

Elevated 20-hydroxyeicosatetraenoic acid (20-HETE) impairs coronary collateral growth (CCG) in metabolic syndrome by eliciting endothelial dysfunction and apoptosis via excessive neutrophil infiltration. 20-HETE antagonists completely restore coronary collateral growth in metabolic syndrome. microRNA-145 (miR-145) is an upstream regulator of 20-HETE production in metabolic syndrome; low expression of miR-145 in metabolic syndrome promotes elevated production of 20-HETE.
Keywords: 20-HETE, miR-145, arteriogenesis, endothelial dysfunction, neutrophils
Abstract
Coronary collateral growth (CCG) is impaired in metabolic syndrome (MetS). microRNA-145 (miR-145-Adv) delivery to our rat model of MetS (JCR) completely restored and neutrophil depletion significantly improved CCG. We determined whether low endogenous levels of miR-145 in MetS allowed for elevated production of 20-hydroxyeicosatetraenoic acid (20-HETE), which, in turn, resulted in excessive neutrophil accumulation and endothelial dysfunction leading to impaired CCG. Rats underwent 0–9 days of repetitive ischemia (RI). RI-induced cardiac CYP4F (neutrophil-specific 20-HETE synthase) expression and 20-HETE levels were increased (4-fold) in JCR vs. normal rats. miR-145-Adv and 20-HETE antagonists abolished and neutrophil depletion (blocking antibodies) reduced (~60%) RI-induced increases in CYP4F expression and 20-HETE production in JCR rats. Impaired CCG in JCR rats (collateral-dependent blood flow using microspheres) was completely restored by 20-HETE antagonists [collateral-dependent zone (CZ)/normal zone (NZ) flow ratio was 0.76 ± 0.07 in JCR + 20-SOLA, 0.84 ± 0.05 in JCR + 20-HEDGE vs. 0.11 ± 0.02 in JCR vs. 0.84 ± 0.03 in normal rats]. In JCR rats, elevated 20-HETE was associated with excessive expression of endothelial adhesion molecules and neutrophil infiltration, which were reversed by miR-145-Adv. Endothelium-dependent vasodilation of coronary arteries, endothelial nitric oxide synthase (eNOS) Ser1179 phosphorylation, eNOS-dependent NO·− production and endothelial cell survival were compromised in JCR rats. These parameters of endothelial dysfunction were completely reversed by 20-HETE antagonism or miR-145-Adv delivery, whereas neutrophil depletion resulted in partial reversal (~70%). We conclude that low miR-145 in MetS allows for increased 20-HETE, mainly from neutrophils, which compromises endothelial cell survival and function leading to impaired CCG. 20-HETE antagonists could provide viable therapy for restoration of CCG in MetS.
NEW & NOTEWORTHY Elevated 20-hydroxyeicosatetraenoic acid (20-HETE) impairs coronary collateral growth (CCG) in metabolic syndrome by eliciting endothelial dysfunction and apoptosis via excessive neutrophil infiltration. 20-HETE antagonists completely restore coronary collateral growth in metabolic syndrome. microRNA-145 (miR-145) is an upstream regulator of 20-HETE production in metabolic syndrome; low expression of miR-145 in metabolic syndrome promotes elevated production of 20-HETE.
the metabolic syndrome (MetS) describes a collection of risk factors that predisposes individuals to cardiovascular disease (57, 69). Moreover, current revascularization therapies, coronary artery bypass grafting (CABG), and percutaneous transluminal coronary angioplasty (PTCA) are associated with higher procedural risk and poorer long-term outcomes in patients with MetS (64).
In response to transient, repetitive coronary artery occlusion and resultant tissue ischemia (RI), native coronary collateral vessels undergo an adaptive remodeling process called coronary collateral growth (CCG) to form larger conduit arteries, which protect the heart from ischemic damage by reestablishing blood supply (9, 10, 66, 72, 73). However, CCG is impaired in patients with and animal models of MetS (11, 24, 39, 41, 42, 57, 61, 62, 65). Although many factors involved in collateral growth have been known for decades, clinical trials targeting the induction of collateral growth in humans remain largely disappointing (8, 19, 25). We have recently achieved complete recovery of CCG in a MetS rat model (JCR:LA-cp, JCR) via adenovirus-mediated (Adv) delivery of microRNA-145 (miR-145) (41).
Adv-mediated delivery of miR-145 at normal, physiological levels [measured in normal Sprague-Dawley (SD) rats] to JCR rats restored the contractile vascular smooth muscle cell (VSMC) phenotype and resulted in complete restoration of CCG (41). As the only intervention capable of completely restoring CCG in an animal model of MetS, this is a significant achievement. However, Adv-mediated intervention is not feasible for induction of CCG in humans due to inflammation and heart failure associated with Advs (58). No other reliable method of miR delivery in the cardiovascular system has yet been devised. Thus identification of alternative approaches capable of complete CCG recovery in MetS is of interest.
Endothelial dysfunction and a chronic proinflammatory state with leukocyte recruitment and upregulation of endothelial adhesion molecules are also hallmarks of MetS (43), and endothelial dysfunction is a characteristic of the JCR phenotype. JCR rats mimic not only the metabolic parameters but also the cardiovascular dysfunction characteristic of the human MetS (23, 31, 39, 41, 42, 62, 75). Inflammatory cells, including neutrophils, have been associated with endothelial dysfunction (70, 77). We have recently shown that while in normal animals RI induces transient infiltration of monocytes, which correlates with successful CCG, in MetS, RI induces excessive and sustained accumulation of neutrophils, which plays a causative role in impairment of CCG (42).
20-Hydroxyeicosatetraenoic acid (20-HETE) is a cytochrome P450 (CYP)-derived metabolite of arachidonic acid, synthesized, in the rat, by the CYP4A and 4F gene families of which the 4F isoform is expressed in rat neutrophils (38, 74, 84). 20-HETE plays an important role in the regulation of vascular tone (2, 29, 47), renal function (26, 67, 88), and blood pressure (22, 29, 84). 20-HETE synthesis has been reported to predominantly occur in the microcirculation in VSMCs, neutrophils, and platelets (29, 37, 38). Increasing vascular 20-HETE has been linked with endothelial dysfunction and activation in animal models of hypertension and in hypertensive patients (18, 76, 82). 20-HETE is also elevated in obese patients and animal models (50, 76), correlates with hyperglycemia (51), and is associated with increased triglycerides and decreased HDL in MetS patients (27).
In cell culture, 20-HETE has been reported to be a proangiogenic factor shown to decrease endothelial cell (EC) apoptosis and enhance EC and endothelial progenitor cell (EPC) survival (20), proliferation and migration (32, 33), ultimately stimulating angiogenic processes (13, 15, 20). Conversely, increased 20-HETE was shown to induce endothelial nitric oxide synthase (eNOS) uncoupling and decrease nitric oxide (NO·−) biosynthesis in ECs (18), which would be detrimental to angiogenesis and collateral growth. However, the role of 20-HETE in the regulation of collateral growth never been studied. Likewise, the mechanism(s) by which it may do so are entirely unknown.
Here, we explored the hypothesis that in contrast to low levels of 20-HETE, which are produced in response to RI in normal animals and are conducive to angiogenesis and collateral growth, in MetS low levels of miR-145 allow for production of too high levels of 20-HETE mainly due to excessive neutrophil accumulation. Thigh levels of 20-HETE result in EC apoptosis, endothelial dysfunction, and impaired CCG. We further hypothesize that specific pharmacological 20-HETE antagonists would be able to completely restore CCG in MetS.
MATERIALS AND METHODS
20-HETE antagonists.
JCR and SD rats were treated with 20-HETE antagonists: 20-SOLA, [2,5,8,11,14,17-hexaoxanonadecan-19-yl 20-hydroxyicosa-6(z),15(z)-dienoate] and 20-HEDGE, N-[20-hydroxyeicosa-6(Z),15(Z)-dienoyl] glycine, administered via intraperitoneal injection at 10 mg·kg−1·day−1 starting 2 days before initiation of the RI protocol (day 2 RI) and through the duration of the protocol (days 2–9 RI). 20-SOLA and 20-HEDGE were synthesized by Dr. John R. Falck. The compounds are competitive antagonists of 20-HETE and do not decrease 20-HETE levels (14, 28, 29).
20-HETE analogue.
JCR rats were treated with a 20-HETE analogue, 5,14,20-HEDGE, N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine, administered via intraperitoneal injection (ip) at 10 mg·kg−1·day−1 starting 2 days before initiation of the RI protocol (day 2 RI) and through the duration of the protocol (days 2–9 RI). 5,14,20-HEDGE is synthesized by Dr. John R. Falck and is a partially saturated form of 20-HETE in which two double bonds located between carbon positions 8–9 and 11–12 are removed to make it resistant to metabolism by cyclooxygenase. It is further modified at the COOH terminus to make it more resistant to esterification and β-oxidation (1). 5,14,20-HEDGE does not alter endogenous 20-HETE levels; instead, it mimics the actions of 20-HETE (63).
Adenoviral constructs.
The miR-145-Adv construct was made and purified by ViraQuest (North Liberty, IA) as described in our previous study (41). The miR-145 precursor sequence CACCTTGTCC TCACGGTCCA GTTTTCCCAG GAATCCCTTA GATGCTAAGA TGGGGATTCC TGGAAATACT GTTCTTGAGG TCATGGTT was inserted into an Adv vector behind the smooth muscle (SM)22α promoter (gift from Dr. Thomas Lincoln, University of South Alabama, Mobile, AL) to ensure SM-specific delivery. The enhanced green fluorescent protein (EGFP)-Adv was on an identical viral backbone with EGFP expression under the direction of the SM22α promoter. The Adv constructs were injected at 1.5 × 1012 plaque-forming units (PFU) in isotonic saline (100 µl) by direct injection into the left ventricular (LV) cavity followed by a 40-s left anterior descending coronary artery (LAD) occlusion as described previously (41) on day 4 of RI to express maximal levels of mature miR-145 beginning on day 6 of RI and through the duration of the RI protocol.
Anti-miR-145.
Locked nucleic acid (LNA)-modified anti-miR-145 (Exiqon, Woburn, MA) was delivered at 2 mg/kg in 100 µl of sterile saline via intracardiac injection directly into the LV cavity on day 6 of RI as described in our previous studies (41). Scrambled LNA-anti-miR sequence was used as control.
Blocking antibodies.
