ice, and thoracotomies were performed to expose the heart and aorta. After injection of heparin into the ventricle, mice were perfused with ice-cold PBS for 5 min to flush out the blood before fixation of tissues with ice-cold Stefanini’s/Zamboni’s buffer (2% paraformaldehyde and 15% picric acid in 0.1 M phosphate buffer, pH 7.4). After 15 min of perfusion, aortas from the renal arteries to the heart were carefully collected and placed into ice-cold PBS. Aortic arches were cleaned off, cut into smaller segments, and opened longitudinally. After an additional 20 min of fixation, tissues were washed with PBS and then permeabilized with 0.3% Triton X-100 in PBS for 20 min. Tissues were then washed and incubated with 0.2% sodium borohydride in PBS for 10 min to reduce autofluorescence. For immunostaining, tissue segments were incubated overnight at 4°C with rabbit anti-COX-2 (1:50; Cayman Chemical) and rat anti-VE-cadherin (1:50; BD Biosciences) after an initial 1-h blocking step with a PBS solution containing normal goat serum and BSA. Tissue segments were washed thoroughly with PBS and incubated for 1 h at room temperature with Alexa Fluor 488-conjugated anti-rat IgG and 568-conjugated anti-rabbit IgG secondary antibodies (1:200; Life Technologies). After final PBS washes, vessels were flat mounted with luminal side facing up between a glass slide and coverslip with VECTASHIELD aqueous mounting medium (Vector Laboratories). Images were acquired on a LSM5 PASCAL confocal fluorescence microscope (Carl Zeiss) equipped with a Plan Apochromatic 63/1.4 numerical aperture oil immersion objective.
COX-2 intensity measurement.
Because COX-2 is expected to be low or absent in the junction area, background intensity was measured in the junction as shown in (Fig. 1). For each image, the junction area was selected in the VE-cadherin channel using ImageJ’s threshold function. The selection was then transferred to the COX-2 channel and the mean intensity within the selection was taken as the background intensity. After the background intensity was subtracted, the mean intensity was measured in the COX-2 channel using ImageJ.
Fig. 1.

Measurement of background intensity. A: VE-cadherin channel. B: original cyclooxygenase-2 (COX-2) channel. C: using the threshold function in ImageJ, the junction area is selected in the VE-cadherin channel. In the junction area, COX-2 is expected to be low or absent. D: the selection is then transferred to the COX-2 channel and the mean intensity within the selection is taken as the background intensity. E: background-corrected COX-2 channel.
PGI2 measurement.
Acute β-adrenergic stimulation with dobutamine (Dob) has been shown to increase heart rate and cardiac output in murine hearts (51). Dobutamine hydrochloride (cat. no. 0515; Tocris, Bristol, UK) was dissolved in sterile distilled H2O to a stock concentration of 3 mg/ml by gently heating at 37°C for 15 min and vortexing, which was then diluted 1:10 in sterile 0.9% NaCl to a working concentration of 0.3 μg/μl. Mice were given a single bolus of Dob via intraperitoneal injection at a dose of 1.5 μg/g body weight or an equivalent volume of 0.9% saline at 5 μl/g body weight as a vehicle control. After 30 min, blood was collected retro-orbitally into microcentrifuge tubes containing 3.8% sodium citrate (final blood to sodium citrate ratio of 9:1) by using heparinized capillary collection tubes. Citrated blood samples were then centrifuged at 2,000 g for 10 min at 4°C to remove red blood cells. Supernatants were carefully taken and centrifuged at 13,000 g for 3 min at 4°C to produce platelet-free plasma. The levels of prostacyclin in WT and KO mice plasma samples were measured by prostacyclin breakdown product 6-keto-prostaglandin F1α using an enzyme immunoassay kit (Abcam).
Statistical analysis.
All experimental data are presented as means ± SE from at least three independent experiments. Data sets were analyzed for statistical significance using a Student's t-test with a two-tailed distribution and P < 0.05 was considered statistically significant. Comparisons of interactions between groups were accomplished using two-way ANOVA analysis with Tukey’s post hoc correction test; again P < 0.05 was considered significant.
RESULTS
Selective removal of HS, HA, and CS using EGL-degrading enzymes had no inhibitory effect on the FSS upregulation of COX-2 expression and release of PGI2.
We first observed the effects of degrading EGL components CS, HA, and HS using moderate and high doses on COX-2 protein expression (Fig. 2A). Western blots revealed that enzymatic removal of CS, HA, and HS had no inhibitory effect on shear-induced COX-2 (Fig. 2B). To confirm these results, the shear-induced release of PGI2 after EGL degradation was measured for high-dose samples and compared with untreated static controls. Similar to our earlier findings using lower doses of each enzyme (45), treatment with higher doses of Hep III, Hyal., and Chond. had no significant inhibitory effect (Fig. 2C). Thus our findings provide further evidence that the EGL does not play a role in PGI2 mechanotransduction. Surprisingly, cells treated with a higher dose of CS enzyme displayed a significantly increased shear-induced COX-2 protein expression and PGI2 release (Fig. 2, B and C).
Fig. 2.

Effects of degrading endothelial glycocalyx layer (EGL) components chondroitin sulfate (CS), hyaluronic acid (HA), and heparan sulfate (HS) with low and high doses of EGL enzymes on shear-induced COX-2 protein expression and prostaglandin I2 (PGI2) release in response to fluid shear stress (FSS). A: Western blots showing FSS induction COX-2 protein in bovine aortic endothelial cells (BAECs) under all treatment conditions compared with their static counterparts (n = 3–4). B: densitometric analyses of westerns under all treatment conditions compared with untreated static controls (n = 3–4). C: FSS-induced PGI2 production in BAECs treated with EGL-degrading enzymes (n = 3–4). Values are means ± SE. *P < 0.05 vs. untreated static (S) controls. #P < 0.05 vs. untreated flow (F) controls.
HS proteoglycan SDC4 knockdown has no inhibitory effect on the FSS upregulation of COX-2.
SDC4 was knocked down in BAECs by siRNA and COX-2 protein expression was measured. BAECs treated with SDC4 siRNA followed by exposure to 5 h of FSS resulted in no significant inhibitory effect on the flow-induced upregulation of COX-2 (Fig. 3C). These results indicate that SDC4 is not involved in the integrin-mediated FSS upregulation of COX-2.
Fig. 3.

Syndecan-4 (SDC4) is not involved in the integrin-mediated FSS induction of COX-2 protein expression in endothelial cells (ECs). A: BAECs were transiently transfected with negative control siRNA or SDC4 siRNA sequences using Lipofecatamine RNAiMAX. RT-PCR was used to measure the efficiency of SDC4 knockdown using the three designed siRNA sequences. All SDC4 siRNA sequences were highly effective in inhibiting SDC4 expression. However, duplex 1 inhibited SDC4 expression by 92%, and was used in all experiments. B: Western blots showing FSS induction of COX-2 protein in BAECs treated with control siRNA (left) and SDC4 siRNA (right) compared with their static counterparts. C: densitometric analyses of the Western blots compared with control siRNA-treated controls (n = 3). Densitometric analysis was accomplished by dividing the target protein densitometric units by β-actin (loading control) densitometric units to normalize for loading and then normalizing by the control siRNA static (S) controls. Values are means ± SE. *P < 0.05 vs. control siRNA or static (S) controls.
Primary Cilia are not required for FSS-induced upregulation of COX-2 expression.
To evaluate whether the primary cilium is a mechanosensor involved in the shear stress-mediated upregulation of COX-2 protein expression in ECs, BAECs were transfected with siRNA that targeted the IFT88/Polaris gene to prevent ciliogensis. Analysis of Western blots confirmed that IFT88/polaris protein expression was reduced by ~99% compared with controls (Fig. 4A). Immunofuorescence analysis confirmed that IFT88/Polaris depletion inhibited ciliogensis by >60%, compared with controls (Fig. 4, B–D). We then investigated the effects of IFT88/Polaris knockdown on the FSS induction of COX-2. BAECs treated with IFT88/Polaris siRNA displayed no inhibition of the FSS induction of COX-2 (Fig. 5, A and B). These results demonstrate that primary cilia are not involved in the induction of COX-2 in response to FSS in ECs.
Fig. 4.

Knockdown of IFT88/Polaris prevents ciliogensis in cultured ECs. A: Western blot images showing effect depletion of IFT88 protein levels in ECs transfected with IFT88 siRNA compared with negative control siRNA. B: quantification of immunofluorescent images showing number of ciliated cells 72 h following transfection. C: maximum projection of a confocal z-stack shows control siRNA-treated ECs stained for primary cilia extending from the apical surface marked by acetylated α-tubulin (green), and DNA is stained with DAPI (blue). D: maximum projection of IFT88 siRNA-transfected cells showing the absence of primary cilia on the cell surface as marked by acetylated α-tubulin staining (n = 3). Values are means ± SE. ****P < 0.0001 vs. control.
Fig. 5.
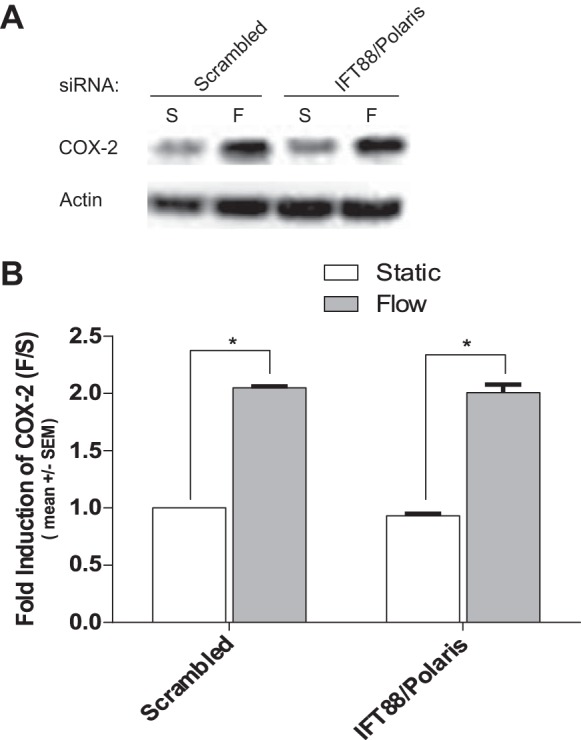
Primary cilia are not required for the FSS induction of COX-2 in ECs. A: Western blots showing FSS induction COX-2 protein expression in BAECs occurred in the presence and absence of primary cilia. BAECs were exposed to 10 dyn/cm2 of FSS for 5 h post 72 h of transfection with IFT88 siRNA. B: densitometric analysis of Western blots showing depletion of primary cilia had no significant inhibitory effect on the FSS induction of COX-2 in BAECs. Densitometric analysis was accomplished by dividing the target protein densitometric units by β-actin (loading control) densitometric units to normalize for loading, and then normalizing by the control siRNA-treated controls. Values are means ± SE. *P < 0.05 vs. control siRNA or static (S) controls.
PECAM-1 is required for the FSS induction of COX-2 and PGI2.
To investigate the possible role of PECAM-1 in the FSS induction of COX-2 and PGI2, BAECs were transfected with PECAM-1 siRNA sequences for bovine (58) and exposed to 5 h of FSS. As demonstrated in Fig. 6A, PECAM-1 siRNA reduced PECAM-1 protein expression by >90% compared with controls. We then determined the effects of PECAM-1 knockdown on the FSS induction of COX-2 protein. Western blot analysis demonstrated that COX-2 protein expression was significantly inhibited compared with flow-treated siRNA controls (Fig. 6B). These results indicate that PECAM-1 is a sensor in the mechanotransduction cascade responsible for FSS-induced COX-2 upregulation.
Fig. 6.

Knockdown of platelet endothelial cell adhesion molecule-1 (PECAM-1) expression inhibits COX-2 protein expression in BAECs and human umbilical vein endothelial cells (HUVECs). A: Western blots showing the expression of PECAM-1 after treatment with control and PECAM-1 siRNA in BAECs (n = 3). B: Western blots showing COX-2 expression in BAECs treated with control or PECAM-1 siRNA for 72 h before exposure to FSS for 5 h (n = 4). C: Western blots showing the expression of PECAM-1 after treatment with control and PECAM-1 siRNA, compared with HUVECs treated with control siRNA (n = 3). D: COX-2 expression in HUVECs treated with control or PECAM-1 siRNA for 72 h before exposure to FSS for 5 h (n = 4) compared with controls. Densitometric analysis was accomplished by dividing the target protein densitometric units by β-actin (loading control) densitometric units to normalize for loading and then normalizing by the control siRNA-treated controls. Values are means ± SE. *P < 0.05 vs. untreated static (S) controls. #P < 0.05 vs. untreated flow (F) controls.
Next, we tested the robustness of this conclusion in HUVECs, using a human PECAM-1 siRNA sequence previously described (58). Similarly, PECAM-1 expression was reduced by >80% in transfected HUVECs (Fig. 6C). HUVECs were then exposed to 5 h of FSS, and analysis of Western blots revealed that COX-2 expression was also significantly inhibited in HUVECs (Fig. 6D). We then confirmed that the inhibition of COX-2 after PECAM-1 knockdown also resulted in a significant reduction in PGI2 release in both BAECs (Fig. 7A) and HUVECs (Fig. 7B).
Fig. 7.

PECAM-1 is required for the FSS-induced production of PGI2 ECs. Cells were transfected with PECAM-1 siRNA 72 h before exposure to FSS. Experimental media were collected and assayed for PGI2 stable metabolite 6-keto-prostaglandin F1α. A: FSS-induced PGI2 production in BAECs treated with PECAM-1 siRNA (n = 4). B: FSS-induced PGI2 production in HUVECs treated with PECAM-1 siRNA (n = 4). Values are means ± SE. *P < 0.05 vs. untreated static (S) controls. #P < 0.05 vs. untreated flow (F) controls.
The results described above demonstrate, for the first time, that PECAM-1 is a mechanosenor responsible for transmission of FSS across the plasma membrane, leading to the activation of downstream, intracellular pathways that upregulate COX-2 expression and release of PGI2 in ECs.
The PI3K pathway is downstream of PECAM-1.