JCR rats were treated with blocking antibodies against the major monocyte/neutrophil adhesion receptor CD11b/CD18 (also known as Mac-1 or αMβ2 integrin, which binds to ICAM on endothelial cells, mAb clones M1/70/M18/2; Abcam, Cambridge, MA) and with the blocking antibody against CD44 (receptor for hyaluronic acid isoform 10, lectin-like LINK domain, mAb clone IM7; Abcam), at the dose of 1 mg·kg−1·day−1 by direct LV injection on day 6 through day 9 of RI.
Rat model of CCG and RI.
Male, 10- to 12-wk-old SD (Charles Rivers, Wilmington, MA) (300–350 g) or JCR:LA-cp rats (JCR; S. Proctor, University of Alberta, Edmonton, Canada) (650–700 g) were used for chronic (0–9 days) implantation of a pneumatic occluder over the LAD as described previously (39, 41, 62). Only male animals are used because CCG impairment in female JCR rats is not as severe as in males (Rocic P, unpublished observations). A suture was passed under the proximal portion of the LAD and the occluder was implanted into the surface of the heart. The occluder catheter was externalized between the scapulae. When the occluder is inflated, the suture is pulled toward the surface of the heart and the LAD is occluded. The LAD perfusion territory is termed the collateral-dependent zone (CZ) because perfusion in this area, while the LAD is occluded, depends on the development of coronary collaterals. The animals underwent the RI protocol consisting of eight 40-s occlusions, once every 20 min (2 h, 20 min total) followed by a rest period of 5 h, 40 min. This 8-h cycle was repeated three times per day for 0–9 days. Surgical procedures were performed in accordance with the Animal Welfare Act and were approved by the Institutional Animal Care and Use Committees of New York Medical College. CZs were separated from normal, nonischemic zones (NZ) on the basis of visual assessment of blanching during LAD occlusion (occluder inflation), which is indicative of ischemia and hyperemia upon reperfusion in the CZ at the time of occluder implantation (initial surgery), before collaterals have formed. It was confirmed by measurements of baseline (day 0 RI) blood flows. Notes delineating the CZ and the NZ were made for each animal and referred to at time of death and the border zone was excluded from tissue samples.
Myocardial and collateral-dependent blood flow measurements.
CCG was assessed by measuring CZ and NZ coronary blood flow using microspheres as previously described (24, 39, 41, 62). Neutron-activated microspheres (5 × 105, 15-µM diameter) labeled with samarium [day 0 RI (initial surgery))] lutetium (day 7 RI), or gold (day 9 RI) were injected into the LV lumen during LAD occlusion, with or without adenosine, and with or without nitroglycerine (20 mg/kg iv) (5). Fifteen-micromolar microspheres were retained in the heart tissue and when neutron-activated emit at different frequencies; therefore, blood flows on days 0, 7, and 9 can be differentiated. Arterial reference blood samples (carotid) and heart tissue from the NZ and the CZ were collected, weighed, and sent to BioPal (Worcester, MA) for analysis. Blood flows to the NZ and the CZ were calculated from the formula: blood flow (ml·min−1·g−1) = {[radioactive counts in myocardial tissue (dpm)] × (0.34 ml/min)/[radioactive count in blood reference (dpm)]}/[weight of myocardial tissue (g)], and results are expressed as flow ratio CZ (ml·min−1·g−1)/NZ (ml·min−1·g−1).
Arteriolar and capillary density.
Arteriolar and capillary density were measured in cardiac cross-sections on day 9 of RI as in our previous studies (41). Formalin-fixed, parafin-imbedded cardiac tissue was cut into 5-µm sections. For measurements of arteriolar and capillary densities, a 1-mm2 grid was superimposed over smooth muscle-mysoin heavy chain (SM-MHC)-stained (for arterioles/arteries) and hematoxylin/eosin-stained (for capillaries and arterioles/arteries) cardiac cross sections and vessels <20 µM in diameter and >20 µM in diameter inside the grid were counted as capillaries and arterioles/arteries, respectivelly. Fully formed arterioles/arteries were additionally identified by the presence SM-MHC-positive VSMCs. Larger arteries were distinguished from veins by assessement of vessel wall thickness, where a wall thickness/lumen diameter ratio >0.25 signified an artery and <0.25 a vein5. Data were collected from n = 8 animals per group from five consecutive cross sections per animal and five separate 1-mm2 grids per slide.
Measurements of 20-HETE.
20-HETE levels were measured in the CZ and NZ on days 0 and 9 of RI. CZ and NZ were isolated and incubated in oxygenated Krebs bicarbonate buffer, pH 7.4, with 1 mM NADPH for 1 h at 37°C. Tissue incubations were terminated with 2 vol cold methanol, internal standards were added, and samples were kept at −80°C. Eicosanoids were extracted using solid phase Strata-X Polymeric Reversed Phase 60 mg cartridges (Phenomenex, Torrance, CA). In brief, each sample was centrifuged for 15 min at 4°C. The supernatant was then collected, diluted with 5 vol of water, and acidified to pH 5.0 with HCl. Cartridges were primed with 6 ml of methanol followed by 6 ml of water. Samples were loaded, washed with 2 ml 20% methanol, and eluted with 1.5 ml of ethyl acetate. The collected ethyl acetate fraction was dried under nitrogen, resuspended in 50 µl of methanol, and stored at −80°C until analysis. Protein pellets were saved and quantified by Bradford protein assay. Identification and quantification of Eicosanoids were performed on a Shimadzu Triple Quadrupole Mass Spectrometer LCMS-8050 equipped with a Nexera UHPLC using multiple reaction monitoring mode. MS conditions were ionization mode: negative heated electrospray (HESI), applied voltage: −4.5 to approximately −3 kV; nebulizer gas: 3.0 l/min N2; drying gas: 5.0 l/min N2; heating gas: 12.0 l/min air; interface temp.: 400°C; DL temp.: 100°C; heat block temp.: 500°C; internal standards: d6 20-HETE, d8 5-HETE, d4 PGE2, d11 14(15)-EET, and d11 11,12-DHET; UHPLC conditions: analytical column: Zorbax Eclipse Plus C18 RRHD (50-mm length × 2.1-mm inner diameter, 1.8 μm); mobile phase A: 95% water 5% acetonitrile 0.05% acetic acid; mobile phase B: acetonitrile 0.05%; time program 40% B. (0 min)→75% B. (3 min)→85% B. (7.5 min); flow rate: 0.4 ml/min.; injection volume: 5 μl; and column oven temp.: 40°C. Synthetic standards were used to obtain standard curves (0.5–500 pg) for each compound. These standard curves were used to calculate the final concentrations of the Eicosanoids. All solvents were HPLC grade or higher.
Measurements of NO·−.
NO·− was measured by X-band electroparamagnetic resonance (EPR) as previously described for superoxide (O2·−) in our studies (61, 62). Fe-DETC was used as a spin-trap. A DETC·3H2O solution (400 mg/kg; Aldrich Chemical, Milwaukee, WI) and an Fe-citrate mixture (40 mg/kg of FeSO4·7H2O and 200 mg/ml of sodium citrate; Sigma-Aldrich, St. Louis, MO) were injected intraperitoneally to form Fe-DETC complexes which trap endogenously produced NO·− to yield NO·−-Fe-DETC complexes as previously described (79). Rats underwent two consecutive periods of ischemia/reperfusion and were killed, the heart was removed, and the CZ was separated from the NZ. Tissue was then homogenized by sonication on ice and frozen in liquid nitrogen until EPR measurements. NO·− concentration was obtained from the standard curve generated with the NO·− donor SNAP in the solution of FeSO4 and sodium citrate. Tissue NO·− concentration was expressed in nM/g tissue and expressed as fold difference vs. day 0 RI in SD rats. To specifically measure eNOS-derived NO·−, animals were treated with 1,400 W (5 mg/kg iv) to inhibit iNOS and ARL17477 (10 mg/kg) to inhibit nNOS.
Western blot analysis.
Western blot analysis was performed as in our previous studies (39, 42). Unperfused hearts were excised, the LV was dissected, and CZ was separated from the NZ and snap-frozen in liquid nitrogen before homogenization in lysis buffer containing 0.1% SDS and 1% Triton. Equal amounts of protein (30 µg) were separated by SDS-PAGE and transferred to Trans-Blot Turbo Mini PVDF membranes (Bio-Rad, Hercules, CA), blocked with blocking buffer (Li-Cor, Lincoln, NE), followed by incubation with primary and secondary antibodies. Anti-phospho-eNOS (Ser1179) (Cell Signaling, Boston, MA), anti-eNOS (Abcam, Cambridge, MA), anti-CYP4F (US Biological, Salem, MA), anti-P-selectin (Abcam), anti-ICAM-1 (Abcam), anti-β-tubulin (Abcam), and goat anti-rabbit IRDye 800CW (Li-Cor) antibodies were used for Western blotting. Fluorescence-based immunodetection of membranes was performed with the Li-Cor Odyssey Infrared Imaging System (Li-Cor), and band densities were analyzed using the Odyssey Application software version 3.0.21. Data were normalized to β-tubulin (loading control), except p-eNOS, which was also normalized to total eNOS, and expressed as CZ/NZ ratios, which is indicative of changes in response to RI in the CZ.
Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling and eNOS immunohistochemistry.
Unperfused and perfused (isotonic saline followed by 4% paraformaldehyde), formalin-fixed, paraffin-imbedded cardiac tissue was cut into 10-µm sections. Slides were de-araffinized, subjected to mild proteinase K digestion, and terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) labeling was performed using the TUNEL kit (Roche, Indianapolis, IN). The kit contains an optimized terminal deoxynucleotide transferase (TdT) and red fluorescent-labeled dUTP (TMR-dUTP), which was visualized with a Nikon fluorescent microscope equipped with Nikon Elements software. Nuclei were stained with DAPI and the percentage of TUNEL-positive cells was determined from the ratio of TUNEL-positive nuclei/total nuclei. eNOS (1:100) was from Abcam. All slides were costained with anti-smooth muscle-specific α-actin (SM-α-actin; Sigma) to identify blood vessels. Arteries were distinguished from veins by assessment of vessel wall thickness, where the wall thickness/lumen diameter ratio >0.25 signified an artery and <0.25 a vein, as described in our previous studies (41).
To select for collateral arteries vs. preexisting vessels, only arteries that also stained positive for PCNA on adjacent sections were included in data analysis, since proliferating cells in the adult heart are associated only with actively remodeling blood vessels, as described in our previous studies (41).
Flow cytometry.