To investigate which intracellular pathways may be involved, we first examined the PI3K pathway, since previous studies have demonstrated that the activation of PI3K is downstream of PECAM-1 (12, 61) and is also capable of mediating COX-2 expression (59). BAECs were pretreated with the pharmacological PI3K inhibitor LY294002 (LY29, 30 µM) before exposure to 5 h of FSS. Analysis of Western blots confirmed that LY29 significantly inhibited COX-2 protein expression (Fig. 8A) and PGI2 release (Fig. 8C). RT-PCR data also confirmed similar reduction in COX-2 mRNA expression in response to FSS (Fig. 8B). Taken together these results confirm, for the first time, that a PECAM-1/PI3K pathway is a major mechanosensory pathway by which FSS is transduced to upregulate COX-2 and PGI2 in ECs.
Fig. 8.

Inhibition of the phosphatidylinositol 3-kinase (PI3K) pathway inhibits the FSS upregulation of COX-2 protein and mRNA and release of PGI2 in ECs. A: COX-2 protein expression after BAECs were pretreated for 30 min with the PI3K inhibitor LY29 (30 μM) or with DMSO vehicle before exposure to fluid flow for 5 h (n = 4). B: FSS-induced COX-2 mRNA expression after treatment with the PI3K inhibitor LY29 (30 μM) and exposure to FSS for 5 h (n = 4). C: FSS-induced PGI2 production after treatment with the PI3K inhibitor LY29 (30 μM) and exposed to FSS for 5 h (n = 4). Values are means ± SE. *P < 0.05 vs. untreated static (S) controls. #P < 0.05 vs. untreated flow (F) controls.
FSS-induced activation of the α5β1-integrin is dependent on the PECAM-1/PI3K pathway.
Earlier studies have implicated the PECAM-1/PI3K pathway in the activation of integrins in several cell types including ECs exposed FSS for short time points (12, 42, 61). Therefore, we first confirmed that PECAM-1 mediates the FSS-induced activation of α5β1-integrin in BAECs and HUVECs. Activation of α5β1-integrin was assessed by monitoring the binding of GST-FNIII9–11 as previously described (43). Analysis of Western blots demonstrated that knockdown of PECAM-1 in both BAECs (Fig. 9A) and HUVECs (Fig. 9B) significantly reduced GSTFNIII9–11 binding after exposure to FSS, when compared with vehicle-treated controls. We then confirmed that inhibition of PI3K also effectively inhibited integrin activation in BAECs. Cells were pretreated with the PI3K inhibitor LY294002 (LY29; 30 µM) before exposure to 5 h of FSS. Analysis of Western blots confirmed that inhibition of PI3K significantly reduced GSTFNIII9–11 binding after FSS (Fig. 9C). Taken together these results suggest that shear stress activation of PECAM-1/PI3K mediates the activation of integrins leading to the upregulation of COX-2.
Fig. 9.

Knockdown of PECAM-1 and inhibition of PI3K inhibits FSS activation of integrin α5β1 in ECs. A: FSS activates integrins in BAECs that is blocked in PECAM-1 siRNA-treated BAECs. Integrin activation was assayed by measuring GSTFNIII9–11 binding via Western blots (n = 4). B: FSS activates integrins in HUVECs that is blocked in PECAM-1 siRNA-treated HUVECs (n = 4). C: integrin activation in BAECs pretreated for 30 min with the PI3K inhibitor LY29 (30 μM) or with DMSO vehicle before exposure to fluid flow for 5 h (n = 4). Densitometric analysis was accomplished by dividing the target protein densitometric units by β-actin (loading control) densitometric units to normalize for loading and then normalizing by the control siRNA-treated controls. Values are means ± SE. *P < 0.05 vs. untreated static (S) controls. #P < 0.05 vs. untreated flow (F) controls.
Phosphorylation of FAK and p38, but not ERK, mediates the induction of COX-2 protein expression in EC response to FSS.
It is well established that FAK is capable of regulating integrin-mediated signaling in response to FSS (7, 8, 27, 35, 56, 62, 65). As shown in Fig. 10A, phosphorylation of FAK at Tyr-397 peaked at 5 min and returned to basal levels 60 min after the application of FSS, while phosphorylation at Tyr-925 was not activated. FSS-induced phosphorylation of the MAPKs ERK1/2 and p38 peaked at 30 min and returned to basal levels at 60 min after the onset of fluid flow (Fig. 10, B and C). These results indicate that FSS induces phosphorylation of FAK, ERK, and p38.
Fig. 10.

FSS-induced phosphorylation of focal adhesion kinase (FAK), ERK, and p38 is time dependent. BAECs were exposed to a time course of FSS lysed and analyzed by Western blotting. Western blots were probed for both phosphorylated and unphosphorylated kinase levels of the following: A: FAK at phosphorylation sites Tyr397 and Tyr925 in the presence and absence of the inhibitor PF-228 (10 uM) (n = 3, each time point); B: ERK in the presence and absence of the inhibitor PD980059 (10 uM) (n = 5, each time point); C: p38 in the presence and absence of the inhibitor SB203580 (10 uM) (n = 3, each time point). Densitometric analysis was accomplished by dividing the target protein densitometric units by β -actin (loading control) densitometric units to normalize for loading and then normalizing by the control (time 0). Values are means ± SE. *P < 0.05 vs. untreated static (control 0 min) controls. #P < 0.05 vs. untreated flow (F) controls.
To further identify the relevant pathways involved in the FSS upregulation of COX-2 at 5 h, we turned our attention to the early signaling pathways downstream of flow-induced integrin activation. Previous studies have shown that both MAPKs ERK1/2 and p38 are phosphorylated downstream of FAK activation (38, 39) and are also involved in the FSS upregulation of COX-2 in ECs (4, 21, 35, 40, 62, 65). Additionally, FAK inhibitors have been shown to inhibit the activation of MAPKs (57, 63). To determine whether the FSS-induced activation of FAK and phosphorylation of p38 and ERK1/2 are involved, BAECs were pretreated with the FAK inhibitor PF-228, MEK inhibitor PD58059, and p38 inhibitor SB203580 before exposure to 5 h of FSS to inhibit flow-induced phosphorylation. Pretreatment of BAECs with the FAK inhibitor PF-228 (10 µM) completely inhibited the FSS induction of COX-2 in BAECs (Fig. 11A), and the p38 inhibitor SB203580 (10 µM) partially, but significantly, inhibited COX-2 expression (Fig. 11B). In contrast, treatment with ERK1/2 MEK inhibitor PD98059 (10 μM) had no inhibitory effect (Fig. 11C). These results suggest that the rapid and transient phosphorylation of FAK and p38 is sufficient to regulate the transcriptional induction of COX-2 mRNA (4, 40, 62) (Fig. 8B) and translation of COX-2 protein (8, 65) (Fig. 8A) in ECs exposed to 5 h of FSS.
Fig. 11.

FAK, and p38 MAPK, but not ERK are required for FSS induction of COX-2 in BAECs. BAECs were pretreated with the following inhibitors 1-h before exposure to 5 h of FSS. A: COX-2 protein expression in BAECs pretreated with the FAK inhibitor PF-228 (10 μM) before exposure to 5 h of FSS. B: the p38 MAPK inhibitor SB2003980 (10 μM). C: the ERK and MEK inhibitor PD98059 (10 μM). Densitometric analysis was accomplished by dividing the target protein densitometric units by β-actin (loading control) densitometric units to normalize for loading and then normalizing by the vehicle-treated controls. Values are means ± SE. *P < 0.05 vs. untreated static (S) controls. #P < 0.05 vs. un-treated flow (F) controls.
COX-2 expression and PGI2 plasma levels in PECAM-1-deficient mice.
To address the validity of our in vitro results in an animal model, we conducted experiments in PECAM-1-deficient and WT control mice. First, we measured COX-2 expression by immunofluorescence staining in the aortas of KO and WT animals. The results are displayed in Fig. 12 where it is shown that there was a significant 1.37-fold higher level of COX-2 immunofluorescence intensity in the WT vs. the KO animals. PGI2 levels were measured in WT and KO animals after a brief saline injection that served as a control for our dobutamine experiments, and the PGI2 concentration measured in plasma samples was a significant 1.49-fold higher in the WT vs the KO animals (Fig. 13). To simulate a sudden increase in shear stress in the vascular system, we injected the animals with an acute bolus of dobutamine to elevate heart rate and cardiac output (51). After 30 min, plasma samples were drawn for PGI2 analysis. We observed a significant 2.53-fold increase in plasma PGI2 levels for the WT animals injected with dobutamine relative to WT animals injected with a control saline bolus (Fig. 13). For the same experiment in the KO animals, there was no difference in plasma PGI2 levels between dobutamine- and saline-injected animals. These results confirm in an animal model that PECAM-1 plays a role in controlling basal levels of COX-2 and PGI2, and the response of PGI2 to elevated shear stress is controlled by PECAM-1.
Fig. 12.

COX-2 staining in wild-type (WT) vs PECAM-1 knockout (KO) mice. Aortic samples were immunostained for COX-2 (green) and VE-cadherin (red) and background correction performed as indicated in materials and methods. Representative images of immunostaining in WT (A) and PECAM-1 KO (B) are shown. C: mean intensity of COX-2 staining was 1.37 times higher in WT than in than KO. Average ± SE are shown. *P = 0.00182; n = 26 images from 3 animals for WT; n = 38 images from 3 animals for KO.
Fig. 13.
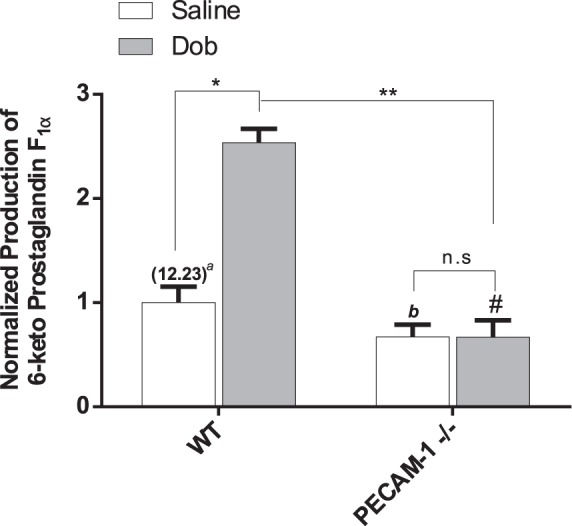
In vivo prostaglandin release in WT and PECAM-1−/− mice. In vivo PGI2 levels determined by measuring 6-keto-prostaglandin F1α levels in plasma 30 min after single bolus intraperitoneal injection of vehicle (Saline) or dobutamine (Dob; 1.5 µg/g body weight). aIn parentheses is the normalizing factor (ng/ml). Representative results from at least 4 separate animals are shown. Values are mean ± SE. *P < 0.0001 vs. WT saline-treated controls. #P < 0.0001 vs. WT saline-treated controls. bP < 0.0001 vs. WT saline-treated controls. **P < 0.0001 vs. WT Dob-treated controls. n.s, Nonsignificant.
DISCUSSION
Flow-induced mechanotransduction is particularly important in arteries, where blood flow regulates vascular structure and function. The goal of the present study was to elucidate the mechanisms involved in the fluid shear stress-mediated mechanotransduction of COX-2 expression and PGI2 release in ECs. Several primary mechanosensors, EGL, SDC4, primary cilia, and PECAM-1, were considered. Downstream activation of the signaling molecules PI3K, FAK, and MAPKs were studied due to their association with FSS-induced cellular signaling in previous studies (1, 29, 30, 48, 61, 65).
Although we demonstrated that almost complete removal of HS, CS, and HA from the EGL by enzymes did not inhibit COX-2 protein expression or PGI2 release (Fig. 2), cells treated with a high dose of CS enzyme actually displayed a significant increase in shear-induced COX-2 protein expression and PGI2 release. This enhancement is consistent with a study in femoral arteries of adult rabbits showing that addition of CS reduced several proinflammatory mediators including COX-2 protein and gene expression (24). Several other studies have shown that CS is a natural GAG with a potent anti-inflammatory influence (18, 23, 60) and that COX-2 mediates an acute inflammatory response (53). Consistent with these studies, selective removal of CS appears to have induced an acute inflammatory response characterized by the significant increase of FSS-induced COX-2 protein expression and PGI2 release observed in the present study.
To further explore the potential role of the glycocalyx in COX-2/PGI2 mechanotransduction, we considered the basal proteoglycan SDC4 whose GAG chains would not be accessible to the GAG-degrading enzymes applied apically to confluent monolayers. In vitro studies have demonstrated the synergistic relationship between the syndecan and integrin cascades in response to nonmechanical stimuli (2, 3, 36). Our observations indicate that SDC4 has no effect on the shear-induced upregulation of COX-2 in ECs (Fig. 3), although it has been shown to play a role in integrin trafficking to the cell membrane, focal adhesion assembly and cell migration, as demonstrated by Bass et al. (3), using mice fibroblast.
Primary cilia are present on EC in vivo and in vitro and have been shown to have potential involvement in shear-induced calcium transients and regulation of Kruppel-like factor 2 (KLF2) gene expression (17). The possible role of primary cilia in shear-induced COX-2/PGI2 expression in ECs has not been considered previously. However, in vitro studies in bone cells demonstrated that shear-induced COX-2 mRNA expression was completely inhibited in the absence of primary cilia (25, 34). Our studies have now shown (Fig. 5) that primary cilia are not involved in shear-induced COX-2/PGI2 expression in ECs.
PI3K has been implicated in the “inside out” activation of integrins in several cell types (26, 43, 61). Tzima et al. (61) exposed sparsely plated BAECs to direct mechanical force using twisting of antibody coated magnetic beads for short exposures up to several minutes and showed that PECAM-1 was required for the activation of αvβ3-integrin. Orr et al. (43) showed that short-term exposures (10–30 min) to shear-induced α5β1-activation requiring PI3K. From these studies PECAM-1 might be predicted to be important for α5β1-activation by flow, but this had not been shown definitively. Now we have observed that knockdown of PECAM-1 or inhibition of PI3K significantly inhibited major α5β1 activation after 5 h of fluid flow (Fig. 9, A–C). Since integrin activation pathways could differ temporally with shear stress, our data at 5 h provide novel information as to the nature of shear-induced integrin signaling. Perhaps more importantly, we also observed that inhibition of PI3K significantly inhibited COX-2 protein and mRNA expression and PGI2 release (Fig. 8, A–C). This finding in ECs exposed to shear is novel but consistent with other studies showing that PI3K suppression inhibited the induction of COX-2 in osteoblasts exposed to ultrasound and in migrating ECs (59, 65).