Excised hearts were flushed with saline (1 ml, 3×) through the aortic root using a 5-cc syringe to create sufficient pressure to dislodge and separate adherent leukocytes without perturbing the integrity of the vascular wall [confirmed by high-power (×100) microscopy for 20–150 µM vessels in every heart]. Flow cytometry was performed on the eluate using a Gr-1 antibody (mAb clone Ly6G; Acris, San Diego, CA) to label peripheral neutrophils as described in our previous study (42).
Measurements of vascular function.
Coronary arteries (~50 μm) from the NZ and CZ were isolated on days 0 and 9 of RI and mounted on wires in the chambers of a multivessel myograph (JP Trading, Aarhus, Denmark) filled with Krebs buffer (37°C) gassed with 95% O2-5% CO2. Coronary arteries were precontracted with phenylephrine (1 µM) to 90% of maximal contraction under the intraluminal pressure of 60 mmHg. Response to treatment with acetylcholine (Ach) (1 × 10−8 to 5 × 10−5 M) was determined. In addition, in coronary arteries isolated from untreated (no miR, anti-miR, blocking antibody, or 20-HETE antagonist or analogue treatment) SD and JCR rats. Ach-induced vasorelaxation was also assessed in the presence of 20-HETE (50 nM) before and after treatment with nitro-l-arginine methyl ester (l-NAME) (50 μM).
Statistical analysis.
All experiments were representative of n = 8 animals per group. Results were analyzed by two-way ANOVA, followed by Bonferroni correction. P < 0.05 determined statistical significance.
RESULTS
20-HETE is elevated in MetS at baseline and in response to RI.
At baseline (day 0 RI), 20-HETE levels and CYP4F (the neutrophil-specific 20-HETE synthase) protein expression were elevated in JCR vs. SD rats (Fig. 1). Nine days of transient, repetitive LAD occlusion (RI) increased 20-HETE levels and protein expression of CYP4F in both SD and JCR rats compared with day 0 RI; however, this increase was greater in JCR vs. SD rats (~2- and 2.5-fold in JCR, respectively, vs. 1.5-fold in SD) (Fig. 1).
Fig. 1.
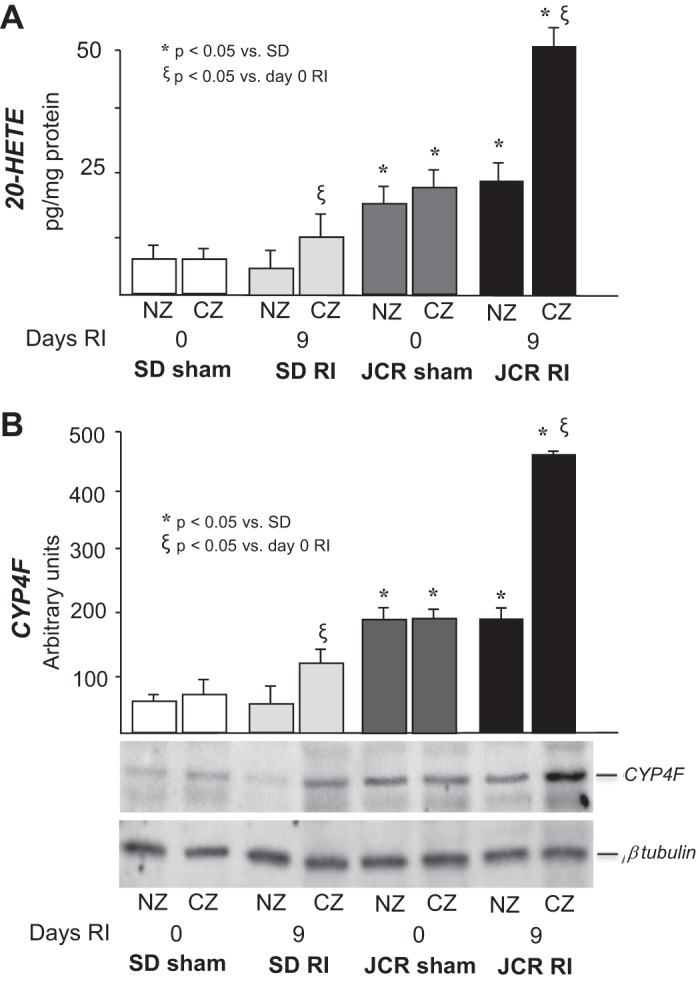
Sprague-Dawley (SD) and JCR rats underwent 0 or 9 days of repetitive ischemia (RI), hearts were excised, and the normal zone (NZ) was separated from the collateral-dependent zone (CZ). A: liquid chromatography-mass spectrometry (LC-MS) quantitation of 20-hydroxyeicosatetraenoic acid (20-HETE) production represented in pg/mg protein. B: representative Western blot of CYP4F protein expression (bottom) and cumulative data (top) in NZ and CZ heart homogenates represented in arbitrary units; n = 8, *ξP < 0.05 as indicated.
20-HETE antagonism completely restores CCG in the MetS.
Since 20-HETE was elevated in our model of MetS, the JCR rat, in response to RI and since we also have previously shown that RI-induced CCG was compromised in JCR rats (11, 23, 24, 39, 41, 42, 46, 62, 65, 75, 87), we next assessed the relationship between elevated 20-HETE and impaired CCG in MetS by using 20-HETE antagonists, 20-SOLA and 20-HEDGE. Administration of either 20-SOLA or 20-HEDGE, fully restored collateral-dependent blood flow in JCR rats (CZ/NZ flow ratios were 0.76 ± 0.07 in JCR + 20-SOLA, 0.84 ± 0.05 in JCR + 20-HEDGE vs. 0.11 ± 0.02 in untreated JCR vs. 0.84 ± 0.03 in SD) (Fig. 2A).
Fig. 2.

SD and JCR rats were treated with 20-SOLA or 20-HEDGE (20-HETE antagonists) or 5,14,20-HEDGE (20-HETE mimetic) on days 2–9 of RI, or with microRNA-145 (miR-145) or anti-miR-145 on day 6 RI as indicated. A and C: coronary flow with or without adenosine and with or without nitroglycerine was measured in the CZ and NZ using microspheres during LAD occlusion and expressed as flow in the CZ in ml·min−1·g−1 over the time course of RI (left) and the flow ratio between the CZ and the NZ on day 9 RI (right). B: collateral arteries [smooth muscle-myosin heavy chain (SM-MHC)/PCNA-positive vessels >20 µM) and capillary (vessels <20 µM) densities were determined in cardiac (CZ) cross sections on day 9 RI; n = 8. *ξϕP < 0.05 as indicated.
As in our previous studies, miR-145 markedly reduced CCG in SD rats (CZ/NZ flow ratios was 0.35 ± 0.02) (Fig. 2C). Anti-miR-145 did not further decrease CCG in JCR rats (CZ/NZ flow ratios was 0.10 ± 0.03 (Fig. 2C), likely because collateral-dependent blood flow in JCR rats is already minimal. We also assessed CCG in two additional groups of animals: JCR + miR-145-Adv + 5,14,20-HEDGE (a 20-HETE analogue), and SD + anti-miR-145 + 20-HEDGE. If 20-HETE were the sole mediator of miR-145’s effects on CCG, then in this group of JCR rats, CCG should be inhibited despite treatment with miR-145, and in the SD rats, CCG should be close to normal despite treatment with anti-miR-145. In the JCR + miR-145-Adv + 5,14,20-HEDGE group, the CZ/NZ flow ratio on day 9 RI was 0.31 ± 0.04 vs. 0.94 ± 0.05 (JCR + miR-145-Adv) vs. 0.11 ± 0.02 (JCR) (Fig. 2C). In SD + anti-miR-145 + 20-HEDGE animals, the CZ/NZ flow ratio on day 9 RI was 0.69 ± 0.05 vs. 0.25 ± 0.03 (SD + anti-miR-145) vs. 0.84 ± 0.03 (SD) (Fig. 2C).
Maximal vasodilation using adenosine (5 ×105 M) or nitroglycerin (20 mg/kg iv) did not significantly alter coronary blood flow in any group (Fig. 2, A and C), as in all our previous studies (11, 23, 24, 39, 41, 42, 46, 62, 65, 75, 87) indicating that the observed increases in collateral-dependent blood flow are a result of CCG and not vasodilation of preexisting arteries. Vasodilation does not play a significant role in our rat model of CCG because >80% occlusion of the LAD at the time of blood flow measurements dictates that the coronary circulation, preexisting and collateral, is already maximally dilated, i.e., coronary flow reserve approaches zero (87). Likewise, as in our previous studies, while capillary number is lower in JCR rats at baseline, neither RI nor treatment with 20-HEDGE or 20-SOLA affected capillary density confirming that increase in collateral-dependent blood flow was due to arteriogenesis and not angiogenesis (Fig. 2B). These results support the idea that 20-HETE is a major negative regulator of RI-induced CCG in MetS.
miRNA-145 is a key regulator of 20-HETE synthesis.
We have previously demonstrated that decreased expression of miR-145 in MetS correlated with impaired CCG (41). Furthermore, miR-145 delivery to JCR rats at physiological levels resulted in complete restoration of CCG in these animals (41). In our present study, the RI-induced decrease of miR-145 expression (~5-fold vs. SD) in MetS animals on day 9 RI (41), correlated with elevated 20-HETE levels (~4-fold vs. SD) and CYP4F expression (~4.5-fold vs. SD) (Fig. 3). Conversely, elevated miR-145 in normal animals (2-fold on day 9 RI vs. day 0 RI) (41) correlated with low 20-HETE levels and CYP4F expression in response to RI (~2-fold increase vs. day 0 RI) (Fig. 3).
Fig. 3.

JCR rats were treated with miR-145-Adv or EGFP-Adv on day 4 of RI or anti-miR-145 on day 6 RI and SD rats were treated with anti-miR-145 or scrambled anti-miR on day 6 of RI or miR-145-Adv on day 4 RI where indicated, and SD and JCR rats underwent 0 or 9 days of RI. Rats were killed on days 0 and 9 RI, hearts were excised, and the CZ was separated from NZ. A: LC-MS quantitation of 20-HETE production represented in pg/mg protein. B: representative Western blot of CYP4F protein expression (bottom) and cumulative data (top) in NZ and CZ heart homogenates represented in arbitrary units; n = 8. *ξϕP < 0.05 as indicated.