Additionally, we observed that PECAM-1 knockdown significantly attenuated shear-induced COX-2 protein expression and PGI2 production in both BAECs and HUVECs (Figs. 6 and 7). It is noticeable that the reduction in PGI2 production is less in HUVECs than in BAECs. This may be due to differences in PGI2 production mediated by COX-2 and COX-1 enzymes in the two cell types. For example, Bolego et al. (6) showed that COX-1 is a major contributor to PGI2 production in HUVECs.
Integrins mediate both “inside out” and “outside in” signaling by activating FAK and MAPK in response to FSS in several cell types including ECs (7, 8, 27, 56, 62, 65). More directly, early studies have demonstrated the involvement of FAK in the activation of MAPK via flow-induced activation of β1-integrin in ECs (29). Additionally, a recent study by Yurdagul et al. (64) demonstrated that oxidized low-density lipoprotein-mediated autophosphorylation of FAK at the Tyr397 site was completely inhibited in ECs treated with α5β1-blocking antibodies, suggesting that FAK signaling is integrin mediated. In ECs, FSS has been shown to cause rapid and transient phosphorylation of FAK and MAPKs ERK and p38 (29, 35). Consistent with these studies, we found that the FSS-induced phosphorylation of FAK peaked at 5 min and returned to basal levels by 60 min, while MAPKs ERK and p38 both peaked at 30 min and returned to basal levels by 60 min (Fig. 10).
There is abundant evidence in the literature supporting the involvement of the FAK and p38 MAPK signaling pathways in the transcription and translation of COX-2 in ECs (4, 21, 35, 40, 62, 65). More directly, previous studies have provided evidence that COX-2 gene and protein expression is dependent on a fibronectin-α5β1/FAK/p38-signaling pathway in ECs (21, 62, 65), although these studies did not investigate the effects of FSS on the upregulation of these pathways. We observed that inhibition of the FAK signaling pathway also completely blocked the FSS-induced expression of COX-2 at 5 h (Fig. 11A). Furthermore, the shear-induced upregulation of COX-2 was attributable to MAPK p38, as inhibition of this pathway (Fig. 11B), but not ERK (Fig. 11C), inhibited COX-2 protein expression in response to FSS. Thus it appears that the FSS upregulation of COX-2 expression proceeds through a mechanism involving integrin-associated FAK and the p38 MAPK. Interestingly, this same mechanism (integrin/FAK/MAPK) has been shown to activate COX-2 protein and gene expression in ECs exposed to chemical stimuli (8, 21, 40) and in studies monitoring integrin-extracellular matrix mechanotransduction (62, 65). Hence, our data suggest that PI3K regulated activation of integrins leads to the downstream activation of integrin-associated FAK, followed by the activation of p38 MAPK and subsequent upregulation of COX-2 gene and protein expression in response to FSS in ECs.
The results for COX-2 immunostaining and 6-keto-prostaglandin F1α plasma concentrations in PECAM-1 KO and WT mice provide further support for our overall claim that PECAM-1 is a mechanotransducer that mediates shear (flow)-induced upregulation of COX-2 and PGI2. Immunostaining (Fig. 12) shows the characteristic nuclear/perinuclear localization of COX-2 that has been described in previous studies (46). The basal level of 6-keto-prostaglandin F1α plasma concentrations that we report in Fig. 13 (12.3 pg/ml) is of the same order of magnitude as reported in healthy humans (3 pg/ml) using precise mass spectrophotometry measurements (5). Our observation that increased flow induced by dobutamine significantly increased plasma PGI2 levels in WT animals is consistent with other studies in humans (54) and in mice (50). Our finding that this flow-induced increase is completely inhibited in the PECAM-1 KO animals is, however, completely novel.
These results demonstrate for the first time that PECAM-1 is an important mechanosensor responsible for the FSS upregulation of COX-2 and PGI2 as summarized in Fig. 14. Integrin α5β1 is activated by a PI3K-mediated “inside out” signaling cascade leading to early activation of FAK/MAP:p38, sustained transcription of COX-2 mRNA, and translation of COX-2 protein expression and PGI2 production. Hence, we propose that FSS activation of this PECAM-1-initiated pathway is the major mechanotransduction route responsible for the FSS-induced transcription and translation of COX-2 expression and release of PGI2 in ECs.
Fig. 14.

Proposed model for PECAM-1/PI3K-mediated activation of the integrins FAK, p38, COX-2, and PGI2 in ECs exposed to FSS. Based on our findings we propose that the shear-induced upregulation of COX-2 and release of PGI2 is mediated by a PECAM-1-mediated/PI3K integrin activation complex. FSS activation of PECAM-1 results in the phosphorylation of PI3K, which subsequently activates integrins. Upon integrin activation FAK is phosphorylated, leading to the downstream phosphorylation of MAPK p38. Phosphorylation of the MAPK signaling pathway leads to the transcription of COX-2 mRNA and posttranscriptional synthesis of COX-2 protein and PGI2 release in ECs exposed to FSS.
However, further studies will be required to determine the mechanism by which mechanical forces applied to the cell surface lead to the activation of cell junction protein PECAM-1 in confluent ECs. One possibility was described by dela Paz et al. (14), who showed that the mechanosensitive G protein subunits G-αq and -11 (Gαq/11) that have been independently proposed as a mechanosensor form a mechanosensitive PECAM-1-Gαq/11 complex that contains an endogenous HSPG with HS chains that are critical for junctional complex assembly and regulation of the flow response. However, they also showed that treatment with heparinase disrupted the mechanosensitive complex. Since we have shown that heparinase treatment does not affect shear-induced COX-2/PGI2, it is not likely that this complex is important for the shear response of COX-2/PGI2. Osawa et al. (44) showed that pulling on PECAM-1 with magnetic beads coated with antibody to the external domain of PECAM-1 in sparsely coated EC surfaces induced ERK1/2 activation requiring tyrosine phosphorylation of PECAM-1. Our studies showed that although shear stress applied to confluent cell monolayers induced ERK1/2 activation (Fig. 9), this was not necessary for the shear-induced upregulation of COX-2 (Fig. 10C) that is mediated by PECAM-1 (Fig. 6, B and D). This suggests, but does not prove, that shear-induced tyrosine phosphorylation of PECAM-1 is not required for COX-2/PGI2 induction. Because ERK 1/2 activation is only one possible consequence of PECAM-1 phosphorylation, another arm of PECAM-1 signaling activated in response to PECAM-1 phosphorylation could be operative. Alternatively, there may be significant differences in intracellular signaling mechanisms in sparse and confluent monolayers that have well-formed intercellular junctions where PECAM-1 is enriched. To address this point, it would be valuable to study the response of confluent EC to direct force application to PECAM-1.