Furthermore, restoration of physiological miR-145 in JCR rats by miR-145-Adv delivery resulted in a reduction in 20-HETE levels and CYP4F expression similar to that found in SD rats (Fig. 3). Anti-miR-145 administration to further downregulate miR-145 in JCR rats resulted in a further increase in 20-HETE (Fig. 3A). Conversely, miR-145 downregulation by anti-miR-145 in SD rats increased 20-HETE levels and CYP4F expression in SD rats to levels comparable to those found in JCR rats (Fig. 3). miR-145 administration in SD rats led to a decrease in 20-HETE (Fig. 3A). These results indicate that while RI induces CYP4F expression and 20-HETE production in both normal and MetS animals, high levels of miR-145 in normal animals keep CYP4F expression and 20-HETE levels low. In contrast, low levels of miR-145 in MetS allow for their striking increase.
Concentrations of other arachidonic acid metabolites in SD vs. JCR rats measured in response to RI and manipulation of miR-145 levels are recorded in Table 1. Of note, the eicosanoids, which are generally thought to be proinflammatory, 12- and 15-HETE were elevated in JCR rats, while 11(12)- and 14(15)-EETs, reported as endothelial-dependent relaxing factor (EDHF) (4, 30), were decreased. miR-145 also, unsurprisingly, regulated 12-HETE and 15-HETE levels, consistent with miR-145-dependent regulation of VSMC phenotype. 12-HETE, which is especially high in JCR hearts and further elevated by RI (Table 1), is produced by 12-lipoxygenases the expression and activity of which are likely elevated in JCR VSMC, since it has been shown that elevated glucose and angiotensin II, which are elevated in JCR rats (46), increase its expression and activity (59).
Table 1.
Arachidonic acid metabolites were measured by LC-MS
| SD Sham | SD RI | SD RI + miR-145 | SD RI + Anti-miR-145 | JCR Sham | JCR RI | JCR RI + miR-145 | JCR RI + Anti-miR-145 | JCR RI + Block Ab | |
|---|---|---|---|---|---|---|---|---|---|
| 20-HETE | 7.2 ± 3 | 19.5 ± 5† | 5.0 ± 1* | 39.6 ± 2*† | 24.3 ± 4* | 50.7 ± 5*† | 22.5 ± 4‡ | 74.4 ± 3*†‡ | 24.8 ± 6*‡ |
| 15-HETE | 216.2 ± 2 | 211.3 ± 2 | 186.8 ± 6 | 266.8 ± 3* | 312.3 ± 8* | 513.8 ± 4*† | 108.6 ± 8‡ | 523.4 ± 9*† | 519.5 ± 2*† |
| 12-HETE | 1098.1 ± 3 | 1114.1 ± 4 | 1000.6 ± 4 | 1564.3 ± 5* | 5173.5 ± 12* | 7232.8 ± 11* | 2225.9 ± 8*‡ | 7251.6 ± 12* | 7239.2 ± 9* |
| 5-HETE | 3.8 ± 2 | 4.2 ± 2 | 4.1 ± 3 | 14.4 ± 5* | 158.7 ± 8* | 208.4 ± 6* | 111.2 ± 7*‡ | 206.4 ± 3* | 211.4 ± 1* |
| 14(15)-EET | 299.0 ± 8 | 304.4 ± 6 | 301.4 ± 6 | 303.4 ± 2 | 139.3 ± 8* | 79.3 ± 5* | 80.4 ± 3* | 78.2 ± 4* | 81.3 ± 1* |
| 11(12)-EET | 634.0 ± 6 | 629.6 ± 9 | 641.1 ± 9 | 622.2 ± 12 | 101.3 ± 6* | 55.2 ± 8*† | 56.6 ± 2*† | 57.6 ± 4*† | 55.6 ± 4*† |
| 8(9)-EET | 101.1 ± 4 | 89.6 ± 7 | 108.5 ± 2 | 103.5 ± 7 | 102.5 ± 9 | 81.4 ± 4 | 85.3 ± 9 | 85.4 ± 3 | 83.0 ± 3 |
| 5(6)-EET | 245.6 ± 11 | 286.2 ± 6 | 249.2 ± 8 | 273.248 | 275.3 ± 3 | 183.7 ± 5* | 190.1 ± 9* | 192.7 ± 11* | 181.2 ± 5* |
Values are means ± SE in pg/mg. Concentrations in pg/mg tissue in the collateral-dependent zone (CZ) are shown. LC-MS, liquid chromatography-mass spectrometry; SD, Sprague Dawley; NZ, normal zone; RI, repetitive ischemia.
P < 0.05 vs. SD.
P < 0.05 vs. day 0 RI.
P < 0.05 vs. JCR.
Neutrophils are a major source of 20-HETE produced in response to RI in MetS.
In our recent study, we showed that neutrophil accumulation was significantly higher and remained elevated for the duration of the RI protocol in the CZ of JCR vs. SD rats, where infiltration was low and transient (confined to the early stage of CCG) (42). Furthermore, we demonstrated that neutrophil depletion in JCR rats improved CCG (42).
In our present study, we sought to determine if neutrophils were a major source of elevated 20-HETE in MetS. To determine the contribution of neutrophils, neutrophils were depleted using blocking antibodies against the major monocyte/neutrophil adhesion receptor, CD11b/CD18 and against CD44, a major receptor that regulates adhesion to the inflammatory extracellular matrix components hyaluronan and osteopontin. Anti-CD11b/CD18 blocking antibodies target both neutrophils and monocytes; however, at the time point of their delivery (day 6 of RI), monocytes are nondetectable in JCR rats, and thus this cross-reactivity is not a concern in this study (42). As in our previous study (42), neutrophil accumulation was ~3.5-fold higher and persisted for the duration of the RI protocol in the CZ of JCR rats (Fig. 4A). This RI-induced, elevated neutrophil infiltration in the CZ of JCR rats correlated with the ~4.5-fold increase in CYP4F expression and the ~4-fold increase in 20-HETE levels in JCR vs. SD rats (Fig. 4, B and C). Importantly, neutrophil depletion by blocking antibodies completely blocked the RI-induced increase in CYP4F expression (as expected, since the CYP4F isoform is neutrophil-specific in rats) (Fig. 4C). This resulted in a ~60% reduction in 20-HETE levels in JCR rats (Fig. 4B). These results suggest that ~60% of 20-HETE produced in response to RI in MetS is neutrophil derived.
Fig. 4.

JCR rats were treated with anti-CD11b/CD18/CD44 antibodies (to deplete neutrophils) on days 6–9 of RI. SD and JCR rats underwent 0, 3, 6, or 9 days of RI. A: flow cytometry was performed on CZ homogenates using Gr-1 (Ly6G) antibodies to label neutrophils and quantified. B: LC-MS quantitation of 20-HETE production represented in pg/mg protein. C: representative Western blot of CYP4F protein expression (bottom) and cumulative data (top) in NZ and CZ heart homogenates represented in arbitrary units. Note: cumulative data for SD RI and JCR RI groups are identical to those in Fig. 3B and are derived from the same data set; n = 8. *ξϕP < 0.05 as indicated.
miR-145 downregulation promotes neutrophil accumulation in MetS.
The effect of miR-145 on inflammatory cell numbers, particularly neutrophils, was evaluated by flow cytometry. miR-145 delivery completely prevented RI-induced neutrophil accumulation in MetS animals, which correlated with outward collateral remodeling (Fig. 5, A and B). In contrast, anti-miR-145 delivery to SD rats increased neutrophil accumulation, which correlated with lack of outward collateral remodeling (Fig. 5, A and B). These results demonstrate that low miR-145 expression in MetS plays a causative role in increased neutrophil accumulation in MetS.
Fig. 5.

JCR rats were treated with miR-145-Adv on day 4 of RI or 20-SOLA or 20-HEDGE (20-HETE antagonists) on days 6–9 of RI and SD rats were treated with anti-miR-145 on day 6 of RI where indicated, and SD and JCR rats underwent 0, 3, 6, or 9 days of RI. A: flow cytometry was performed on CZ homogenates using Gr-1 (Ly6G) antibodies to label neutrophils and quantified. B: representative hematoxylin-and-eosin images (×100) of remodeling collateral arteries. White arrows point to neutrophils. Note differences in size of scale bars; n = 8. *ξϕP < 0.05 as indicated.
In addition, we also demonstrate that treatment of JCR rats with the 20-HETE antagonists 20-HEDGE or 20-SOLA results in a similar prevention of neutrophil accumulation (Fig. 5, A and B) connected with outward collateral remodeling. In conjunction with results in Fig. 4, these results suggest an existence of a positive feedback loop between neutrophil infiltration and 20-HETE production in MetS. In further support of this idea, 20-HETE antagonists and miR-145 decreased RI-induced overexpression of endothelial adhesion molecules P-selectin and ICAM-1 in JCR rats (Fig. 6A). While ICAM-1 serves as an adhesion receptor for many inflammatory cells and its increased expression marks endothelial dysfunction, P-selectin is a relatively specific mediator of neutrophil adhesion. As expected, neutrophil adhesion blocking antibodies had no effect on adhesion molecule expression (Fig. 6A).
Fig. 6.

JCR rats were treated with miR-145-Adv on day 4 of RI, 20-SOLA or 20-HEDGE (20-HETE antagonists) on days 2–9 of RI, or anti-CD11b/CD18/CD44 antibodies (to deplete neutrophils) on days 2–9 of RI and SD rats were treated with anti-miR-145 on day 6 of RI. SD and JCR rats underwent 9 days of RI. A: P-selectin and ICAM-1 expression were determined by Western blot. B: coronary arteries (~50 μM) were isolated from SD (left) and JCR (right) rats on day 9 of RI and Ach-dependent dilation was measured as %increase in artery diameter. In addition, arteries isolated from SD (left) and JCR (right) rats on day 0 RI were treated with 20-HETE or 20-HETE + nitro-l-arginine methyl ester (l-NAME) and Ach-dependent dilation was measured as % increase in artery diameter. C, left: pSer1179-endothelial nitric oxide synthase (eNOS) and total eNOS expression were determined by Western blot (left) and NO·− production by EPR (right). C, right: representative images of cardiac cross-sections stained with anti-eNOS (total) from the CZ are shown. D, top: representative images of cardiac cross-sections costained with anti-SM-α-actin and terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) from the CZ are shown. D, bottom: apoptosis was quantified as %TUNEL-positive EC nuclei; n = 8. *ξϕP < 0.05 as indicated. Note: all bands on the representative Western blots come from the same gel; single lane presentation (where applicable) is a result of cropping the images to maintain consistent order of presentation with regard to treatment groups.
miR-145 delivery or 20-HETE antagonism restores endothelial function in MetS.
Normal EC function is vital to successful CCG. Mediators released from healthy ECs, including NO, have been shown to play critical roles in the growth of collaterals (54, 85). 20-HETE has been associated with endothelial dysfunction in hypertensive patients and animal models (18, 44, 82), and endothelial dysfunction is a hallmark of MetS (40) and has been reported as a feature of JCR rats (55). Thus we investigated the effects of low miR-145 and elevated 20-HETE on endothelium function within the context of RI and CCG in MetS.
Coronary arteries from JCR rats displayed reduced endothelium-dependent vasodilation at baseline (~35%), which was further worsened on day 9 of RI vs. SD rats (25 ± 1.6% JCR vs. 90 ± 5% SD) (Fig. 6B). eNOS phosphorylation on Ser1179 (p1179), which is a marker of eNOS activation, expression, and NO·− production, was likewise decreased (~60, ~50, and ~70%, respectively) in the CZ of JCR vs. SD rats on day 9 of RI (Fig. 6C). Anti-miR-145 delivery in normal rats significantly diminished endothelium-dependent relaxation (by ~60% vs. untreated SD rats) (Fig. 6B) and reduced eNOS phosphorylation (~60%), expression and NO·− production (~80%) (Fig. 6C). Administration of miR-145 or the 20-HETE antagonists 20-HEDGE or 20-SOLA, in contrast, fully reversed impaired Ach-dependent vasodilation in JCR rats (Fig. 6B) as well as eNOS phosphorylation, expression, and NO·− production (Fig. 6C) on day 9 of RI, while depletion of neutrophils with blocking antibodies resulted in partial attenuation of endothelial dysfunction (~50%) (Fig. 6B), eNOS activation (~75%), and expression (~60%) and restored NO·− production (Fig. 6C).
In ex vivo preparations, exogenously applied 20-HETE (50 nM) inhibited Ach-dependent vasorelaxation in coronary arteries derived from SD rats in agreement with data previously reported for renal arteries (17) and did not markedly reduce already severely compromised vasodilation in coronary arteries derived from JCR rats (Fig. 6B). Treatment with l-NAME (50 µM) in the ex vivo preparations did not result in further reduction in Ach-induced vasodilation in either SD or JCR coronary arteries (Fig. 6B).
We and others have shown that EC apoptosis is increased in MetS (24, 60). Therefore, in this study we also evaluated RI-induced and 20-HETE-dependent EC apoptosis during CCG in normal vs. MetS animals. In our previous study, we demonstrated that neutrophil and VSMC apoptosis is negligible (<5%) by day 9 RI, with major neutrophil apoptosis occurring at days 3–6 RI (42). Here, we show that in response to RI, MetS animals and SD rats treated with anti-miR-145 exhibit marked EC apoptosis (~75% in JCR, ~65% in SD + anti-miR-145 vs. <5% in SD) (Fig. 6D). In contrast, JCR rats treated with 20-HETE antagonists or the miR-145-Adv exhibit negligible apoptosis, similar to untreated SD rats (Fig. 6D). Treatment with neutrophil-blocking antibodies resulted in partial attenuation of apoptosis in MetS animals (~70%) (Fig. 6D).
DISCUSSION
The most important and novel finding of this study is that high 20-HETE, at levels such as are produced in MetS, is a major negative regulator of CCG. While low levels of 20-HETE generated by RI in normal animals correlate with CCG, in agreement with a recent study in which 20-HETE was elevated in response to femoral artery occlusion in a Balb/c mouse model of hindlimb ischemia and this was associated with angiogenesis (14), high levels of 20-HETE induced by RI in MetS are detrimental to CCG. Specific 20-HETE antagonists normalized 20-HETE levels in MetS, which resulted in complete restoration of CCG. Angiogenesis, sprouting of new capillaries from preexisting vessels (12), and arteriogenesis, enlargement of small, native collaterals into conduit arteries (9, 66), despite sharing initiating factors, cytokines, and growth factors, are distinct processes (9, 12, 85). 20-HETE has been reported as a proangiogenic factor in vitro and in angiogenesis in vivo (7, 13, 14). It mediates angiogenesis by prolonging EC and EPC survival (20), proliferation, and migration (13, 15, 20, 32, 33) and upregulating proangiogenic factors HIF-1α and VEGF (32, 34). However, this is the first study to identify 20-HETE as a regulator of arteriogenesis, and the first study to demonstrate that 20-HETE can play a negative regulatory role in vascular growth in vivo under pathological conditions, such as MetS.
We have previously shown that MetS animals also express low levels of miR-145 at baseline and in response to RI and that this was causally related to impaired CCG in MetS (41). In fact, apart from 20-HETE antagonists, restoration of physiological levels of miR-145 in MetS rats was the only other intervention to date that was capable of restoring RI-induced CCG to levels seen in normal animals. Results in this study demonstrate that miR-145 is a key upstream regulator of CYP4F, the neutrophil-specific isoform of the 20-HETE synthase in rat, and of 20-HETE production. Thus the interplay between miR-145 and 20-HETE appears to be a major regulator of CCG so that low levels of miR-145 in MetS allow for excessive RI-induced 20-HETE production, which impairs CCG in MetS, while high miR-145 levels in normal animals allow for a small increase in 20-HETE production by RI, which is conducive to CCG. In further support of this idea, in MetS animals, if 20-HETE levels were elevated by pharmacological means (20-HETE analogue), even if the animals were treated with miR-145 MetS animals, CCG was severely reduced, and vice versa, even if miR-145 levels were reduced in normal animals (anti-miR-145), blocking effects of 20-HETE with 20-HETE antagonists nearly normalized CCG. The <100% efficacy of these treatments (vs. untreated JCR and SD animals, respectively) could be due either to involvement of miR-145-sensitive but 20-HETE-insensitive pathways in the regulation of CCG and/or to the fact that these effects may be dose dependent. In the present as in our previous study (41), miR-145 markedly reduced CCG in SD rats. We had hypothesized that this was due to development of the VSMC hypercontractile phenotype which was not conducive to CCG; however, it is equally likely that very high levels of miR-145 do not allow for the generation of even the low levels of 20-HETE by RI that are necessary for CCG.
Our results further demonstrate that a significant amount of 20-HETE involved in the regulation of CCG in MetS is derived from neutrophils. 20-HETE has been shown to be synthesized from human neutrophils (76). In our study, neutrophil depletion blocked RI-induced CYP4F expression and significantly decreased elevated 20-HETE levels and partially restored CCG in JCR rats. We have recently shown that while in normal animals, RI induces transient infiltration of monocytes, which correlates with successful CCG, in MetS, RI induces excessive and sustained accumulation of neutrophils, which plays a major contribution in impairment of CCG (42). In this study, we demonstrate that low miR-145 levels in JCR rats play a causative role in this excessive and prolonged neutrophil infiltration. Normalization of miR-145 expression via miR-145-Adv delivery blocked RI-induced neutrophil infiltration and elevation in CYP4F expression and significantly decreased 20-HETE levels. We have also recently shown that neutrophil survival was prolonged in MetS and that expression of a proinflammatory extracellular matrix component, osteopontin, was elevated (42). Binding to osteopontin prolongs inflammatory cell, especially neutrophil, survival from hours to days in the periphery (71). Osteopontin is primarily secreted from synthetic VSMCs, which are abundant in coronary arteries of MetS animals and are characterized by low miR-145 expression (41). Thus osteopontin production may provide a link between low miR-145 levels and excessive neutrophil accumulation in MetS during CCG.
In turn, 20-HETE antagonists decreased neutrophil accumulation in MetS animals, suggesting a positive feedback mechanism between 20-HETE production and neutrophil accumulation. 20-HETE has been shown to play a causative role in endothelial activation and proinflammatory states. Increased vascular 20-HETE leads to EC activation by activation of transcription factor NF-κB and MAPK/ERK kinase pathways resulting in increased gene transcription of proinflammatory mediators such as ICAM (45), consequently leading to leukocyte recruitment (80, 83). Endothelial activation is marked by the increase in inflammatory cell adhesion molecules, interaction between endothelial cells and inflammatory cells, and endothelial permeability (36, 84). A hallmark of MetS in humans is a chronic proinflammatory state characterized by the upregulation of adhesion molecules and elevated baseline levels of proinflammatory cytokines (21, 43, 86). In agreement with these observations, ICAM-1 expression and P-selectin expression were increased in MetS animals, and 20-HETE antagonists and miR-145 delivery markedly reduced their expression in our present study.
On the other hand, leukocyte accumulation contributes to further endothelial dysfunction, in part by O2.--mediated scavenging of NO·−. Furthermore, oxidative stress contributes to endothelial cell apoptosis via NF-κB-dependent pathways (3, 48). Inflammatory cells, especially neutrophils, via their NAD(P)H oxidases, are major producers of reactive oxygen species (ROS) (49). In agreement with these studies, in our present study, neutrophil depletion in the MetS was associated with a partial improvement of endothelial function and viability and NO·− production, while normalization of miR-145 levels and 20-HETE antagonism resulted in complete restoration of normal endothelial function, including NO production. Elevated 20-HETE levels in the vasculature have been associated with endothelial dysfunction (16, 81, 82). Increased 20-HETE causes dissociation of HSP90 from eNOS, resulting in eNOS uncoupling and decreased NO·− production in ECs and decreased endothelial-dependent vasodilation (16, 29), O2·− generation, and NO·− scavenging further decreasing its bioavailability (35, 68). Our results in coronary arteries isolated from normal and MetS animals are consistent with the possibility that 20-HETE-mediated reduction in NO·− production from ECs may be involved in CCG impairment in the MetS since in ex vivo preparations coronary arteries from the MetS animals with exogenously added 20-HETE or 20-HETE and l-NAME responded equally to Ach, thus suggesting that NO·− production under all three conditions is similar.
We have shown that high levels of ROS impair CCG in MetS (65), and NO·− has been shown to be required for CCG (54). It is however unlikely that NO·− regulates CCG or coronary blood flow in the CZ through vasodilation (87). A more likely scenario includes NO·−-dependent signaling including regulation of VSMC phenotype (6) and proliferation (78) and VEGF production (52). EC survival, proliferation, and ability to produce growth factors like VEGF are also crucial to collateral development (53, 56). This study introduces 20-HETE as a novel major regulator of CCG in MetS. In conjunction with our previous studies, our new data demonstrate that impaired CCG in MetS is a consequence of low VSMC levels of miR-145, which promote synthetic VSMC phenotype leading to excessive neutrophil accumulation and 20-HETE production from CYP4F, which results in endothelial dysfunction and apoptosis.