The role of PECAM-1 mechanotransduction to produce PGI2 in low shear and disturbed flow regions that are associated with atherosclerosis remains to be elucidated. Potter et al. (49) showed that exposure of porcine ECs in vitro to low levels of shear stress (<2 dyn/cm2) for up to 7 days was characterized by low COX-2 expression, whereas our data in BAECs exposed to a higher shear stress of 10 dyn/cm2 show increasing COX-2 expression up to 5 h (Fig. 15). Conway et al. (13) reported a 16.8-fold increase in COX-2 gene expression for HUVECs exposed to 15 dyn/cm2 steady shear stress compared with static controls at 24 h. One interpretation of these results might be that high shear (antiatherogenic) induces COX-2 expression whereas low shear stress (proatherogenic) suppresses COX-2. This point of view has also been expressed by Gimbrone et al. (22). However, Conway et al. (13) also reported an 11.4-fold increase in COX-2 gene expression after exposure of HUVECs to 24 h reversing oscillatory flow compared with static controls, and Passerini et al. (47) reported a 1.55-fold increase in COX-2 gene expression in the inner curvature of the aortic arch (disturbed flow) compared with the descending aorta (undisturbed flow) in pigs. Taken together, the data summarized above suggest that different mechanisms of COX-2 production may be operative in disturbed/reversing flow and undisturbed/steady flow environments. Another possibility is that the same hemodynamic characteristic is controlling in both cases. For example, even in reversing flow where the mean value of the shear stress is low, the time average of the absolute value may in fact be high. This distinction in shear stress characterization has been discussed thoroughly in Mohamied et al. (37).
GRANTS
This work was supported by the National Institutes of Health Research Supplement to Promote Diversity in Health Related Research Grant PA-08–190 (to S. Russell-Puleri) and the National Heart, Lung, and Blood Institute Grants R01-HL-094889 (to J. M. Tarbell), MERIT Award R37-HL-040696 (to J. A. Frangos), and R01-HL-098435 and R01 HL133497 (to A. W. Orr).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.R.-P. conceived and designed research; S.R.-P., N.G.d.P., D.A., M.C., and L.M.C. performed experiments; S.R.-P. analyzed data; S.R.-P., M.C., A.W.O., and J.A.F. interpreted results of experiments; S.R.-P. and L.M.C. prepared figures; S.R.-P. drafted manuscript; S.R.-P., N.G.d.P., E.E., A.W.O., J.A.F., and J.M.T. edited and revised manuscript; S.R.-P., A.W.O., J.A.F., and J.M.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge Linden Langhorne for design improvements of our rotating disk system and Dr. Gilda Barabino and Shereka Banton for generously providing the HUVECs used in our work.
REFERENCES
- 1.Alenghat FJ, Ingber DE. Mechanotransduction: all signals point to cytoskeleton, matrix, and integrins. Sci STKE 2002: pe6, 2002. doi: 10.1126/stke.2002.119.pe6. [DOI] [PubMed] [Google Scholar]
- 2.Baeyens N, Mulligan-Kehoe MJ, Corti F, Simon DD, Ross TD, Rhodes JM, Wang TZ, Mejean CO, Simons M, Humphrey J, Schwartz MA. Syndecan 4 is required for endothelial alignment in flow and atheroprotective signaling. Proc Natl Acad Sci USA 111: 17308–17313, 2014. doi: 10.1073/pnas.1413725111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bass MD, Williamson RC, Nunan RD, Humphries JD, Byron A, Morgan MR, Martin P, Humphries MJ. A syndecan-4 hair trigger initiates wound healing through caveolin- and RhoG-regulated integrin endocytosis. Dev Cell 23: 1081–1082, 2012. doi: 10.1016/j.devcel.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Binion DG, Otterson MF, Rafiee P. Curcumin inhibits VEGF-mediated angiogenesis in human intestinal microvascular endothelial cells through COX-2 and MAPK inhibition. Gut 57: 1509–1517, 2008. doi: 10.1136/gut.2008.152496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blair IA, Barrow SE, Waddell KA, Lewis PJ, Dollery CT. Prostacyclin is not a circulating hormone in man. Prostaglandins 23: 579–589, 1982. doi: 10.1016/0090-6980(82)90118-6. [DOI] [PubMed] [Google Scholar]
- 6.Bolego C, Buccellati C, Prada A, Gaion RM, Folco G, Sala A. Critical role of COX-1 in prostacyclin production by human endothelial cells under modification of hydroperoxide tone. FASEB J 23: 605–612, 2009. doi: 10.1096/fj.08-106591. [DOI] [PubMed] [Google Scholar]
- 7.Burridge K, Fath K. Focal contacts: transmembrane links between the extracellular matrix and the cytoskeleton. BioEssays 10: 104–108, 1989. doi: 10.1002/bies.950100403. [DOI] [PubMed] [Google Scholar]
- 8.Caughey GE, Cleland LG, Penglis PS, Gamble JR, James MJ. Roles of cyclooxygenase (COX)-1 and COX-2 in prostanoid production by human endothelial cells: selective up-regulation of prostacyclin synthesis by COX-2. J Immunol 167: 2831–2838, 2001. doi: 10.4049/jimmunol.167.5.2831. [DOI] [PubMed] [Google Scholar]
- 9.Chien S, Li S, Shyy YJ. Effects of mechanical forces on signal transduction and gene expression in endothelial cells. Hypertension 31: 162–169, 1998. doi: 10.1161/01.HYP.31.1.162. [DOI] [PubMed] [Google Scholar]
- 10.Chiu YJ, McBeath E, Fujiwara K. Mechanotransduction in an extracted cell model: Fyn drives stretch- and flow-elicited PECAM-1 phosphorylation. J Cell Biol 182: 753–763, 2008. doi: 10.1083/jcb.200801062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chrétien ML, Zhang M, Jackson MR, Kapus A, Langille BL. Mechanotransduction by endothelial cells is locally generated, direction-dependent, and ligand-specific. J Cell Physiol 224: 352–361, 2010. doi: 10.1002/jcp.22125. [DOI] [PubMed] [Google Scholar]
- 12.Collins C, Guilluy C, Welch C, O’Brien ET, Hahn K, Superfine R, Burridge K, Tzima E. Localized tensional forces on PECAM-1 elicit a global mechanotransduction response via the integrin-RhoA pathway. Curr Biol 22: 2087–2094, 2012. doi: 10.1016/j.cub.2012.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conway DE, Williams MR, Eskin SG, McIntire LV. Endothelial cell responses to atheroprone flow are driven by two separate flow components: low time-average shear stress and fluid flow reversal. Am J Physiol Heart Circ Physiol 298: H367–H374, 2010. doi: 10.1152/ajpheart.00565.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.dela Paz NG, Melchior B, Shayo FY, Frangos JA. Heparan sulfates mediate the interaction between platelet endothelial cell adhesion molecule-1 (PECAM-1) and the Gαq/11 subunits of heterotrimeric G proteins. J Biol Chem 289: 7413–7424, 2014. doi: 10.1074/jbc.M113.542514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dogné JM, Hanson J, Pratico D. Thromboxane, prostacyclin and isoprostanes: therapeutic targets in atherogenesis. Trends Pharmacol Sci 26: 639–644, 2005. doi: 10.1016/j.tips.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 16.Ebong EE, Lopez-Quintero SV, Rizzo V, Spray DC, Tarbell JM. Shear-induced endothelial NOS activation and remodeling via heparan sulfate, glypican-1, and syndecan-1. Integr Biol 6: 338–347, 2014. doi: 10.1039/C3IB40199E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Egorova AD, van der Heiden K, Poelmann RE, Hierck BP. Primary cilia as biomechanical sensors in regulating endothelial function. Differentiation 83: S56–S61, 2012. doi: 10.1016/j.diff.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Favretto ME, Wallbrecher R, Schmidt S, van de Putte R, Brock R. Glycosaminoglycans in the cellular uptake of drug delivery vectors–bystanders or active players? J Control Release 180: 81–90, 2014. doi: 10.1016/j.jconrel.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 19.Florian JA, Kosky JR, Ainslie K, Pang Z, Dull RO, Tarbell JM. Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ Res 93: e136–e142, 2003. doi: 10.1161/01.RES.0000101744.47866.D5. [DOI] [PubMed] [Google Scholar]
- 20.Frangos JA, Eskin SG, McIntire LV, Ives CL. Flow effects on prostacyclin production by cultured human endothelial cells. Science 227: 1477–1479, 1985. doi: 10.1126/science.3883488. [DOI] [PubMed] [Google Scholar]
- 21.Garonna E, Botham KM, Birdsey GM, Randi AM, Gonzalez-Perez RR, Wheeler-Jones CP. Vascular endothelial growth factor receptor-2 couples cyclo-oxygenase-2 with pro-angiogenic actions of leptin on human endothelial cells. PLoS One 6: e18823, 2011. doi: 10.1371/journal.pone.0018823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gimbrone MA Jr, Topper JN, Nagel T, Anderson KR, Garcia-Cardeña G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann N Y Acad Sci 902: 230–239, 2000. doi: 10.1111/j.1749-6632.2000.tb06318.x. [DOI] [PubMed] [Google Scholar]
- 23.Halden Y, Rek A, Atzenhofer W, Szilak L, Wabnig A, Kungl AJ. Interleukin-8 binds to syndecan-2 on human endothelial cells. Biochem J 377: 533–538, 2004. doi: 10.1042/bj20030729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herrero-Beaumont G, Marcos ME, Sánchez-Pernaute O, Granados R, Ortega L, Montell E, Vergés J, Egido J, Largo R. Effect of chondroitin sulphate in a rabbit model of atherosclerosis aggravated by chronic arthritis. Br J Pharmacol 154: 843–851, 2008. doi: 10.1038/bjp.2008.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoey DA, Chen JC, Jacobs CR. The primary cilium as a novel extracellular sensor in bone. Front Endocrinol (Lausanne) 3: 75, 2012. doi: 10.3389/fendo.2012.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hughes PE, Pfaff M. Integrin affinity modulation. Trends Cell Biol 8: 359–364, 1998. doi: 10.1016/S0962-8924(98)01339-7. [DOI] [PubMed] [Google Scholar]
- 27.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell 69: 11–25, 1992. doi: 10.1016/0092-8674(92)90115-S. [DOI] [PubMed] [Google Scholar]
- 28.Ingber DE, Dike L, Hansen L, Karp S, Liley H, Maniotis A, McNamee H, Mooney D, Plopper G, Sims J, Wang N. Cellular tensegrity: exploring how mechanical changes in the cytoskeleton regulate cell growth, migration, and tissue pattern during morphogenesis. Int Rev Cytol 150: 173–224, 1994. doi: 10.1016/S0074-7696(08)61542-9. [DOI] [PubMed] [Google Scholar]
- 29.Ishida T, Peterson TE, Kovach NL, Berk BC. MAP kinase activation by flow in endothelial cells. Role of beta 1 integrins and tyrosine kinases. Circ Res 79: 310–316, 1996. doi: 10.1161/01.RES.79.2.310. [DOI] [PubMed] [Google Scholar]
- 30.Jalali S, del Pozo MA, Chen K, Miao H, Li Y, Schwartz MA, Shyy JY, Chien S. Integrin-mediated mechanotransduction requires its dynamic interaction with specific extracellular matrix (ECM) ligands. Proc Natl Acad Sci USA 98: 1042–1046, 2001. doi: 10.1073/pnas.98.3.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeon O, Yoo YM, Kim K, Jacobs C, Kim C. Primary cilia-mediated osteogenic response to fluid flow occurs via increases in focal adhesion and Akt signaling pathway in MC3T3-E1 osteoblastic cells. Cell Mol Bioeng 4: 379–388, 2011. doi: 10.1007/s12195-011-0191-x. [DOI] [Google Scholar]
- 32.Kawabe J, Ushikubi F, Hasebe N. Prostacyclin in vascular diseases.–Recent insights and future perspectives–. Circ J 74: 836–843, 2010. doi: 10.1253/circj.CJ-10-0195. [DOI] [PubMed] [Google Scholar]
- 33.Kirkby NS, Lundberg MH, Harrington LS, Leadbeater PD, Milne GL, Potter CM, Al-Yamani M, Adeyemi O, Warner TD, Mitchell JA. Cyclooxygenase-1, not cyclooxygenase-2, is responsible for physiological production of prostacyclin in the cardiovascular system. Proc Natl Acad Sci USA 109: 17597–17602, 2012. doi: 10.1073/pnas.1209192109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwon RY, Temiyasathit S, Tummala P, Quah CC, Jacobs CR. Primary cilium-dependent mechanosensing is mediated by adenylyl cyclase 6 and cyclic AMP in bone cells. FASEB J 24: 2859–2868, 2010. doi: 10.1096/fj.09-148007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li S, Kim M, Hu YL, Jalali S, Schlaepfer DD, Hunter T, Chien S, Shyy JY. Fluid shear stress activation of focal adhesion kinase. Linking to mitogen-activated protein kinases. J Biol Chem 272: 30455–30462, 1997. doi: 10.1074/jbc.272.48.30455. [DOI] [PubMed] [Google Scholar]
- 36.Longley RL, Woods A, Fleetwood A, Cowling GJ, Gallagher JT, Couchman JR. Control of morphology, cytoskeleton and migration by syndecan-4. J Cell Sci 112: 3421–3431, 1999. [DOI] [PubMed] [Google Scholar]
- 37.Mohamied Y, Rowland EM, Bailey EL, Sherwin SJ, Schwartz MA, Weinberg PD. Change of direction in the biomechanics of atherosclerosis. Ann Biomed Eng 43: 16–25, 2015. doi: 10.1007/s10439-014-1095-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Newman PJ, Berndt MC, Gorski J, White GC II, Lyman S, Paddock C, Muller WA. PECAM-1 (CD31) cloning and relation to adhesion molecules of the immunoglobulin gene superfamily. Science 247: 1219–1222, 1990. doi: 10.1126/science.1690453. [DOI] [PubMed] [Google Scholar]
- 39.Nikmanesh M, Shi ZD, Tarbell JM. Heparan sulfate proteoglycan mediates shear stress-induced endothelial gene expression in mouse embryonic stem cell-derived endothelial cells. Biotechnol Bioeng 109: 583–594, 2012. doi: 10.1002/bit.23302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Norata GD, Callegari E, Inoue H, Catapano AL. HDL3 induces cyclooxygenase-2 expression and prostacyclin release in human endothelial cells via a p38 MAPK/CRE-dependent pathway: effects on COX-2/PGI-synthase coupling. Arterioscler Thromb Vasc Biol 24: 871–877, 2004. doi: 10.1161/01.ATV.zhq0504.1403. [DOI] [PubMed] [Google Scholar]