We have also previously shown that miR-21 is involved in the regulation of CCG via regulation of neutrophil accumulation in MetS (42). Our results to date are consistent with the idea that miR-21 and miR-145 both regulate CCG. miR-21 is overexpressed in MetS (JCR rats) (39), while miR-145 is underexpressed (41). High levels of miR-21 in MetS promote excessive proliferation of VSMCs and prolonged survival of neutrophils (42). Low levels of miR-145 prevent conversion of those VSMCs to the normal contractile phenotype (41) and facilitate neutrophil infiltration by allowing for high 20-HETE production. Thus the expression patterns of these two miRs in MetS promote accumulation of neutrophils and synthetic, proliferative VSMCs, which are incompatible with CCG, whereas, in the normal animals (SD rats), low miR-21 levels and high miR-145 levels promote contractile, quiescent VSMC phenotype (39, 41) and clearance of neutrophils by preventing their adhesion (miR-145) and promoting their apoptosis (miR-21) (42).
Growth factor-, cytokine-, and EPC-based therapies targeting the induction of collateral growth in humans have been mostly ineffective (8, 19, 25). Adv-mediated intervention in humans is not feasible because Advs cause inflammation and heart failure (58). Therefore, alternative pathways to restore of CCG in MetS are desirable. This study introduces 20-HETE antagonists as potential therapy for induction of collateral growth in MetS in humans.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants R01-HL-093052 (to P. Rocic) and P01-HL-034300 (to M. L. Schwartzman).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
G.J. and P.R. conceived and designed research; G.J., A.S., R.H., I.H., C.B., B.H., K.H.G., H.J., J.R.F., S.P., M.L.S., and P.R. performed experiments; G.J., A.S., R.H., C.B., K.H.G., H.J., and P.R. analyzed data; G.J., R.H., H.J., and P.R. interpreted results of experiments; G.J. and P.R. prepared figures; G.J. drafted manuscript; G.J., K.H.G., S.P., M.L.S., and P.R. edited and revised manuscript; G.J., A.S., R.H., I.H., C.B., B.H., K.H.G., H.J., J.R.F., S.P., M.L.S., and P.R. approved final version of manuscript.
REFERENCES
- 1.Akbulut T, Regner KR, Roman RJ, Avner ED, Falck JR, Park F. 20-HETE activates the Raf/MEK/ERK pathway in renal epithelial cells through an EGFR- and c-Src-dependent mechanism. Am J Physiol Renal Physiol 297: F662–F670, 2009. doi: 10.1152/ajprenal.00146.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alonso-Galicia M, Drummond HA, Reddy KK, Falck JR, Roman RJ. Inhibition of 20-HETE production contributes to the vascular responses to nitric oxide. Hypertension 29: 320–325, 1997. doi: 10.1161/01.HYP.29.1.320. [DOI] [PubMed] [Google Scholar]
- 3.Aoki M, Nata T, Morishita R, Matsushita H, Nakagami H, Yamamoto K, Yamazaki K, Nakabayashi M, Ogihara T, Kaneda Y. Endothelial apoptosis induced by oxidative stress through activation of NF-kappaB: antiapoptotic effect of antioxidant agents on endothelial cells. Hypertension 38: 48–55, 2001. doi: 10.1161/01.HYP.38.1.48. [DOI] [PubMed] [Google Scholar]
- 4.Archer SL, Gragasin FS, Wu X, Wang S, McMurtry S, Kim DH, Platonov M, Koshal A, Hashimoto K, Campbell WB, Falck JR, Michelakis ED. Endothelium-derived hyperpolarizing factor in human internal mammary artery is 11,12-epoxyeicosatrienoic acid and causes relaxation by activating smooth muscle BK(Ca) channels. Circulation 107: 769–776, 2003. doi: 10.1161/01.CIR.0000047278.28407.C2. [DOI] [PubMed] [Google Scholar]
- 5.Bang L, Boesgaard S, Nielsen-Kudsk JE, Vejlstrup NG, Aldershvile J. Nitroglycerin-mediated vasorelaxation is modulated by endothelial calcium-activated potassium channels. Cardiovasc Res 43: 772–778, 1999. doi: 10.1016/S0008-6363(99)00116-9. [DOI] [PubMed] [Google Scholar]
- 6.Boerth NJ, Dey NB, Cornwell TL, Lincoln TM. Cyclic GMP-dependent protein kinase regulates vascular smooth muscle cell phenotype. J Vasc Res 34: 245–259, 1997. doi: 10.1159/000159231. [DOI] [PubMed] [Google Scholar]
- 7.Borin TF, Zuccari DA, Jardim-Perassi BV, Ferreira LC, Iskander AS, Varma NR, Shankar A, Guo AM, Scicli G, Arbab AS. HET0016, a selective inhibitor of 20-HETE synthesis, decreases pro-angiogenic factors and inhibits growth of triple negative breast cancer in mice. PLoS One 9: e116247, 2014. doi: 10.1371/journal.pone.0116247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buschmann I, Heil M, Jost M, Schaper W. Influence of inflammatory cytokines on arteriogenesis. Microcirculation 10: 371–379, 2003. doi: 10.1080/mic.10.3-4.371.379. [DOI] [PubMed] [Google Scholar]
- 9.Buschmann I, Schaper W. Arteriogenesis versus angiogenesis: two mechanisms of vessel growth. News Physiol Sci 14: 121–125, 1999. [DOI] [PubMed] [Google Scholar]
- 10.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med 6: 389–395, 2000. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 11.Carrão AC, Chilian WM, Yun J, Kolz C, Rocic P, Lehmann K, van den Wijngaard JP, van Horssen P, Spaan JAE, Ohanyan V, Pung YF, Buschmann I. Stimulation of coronary collateral growth by granulocyte stimulating factor: role of reactive oxygen species. Arterioscler Thromb Vasc Biol 29: 1817–1822, 2009. doi: 10.1161/ATVBAHA.109.186445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L, Ackerman R, Guo AM. 20-HETE in neovascularization. Prostaglandins Other Lipid Mediat 98: 63–68, 2012. doi: 10.1016/j.prostaglandins.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 13.Chen L, Ackerman R, Saleh M, Gotlinger KH, Kessler M, Mendelowitz LG, Falck JR, Arbab AS, Scicli AG, Schwartzman ML, Yang J, Guo AM. 20-HETE regulates the angiogenic functions of human endothelial progenitor cells and contributes to angiogenesis in vivo. J Pharmacol Exp Ther 348: 442–451, 2014. doi: 10.1124/jpet.113.210120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen L, Joseph G, Zhang FF, Nguyen H, Jiang H, Gotlinger KH, Falck JR, Yang J, Schwartzman ML, Guo AM. 20-HETE contributes to ischemia-induced angiogenesis. Vascul Pharmacol 83: 57–65, 2016. doi: 10.1016/j.vph.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen P, Guo M, Wygle D, Edwards PA, Falck JR, Roman RJ, Scicli AG. Inhibitors of cytochrome P450 4A suppress angiogenic responses. Am J Pathol 166: 615–624, 2005. doi: 10.1016/S0002-9440(10)62282-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng J, Ou JS, Singh H, Falck JR, Narsimhaswamy D, Pritchard KA Jr, Schwartzman ML. 20-hydroxyeicosatetraenoic acid causes endothelial dysfunction via eNOS uncoupling. Am J Physiol Heart Circ Physiol 294: H1018–H1026, 2008. doi: 10.1152/ajpheart.01172.2007. [DOI] [PubMed] [Google Scholar]
- 17.Cheng J, Ou JS, Singh H, Falck JR, Narsimhaswamy D, Pritchard KA Jr, Schwartzman ML. 20-Hydroxyeicosatetraenoic acid causes endothelial dysfunction via eNOS uncoupling. Am J Physiol Heart Circ Physiol 294: H1018–H1026, 2008. doi: 10.1152/ajpheart.01172.2007. [DOI] [PubMed] [Google Scholar]
- 18.Cheng J, Wu CC, Gotlinger KH, Zhang F, Falck JR, Narsimhaswamy D, Schwartzman ML. 20-hydroxy-5,8,11,14-eicosatetraenoic acid mediates endothelial dysfunction via IkappaB kinase-dependent endothelial nitric-oxide synthase uncoupling. J Pharmacol Exp Ther 332: 57–65, 2010. doi: 10.1124/jpet.109.159863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Degen A, Millenaar D, Schirmer SH. Therapeutic approaches in the stimulation of the coronary collateral circulation. Curr Cardiol Rev 10: 65–72, 2014. doi: 10.2174/1573403X113099990027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dhanasekaran A, Bodiga S, Gruenloh S, Gao Y, Dunn L, Falck JR, Buonaccorsi JN, Medhora M, Jacobs ER. 20-HETE increases survival and decreases apoptosis in pulmonary arteries and pulmonary artery endothelial cells. Am J Physiol Heart Circ Physiol 296: H777–H786, 2009. doi: 10.1152/ajpheart.01087.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ding H, Triggle CR. Endothelial cell dysfunction and the vascular complications associated with type 2 diabetes: assessing the health of the endothelium. Vasc Health Risk Manag 1: 55–71, 2005. doi: 10.2147/vhrm.1.1.55.58939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding Y, Wu CC, Garcia V, Dimitrova I, Weidenhammer A, Joseph G, Zhang F, Manthati VL, Falck JR, Capdevila JH, Schwartzman ML. 20-HETE induces remodeling of renal resistance arteries independent of blood pressure elevation in hypertension. Am J Physiol Renal Physiol 305: F753–F763, 2013. doi: 10.1152/ajprenal.00292.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dodd T, Jadhav R, Wiggins L, Stewart J, Smith E, Russell JC, Rocic P. MMPs 2 and 9 are essential for coronary collateral growth and are prominently regulated by p38 MAPK. J Mol Cell Cardiol 51: 1015–1025, 2011. doi: 10.1016/j.yjmcc.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dodd T, Wiggins L, Hutcheson R, Smith E, Musiyenko A, Hysell B, Russell JC, Rocic P. Impaired coronary collateral growth in the metabolic syndrome is in part mediated by matrix metalloproteinase 12-dependent production of endostatin and angiostatin. Arterioscler Thromb Vasc Biol 33: 1339–1349, 2013. doi: 10.1161/ATVBAHA.113.301533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Epstein SE, Fuchs S, Zhou YF, Baffour R, Kornowski R. Therapeutic interventions for enhancing collateral development by administration of growth factors: basic principles, early results and potential hazards. Cardiovasc Res 49: 532–542, 2001. doi: 10.1016/S0008-6363(00)00217-0. [DOI] [PubMed] [Google Scholar]
- 26.Escalante B, Erlij D, Falck JR, McGiff JC. Cytochrome P-450 arachidonate metabolites affect ion fluxes in rabbit medullary thick ascending limb. Am J Physiol Cell Physiol 266: C1775–C1782, 1994. [DOI] [PubMed] [Google Scholar]
- 27.Fava C, Montagnana M, Danese E, Sjögren M, Almgren P, Guidi GC, Hedblad B, Engström G, Minuz P, Melander O. The functional variant V433M of the CYP4F2 and the metabolic syndrome in Swedes. Prostaglandins Other Lipid Mediat 98: 31–36, 2012. doi: 10.1016/j.prostaglandins.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 28.Gangadhariah MH, Luther JM, Garcia V, Paueksakon P, Zhang MZ, Hayward SW, Love HD, Falck JR, Manthati VL, Imig JD, Schwartzman ML, Zent R, Capdevila JH, Pozzi A. Hypertension is a major contributor to 20-hydroxyeicosatetraenoic acid-mediated kidney injury in diabetic nephropathy. J Am Soc Nephrol 26: 597–610, 2015. doi: 10.1681/ASN.2013090980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia V, Joseph G, Shkolnik B, Ding Y, Zhang FF, Gotlinger K, Falck JR, Dakarapu R, Capdevila JH, Bernstein KE, Schwartzman ML. Angiotensin II receptor blockade or deletion of vascular endothelial ACE does not prevent vascular dysfunction and remodeling in 20-HETE-dependent hypertension. Am J Physiol Regul Integr Comp Physiol 309: R71–R78, 2015. doi: 10.1152/ajpregu.00039.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gauthier KM, Edwards EM, Falck JR, Reddy DS, Campbell WB. 14,15-epoxyeicosatrienoic acid represents a transferable endothelium-dependent relaxing factor in bovine coronary arteries. Hypertens 45: 666–671, 2005. doi: 10.1161/01.HYP.0000153462.06604.5d. . [DOI] [PubMed] [Google Scholar]
- 31.Grundy SM. Metabolic syndrome pandemic. Arterioscler Thromb Vasc Biol 28: 629–636, 2008. doi: 10.1161/ATVBAHA.107.151092. [DOI] [PubMed] [Google Scholar]
- 32.Guo AM, Arbab AS, Falck JR, Chen P, Edwards PA, Roman RJ, Scicli AG. Activation of vascular endothelial growth factor through reactive oxygen species mediates 20-hydroxyeicosatetraenoic acid-induced endothelial cell proliferation. J Pharmacol Exp Ther 321: 18–27, 2007. doi: 10.1124/jpet.106.115360. [DOI] [PubMed] [Google Scholar]
- 33.Guo AM, Janic B, Sheng J, Falck JR, Roman RJ, Edwards PA, Arbab AS, Scicli AG. The cytochrome P450 4A/F-20-hydroxyeicosatetraenoic acid system: a regulator of endothelial precursor cells derived from human umbilical cord blood. J Pharmacol Exp Ther 338: 421–429, 2011. doi: 10.1124/jpet.111.179036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo AM, Scicli G, Sheng J, Falck JC, Edwards PA, Scicli AG. 20-HETE can act as a nonhypoxic regulator of HIF-1α in human microvascular endothelial cells. Am J Physiol Heart Circ Physiol 297: H602–H613, 2009. doi: 10.1152/ajpheart.00874.2008. [DOI] [PubMed] [Google Scholar]
- 35.Guzik TJ, West NEJ, Pillai R, Taggart DP, Channon KM. Nitric oxide modulates superoxide release and peroxynitrite formation in human blood vessels. Hypertension 39: 1088–1094, 2002. doi: 10.1161/01.HYP.0000018041.48432.B5. [DOI] [PubMed] [Google Scholar]
- 36.Hadi HA, Carr CS, Al Suwaidi J. Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vasc Health Risk Manag 1: 183–198, 2005. [PMC free article] [PubMed] [Google Scholar]
- 37.Harder DR, Lange AR, Gebremedhin D, Birks EK, Roman RJ. Cytochrome P450 metabolites of arachidonic acid as intracellular signaling molecules in vascular tissue. J Vasc Res 34: 237–243, 1997. doi: 10.1159/000159228. [DOI] [PubMed] [Google Scholar]
- 38.Hoopes SL, Garcia V, Edin ML, Schwartzman ML, Zeldin DC. Vascular actions of 20-HETE. Prostaglandins Other Lipid Mediat 120: 9–16, 2015. doi: 10.1016/j.prostaglandins.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hutcheson R, Chaplin J, Hutcheson B, Borthwick F, Proctor S, Gebb S, Jadhav R, Smith E, Russell JC, Rocic P. miR-21 normalizes vascular smooth muscle proliferation and improves coronary collateral growth in metabolic syndrome. FASEB J 28: 4088–4099, 2014. doi: 10.1096/fj.14-251223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hutcheson R, Rocic P. The metabolic syndrome, oxidative stress, environment, and cardiovascular disease: the great exploration. Exp Diabetes Res 2012: 271028, 2012. doi: 10.1155/2012/271028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hutcheson R, Terry R, Chaplin J, Smith E, Musiyenko A, Russell JC, Lincoln T, Rocic P. MicroRNA-145 restores contractile vascular smooth muscle phenotype and coronary collateral growth in the metabolic syndrome. Arterioscler Thromb Vasc Biol 33: 727–736, 2013. doi: 10.1161/ATVBAHA.112.301116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hutcheson R, Terry R, Hutcheson B, Jadhav R, Chaplin J, Smith E, Barrington R, Proctor SD, Rocic P. miR-21-mediated decreased neutrophil apoptosis is a determinant of impaired coronary collateral growth in metabolic syndrome. Am J Physiol Heart Circ Physiol 308: H1323–H1335, 2015. doi: 10.1152/ajpheart.00654.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iantorno M, Campia U, Di Daniele N, Nistico S, Forleo GB, Cardillo C, Tesauro M. Obesity, inflammation and endothelial dysfunction. J Biol Regul Homeost Agents 28: 169–176, 2014. [PubMed] [Google Scholar]
- 44.Inoue K, Sodhi K, Puri N, Gotlinger KH, Cao J, Rezzani R, Falck JR, Abraham NG, Laniado-Schwartzman M. Endothelial-specific CYP4A2 overexpression leads to renal injury and hypertension via increased production of 20-HETE. Am J Physiol Renal Physiol 297: F875–F884, 2009. doi: 10.1152/ajprenal.00364.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ishizuka T, Cheng J, Singh H, Vitto MD, Manthati VL, Falck JR, Laniado-Schwartzman M. 20-Hydroxyeicosatetraenoic acid stimulates nuclear factor-kappaB activation and the production of inflammatory cytokines in human endothelial cells. J Pharmacol Exp Ther 324: 103–110, 2008. doi: 10.1124/jpet.107.130336. [DOI] [PubMed] [Google Scholar]
- 46.Jadhav R, Dodd T, Smith E, Bailey E, Delucia AL, Russell JC, Madison R, Potter B, Walsh K, Jo H, Rocic P. Angiotensin type I receptor blockade in conjunction with enhanced Akt activation restores coronary collateral growth in the metabolic syndrome. Am J Physiol Heart Circ Physiol 300: H1938–H1949, 2011. doi: 10.1152/ajpheart.00282.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kauser K, Clark JE, Masters BS, Ortiz de Montellano PR, Ma YH, Harder DR, Roman RJ. Inhibitors of cytochrome P-450 attenuate the myogenic response of dog renal arcuate arteries. Circ Res 68: 1154–1163, 1991. doi: 10.1161/01.RES.68.4.1154. [DOI] [PubMed] [Google Scholar]
- 48.Kobayashi N, DeLano FA, Schmid-Schönbein GW. Oxidative stress promotes endothelial cell apoptosis and loss of microvessels in the spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol 25: 2114–2121, 2005. doi: 10.1161/01.ATV.0000178993.13222.f2. [DOI] [PubMed] [Google Scholar]
- 49.Kojda G, Harrison D. Interactions between NO and reactive oxygen species: pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovasc Res 43: 562–571, 1999. doi: 10.1016/S0008-6363(99)00169-8. [DOI] [PubMed] [Google Scholar]
- 50.Laffer CL, Laniado-Schwartzman M, Nasjletti A, Elijovich F. 20-HETE and circulating insulin in essential hypertension with obesity. Hypertension 43: 388–392, 2004. doi: 10.1161/01.HYP.0000112224.87290.3a. [DOI] [PubMed] [Google Scholar]
- 51.Lai G, Wu J, Liu X, Zhao Y. 20-HETE induces hyperglycemia through the cAMP/PKA-PhK-GP pathway. Mol Endocrinol 26: 1907–1916, 2012. doi: 10.1210/me.2012-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liang CJ, Ives HE, Yang CM, Ma YH. 20-HETE inhibits the proliferation of vascular smooth muscle cells via transforming growth factor-beta. J Lipid Res 49: 66–73, 2008. doi: 10.1194/jlr.M700155-JLR200. [DOI] [PubMed] [Google Scholar]
- 53.Liu ZJ, Shirakawa T, Li Y, Soma A, Oka M, Dotto GP, Fairman RM, Velazquez OC, Herlyn M. Regulation of Notch1 and Dll4 by vascular endothelial growth factor in arterial endothelial cells: implications for modulating arteriogenesis and angiogenesis. Mol Cell Biol 23: 14–25, 2003. doi: 10.1128/MCB.23.1.14-25.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsunaga T, Warltier DC, Weihrauch DW, Moniz M, Tessmer J, Chilian WM. Ischemia-induced coronary collateral growth is dependent on vascular endothelial growth factor and nitric oxide. Circulation 102: 3098–3103, 2000. doi: 10.1161/01.CIR.102.25.3098. [DOI] [PubMed] [Google Scholar]
- 55.McNamee CJ, Kappagoda CT, Kunjara R, Russell JC. Defective endothelium-dependent relaxation in the JCR:LA-corpulent rat. Circ Res 74: 1126–1132, 1994. doi: 10.1161/01.RES.74.6.1126. [DOI] [PubMed] [Google Scholar]