- 41.Oohira A, Wight TN, Bornstein P. Sulfated proteoglycans synthesized by vascular endothelial cells in culture. J Biol Chem 258: 2014–2021, 1983. [PubMed] [Google Scholar]
- 42.Orr AW. Integrin Signaling in Shear Stress-Induced Endothelial Cell Dysfunction (Online). Cambridge, MA: Abcam plc; http://www.abcam.com/index.html?pageconfig=resource&rid=12379. [Google Scholar]
- 43.Orr AW, Ginsberg MH, Shattil SJ, Deckmyn H, Schwartz MA. Matrix-specific suppression of integrin activation in shear stress signaling. Mol Biol Cell 17: 4686–4697, 2006. doi: 10.1091/mbc.E06-04-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Osawa M, Masuda M, Kusano K, Fujiwara K. Evidence for a role of platelet endothelial cell adhesion molecule-1 in endothelial cell mechanosignal transduction: is it a mechanoresponsive molecule? J Cell Biol 158: 773–785, 2002. doi: 10.1083/jcb.200205049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pahakis MY, Kosky JR, Dull RO, Tarbell JM. The role of endothelial glycocalyx components in mechanotransduction of fluid shear stress. Biochem Biophys Res Commun 355: 228–233, 2007. doi: 10.1016/j.bbrc.2007.01.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parfenova H, Parfenov VN, Shlopov BV, Levine V, Falkos S, Pourcyrous M, Leffler CW. Dynamics of nuclear localization sites for COX-2 in vascular endothelial cells. Am J Physiol Cell Physiol 281: C166–C178, 2001. [DOI] [PubMed] [Google Scholar]
- 47.Passerini AG, Polacek DC, Shi C, Francesco NM, Manduchi E, Grant GR, Pritchard WF, Powell S, Chang GY, Stoeckert CJ Jr, Davies PF. Coexisting proinflammatory and antioxidative endothelial transcription profiles in a disturbed flow region of the adult porcine aorta. Proc Natl Acad Sci USA 101: 2482–2487, 2004. doi: 10.1073/pnas.0305938101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ponik SM, Pavalko FM. Formation of focal adhesions on fibronectin promotes fluid shear stress induction of COX-2 and PGE2 release in MC3T3-E1 osteoblasts. J Appl Physiol (1985) 97: 135–142, 2004. doi: 10.1152/japplphysiol.01260.2003. [DOI] [PubMed] [Google Scholar]
- 49.Potter CM, Lundberg MH, Harrington LS, Warboys CM, Warner TD, Berson RE, Moshkov AV, Gorelik J, Weinberg PD, Mitchell JA. Role of shear stress in endothelial cell morphology and expression of cyclooxygenase isoforms. Arterioscler Thromb Vasc Biol 31: 384–391, 2011. doi: 10.1161/ATVBAHA.110.214031. [DOI] [PubMed] [Google Scholar]
- 50.Przyborowski K, Wojewoda M, Sitek B, Zakrzewska A, Kij A, Wandzel K, Zoladz JA, Chlopicki S. Effects of 1-methylnicotinamide (mna) on exercise capacity and endothelial response in diabetic mice. PLoS One 10: e0130908, 2015. doi: 10.1371/journal.pone.0130908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Puhl SL, Weeks KL, Ranieri A, Avkiran M. Assessing structural and functional responses of murine hearts to acute and sustained β-adrenergic stimulation in vivo. J Pharmacol Toxicol Methods 79: 60–71, 2016. doi: 10.1016/j.vascn.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Resnick N, Gimbrone MA Jr. Hemodynamic forces are complex regulators of endothelial gene expression. FASEB J 9: 874–882, 1995. [DOI] [PubMed] [Google Scholar]
- 53.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 31: 986–1000, 2011. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ritter JM, Barrow SE, Blair IA, Dollery CT. Release of prostacyclin in vivo and its role in man. Lancet 1: 317–319, 1983. doi: 10.1016/S0140-6736(83)91626-4. [DOI] [PubMed] [Google Scholar]
- 55.Saoncella S, Echtermeyer F, Denhez F, Nowlen JK, Mosher DF, Robinson SD, Hynes RO, Goetinck PF. Syndecan-4 signals cooperatively with integrins in a Rho-dependent manner in the assembly of focal adhesions and actin stress fibers. Proc Natl Acad Sci USA 96: 2805–2810, 1999. doi: 10.1073/pnas.96.6.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwartz MA, Schaller MD, Ginsberg MH. Integrins: emerging paradigms of signal transduction. Annu Rev Cell Dev Biol 11: 549–599, 1995. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- 57.Shi ZD, Wang H, Tarbell JM. Heparan sulfate proteoglycans mediate interstitial flow mechanotransduction regulating MMP-13 expression and cell motility via FAK-ERK in 3D collagen. PLoS One 6: e15956, 2011. doi: 10.1371/journal.pone.0015956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tai LK, Zheng Q, Pan S, Jin ZG, Berk BC. Flow activates ERK1/2 and endothelial nitric oxide synthase via a pathway involving PECAM1, SHP2, and Tie2. J Biol Chem 280: 29620–29624, 2005. doi: 10.1074/jbc.M501243200. [DOI] [PubMed] [Google Scholar]
- 59.Tang CH, Yang RS, Huang TH, Lu DY, Chuang WJ, Huang TF, Fu WM. Ultrasound stimulates cyclooxygenase-2 expression and increases bone formation through integrin, focal adhesion kinase, phosphatidylinositol 3-kinase, and Akt pathway in osteoblasts. Mol Pharmacol 69: 2047–2057, 2006. doi: 10.1124/mol.105.022160. [DOI] [PubMed] [Google Scholar]
- 60.Tkachenko E, Rhodes JM, Simons M. Syndecans: new kids on the signaling block. Circ Res 96: 488–500, 2005. doi: 10.1161/01.RES.0000159708.71142.c8. [DOI] [PubMed] [Google Scholar]
- 61.Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437: 426–431, 2005. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 62.Viji RI, Kumar VB, Kiran MS, Sudhakaran PR. Modulation of cyclooxygenase in endothelial cells by fibronectin: relevance to angiogenesis. J Cell Biochem 105: 158–166, 2008. doi: 10.1002/jcb.21808. [DOI] [PubMed] [Google Scholar]
- 63.Wang JG, Miyazu M, Matsushita E, Sokabe M, Naruse K. Uniaxial cyclic stretch induces focal adhesion kinase (FAK) tyrosine phosphorylation followed by mitogen-activated protein kinase (MAPK) activation. Biochem Biophys Res Commun 288: 356–361, 2001. doi: 10.1006/bbrc.2001.5775. [DOI] [PubMed] [Google Scholar]
- 64.Yurdagul A Jr, Green J, Albert P, McInnis MC, Mazar AP, Orr AW. α5β1 integrin signaling mediates oxidized low-density lipoprotein-induced inflammation and early atherosclerosis. Arterioscler Thromb Vasc Biol 34: 1362–1373, 2014. doi: 10.1161/ATVBAHA.114.303863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zaric J, Rüegg C. Integrin-mediated adhesion and soluble ligand binding stabilize COX-2 protein levels in endothelial cells by inducing expression and preventing degradation. J Biol Chem 280: 1077–1085, 2005. doi: 10.1074/jbc.M410006200. [DOI] [PubMed] [Google Scholar]
- 66.Zeng Y, Ebong EE, Fu BM, Tarbell JM. The structural stability of the endothelial glycocalyx after enzymatic removal of glycosaminoglycans. PLoS One 7: e43168, 2012. doi: 10.1371/journal.pone.0043168. [DOI] [PMC free article] [PubMed] [Google Scholar]