- 56.Moraes F, Paye J, Mac Gabhann F, Zhuang ZW, Zhang J, Lanahan AA, Simons M. Endothelial cell-dependent regulation of arteriogenesis. Circ Res 113: 1076–1086, 2013. doi: 10.1161/CIRCRESAHA.113.301340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mouquet F, Cuilleret F, Susen S, Sautière K, Marboeuf P, Ennezat PV, McFadden E, Pigny P, Richard F, Hennache B, Vantyghem MC, Bertrand M, Dallongeville J, Jude B, Van Belle E. Metabolic syndrome and collateral vessel formation in patients with documented occluded coronary arteries: association with hyperglycaemia, insulin-resistance, adiponectin and plasminogen activator inhibitor-1. Eur Heart J 30: 840–849, 2009. doi: 10.1093/eurheartj/ehn569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Muinck ED. Gene and cell therapy for heart failure. Antioxid Redox Signal 11: 2025–2042, 2009. doi: 10.1089/ars.2009.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Natarajan R, Gu JL, Rossi J, Gonzales N, Lanting L, Xu L, Nadler J. Elevated glucose and angiotensin II increase 12-lipoxygenase activity and expression in porcine aortic smooth muscle cells. Proc Natl Acad Sci USA 90: 4947–4951, 1993. doi: 10.1073/pnas.90.11.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van den Oever IA, Raterman HG, Nurmohamed MT, Simsek S. Endothelial dysfunction, inflammation, and apoptosis in diabetes mellitus. Mediators Inflamm 2010: 792393, 2010. doi: 10.1155/2010/792393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reed R, Kolz C, Potter B, Rocic P. The mechanistic basis for the disparate effects of angiotensin II on coronary collateral growth. Arterioscler Thromb Vasc Biol 28: 61–67, 2008. doi: 10.1161/ATVBAHA.107.154294. [DOI] [PubMed] [Google Scholar]
- 62.Reed R, Potter B, Smith E, Jadhav R, Villalta P, Jo H, Rocic P. Redox-sensitive Akt and Src regulate coronary collateral growth in metabolic syndrome. Am J Physiol Heart Circ Physiol 296: H1811–H1821, 2009. doi: 10.1152/ajpheart.00920.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Regner KR, Zuk A, Van Why SK, Shames BD, Ryan RP, Falck JR, Manthati VL, McMullen ME, Ledbetter SR, Roman RJ. Protective effect of 20-HETE analogues in experimental renal ischemia reperfusion injury. Kidney Int 75: 511–517, 2009. doi: 10.1038/ki.2008.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rocic P. Why is coronary collateral growth impaired in type II diabetes and the metabolic syndrome? Vascul Pharmacol 57: 179–186, 2012. doi: 10.1016/j.vph.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rocic P, Kolz C, Reed R, Potter B, Chilian WM. Optimal reactive oxygen species concentration and p38 MAP kinase are required for coronary collateral growth. Am J Physiol Heart Circ Physiol 292: H2729–H2736, 2007. doi: 10.1152/ajpheart.01330.2006. [DOI] [PubMed] [Google Scholar]
- 66.van Royen N, Piek JJ, Buschmann I, Hoefer I, Voskuil M, Schaper W. Stimulation of arteriogenesis; a new concept for the treatment of arterial occlusive disease. Cardiovasc Res 49: 543–553, 2001. doi: 10.1016/S0008-6363(00)00206-6. [DOI] [PubMed] [Google Scholar]
- 67.Sánchez-Mendoza A, López-Sánchez P, Vázquez-Cruz B, Rios A, Martínez-Ayala S, Escalante B. Angiotensin II modulates ion transport in rat proximal tubules through CYP metabolites. Biochem Biophys Res Commun 272: 423–430, 2000. doi: 10.1006/bbrc.2000.2807. [DOI] [PubMed] [Google Scholar]
- 68.Santhanam AV, d’Uscio LV, Smith LA, Katusic ZS. Uncoupling of eNOS causes superoxide anion production and impairs NO signaling in the cerebral microvessels of hph-1 mice. J Neurochem 122: 1211–1218, 2012. doi: 10.1111/j.1471-4159.2012.07872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sasmaz H, Yilmaz MB. Coronary collaterals in obese patients: impact of metabolic syndrome. Angiology 60: 164–168, 200910.1177/0003319708316007. [DOI] [PubMed] [Google Scholar]
- 70.Savoia C, Sada L, Zezza L, Pucci L, Lauri FM, Befani A, Alonzo A, Volpe M. Vascular inflammation and endothelial dysfunction in experimental hypertension. Int J Hypertens 2011: 281240, 2011. doi: 10.4061/2011/281240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Scatena M, Liaw L, Giachelli CM. Osteopontin: a multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler Thromb Vasc Biol 27: 2302–2309, 2007. doi: 10.1161/ATVBAHA.107.144824. [DOI] [PubMed] [Google Scholar]
- 72.Schaper W. Collateral circulation: past and present. Basic Res Cardiol 104: 5–21, 2009. doi: 10.1007/s00395-008-0760-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schaper W, Scholz D. Factors regulating arteriogenesis. Arterioscler Thromb Vasc Biol 23: 1143–1151, 2003. doi: 10.1161/01.ATV.0000069625.11230.96. [DOI] [PubMed] [Google Scholar]
- 74.Singh H, Cheng J, Deng H, Kemp R, Ishizuka T, Nasjletti A, Schwartzman ML. Vascular cytochrome P450 4A expression and 20-hydroxyeicosatetraenoic acid synthesis contribute to endothelial dysfunction in androgen-induced hypertension. Hypertension 50: 123–129, 2007. doi: 10.1161/HYPERTENSIONAHA.107.089599. [DOI] [PubMed] [Google Scholar]
- 75.Toyota E, Warltier DC, Brock T, Ritman E, Kolz C, O’Malley P, Rocic P, Focardi M, Chilian WM. Vascular endothelial growth factor is required for coronary collateral growth in the rat. Circulation 112: 2108–2113, 2005. doi: 10.1161/CIRCULATIONAHA.104.526954. [DOI] [PubMed] [Google Scholar]
- 76.Tsai IJ, Croft KD, Puddey IB, Beilin LJ, Barden A. 20-Hydroxyeicosatetraenoic acid synthesis is increased in human neutrophils and platelets by angiotensin II and endothelin-1. Am J Physiol Heart Circ Physiol 300: H1194–H1200, 2011. doi: 10.1152/ajpheart.00733.2010. [DOI] [PubMed] [Google Scholar]
- 77.Tsao PS, Ma XL, Lefer AM. Activated neutrophils aggravate endothelial dysfunction after reperfusion of the ischemic feline myocardium. Am Heart J 123: 1464–1471, 1992. doi: 10.1016/0002-8703(92)90796-X. [DOI] [PubMed] [Google Scholar]
- 78.Tsihlis ND, Oustwani CS, Vavra AK, Jiang Q, Keefer LK, Kibbe MR. Nitric oxide inhibits vascular smooth muscle cell proliferation and neointimal hyperplasia by increasing the ubiquitination and degradation of UbcH10. Cell Biochem Biophys 60: 89–97, 2011. doi: 10.1007/s12013-011-9179-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Uno K, Iuchi Y, Fujii J, Sugata H, Iijima K, Kato K, Shimosegawa T, Yoshimura T. In vivo study on cross talk between inducible nitric-oxide synthase and cyclooxygenase in rat gastric mucosa: effect of cyclooxygenase activity on nitric oxide production. J Pharmacol Exp Ther 309: 995–1002, 2004. doi: 10.1124/jpet.103.061283. [DOI] [PubMed] [Google Scholar]
- 80.Wang JH, Sexton DM, Redmond HP, Watson RW, Croke DT, Bouchier-Hayes D. Intercellular adhesion molecule-1 (ICAM-1) is expressed on human neutrophils and is essential for neutrophil adherence and aggregation. Shock 8: 357–361, 1997. doi: 10.1097/00024382-199711000-00007. [DOI] [PubMed] [Google Scholar]
- 81.Wang JS, Singh H, Zhang F, Ishizuka T, Deng H, Kemp R, Wolin MS, Hintze TH, Abraham NG, Nasjletti A, Laniado-Schwartzman M. Endothelial dysfunction and hypertension in rats transduced with CYP4A2 adenovirus. Circ Res 98: 962–969, 2006. doi: 10.1161/01.RES.0000217283.98806.a6. [DOI] [PubMed] [Google Scholar]
- 82.Ward NC, Rivera J, Hodgson J, Puddey IB, Beilin LJ, Falck JR, Croft KD. Urinary 20-hydroxyeicosatetraenoic acid is associated with endothelial dysfunction in humans. Circulation 110: 438–443, 2004. doi: 10.1161/01.CIR.0000136808.72912.D9. [DOI] [PubMed] [Google Scholar]
- 83.Woodfin A, Voisin M-B, Imhof BA, Dejana E, Engelhardt B, Nourshargh S. Endothelial cell activation leads to neutrophil transmigration as supported by the sequential roles of ICAM-2, JAM-A, and PECAM-1. Blood 113: 6246–6257, 2009. doi: 10.1182/blood-2008-11-188375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu CC, Gupta T, Garcia V, Ding Y, Schwartzman ML. 20-HETE and blood pressure regulation: clinical implications. Cardiol Rev 22: 1–12, 2014. doi: 10.1097/CRD.0b013e3182961659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang HT, Yan Z, Abraham JA, Terjung RL. VEGF121- and bFGF-induced increase in collateral blood flow requires normal nitric oxide production. Am J Physiol Heart Circ Physiol 280: H1097–H1104, 2001. [DOI] [PubMed] [Google Scholar]
- 86.Yang J, Park Y, Zhang H, Gao X, Wilson E, Zimmer W, Abbott L, Zhang C. Role of MCP-1 in tumor necrosis factor-α-induced endothelial dysfunction in type 2 diabetic mice. Am J Physiol Heart Circ Physiol 297: H1208–H1216, 2009. doi: 10.1152/ajpheart.00396.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yun J, Rocic P, Pung YF, Belmadani S, Carrao AC, Ohanyan V, Chilian WM. Redox-dependent mechanisms in coronary collateral growth: the “redox window” hypothesis. Antioxid Redox Signal 11: 1961–1974, 2009. doi: 10.1089/ars.2009.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zou AP, Imig JD, Ortiz de Montellano PR, Sui Z, Falck JR, Roman RJ. Effect of P-450 omega-hydroxylase metabolites of arachidonic acid on tubuloglomerular feedback. Am J Physiol 266: F934–F941, 1994. [DOI] [PubMed] [Google Scholar]