

Abstract
We sought to determine if adenosquamous proliferation of early cellular radial sclerosing lesions of the breast harbours hot spot mutations and to help clarify its relationship to low‐grade adenosquamous carcinoma as a potential form of early neoplasia. Four low‐grade adenosquamous carcinomas, early radial sclerosing lesions from 13 individuals, and 4 benign proliferative breast lesions were microdissected and assessed with a 50‐gene Hot‐spot cancer panel. Early radial sclerosing lesions were selectively microdissected concentrating on their adenosquamous proliferation (nidus). Hot spot mutations in PIK3CA were detected in ten (77% of) radial sclerosing lesions, in one low‐grade adenosquamous carcinoma, and in usual ductal hyperplasia and apocrine adenosis. Over three quarters of individuals with cellular (adenosquamous proliferation rich) early radial sclerosing lesions tested harboured somatic mutations in PIK3CA suggesting that adenosquamous proliferation is a clonal lesion. Its relationship to low‐grade adenosquamous carcinoma remains unclear in view of the small sample size and unmatched radial sclerosing lesions and low‐grade adenosquamous carcinomas.
Keywords: adenosquamous proliferation, breast, complex sclerosing lesion, low‐grade adenosquamous carcinoma, radial sclerosing lesion, radial scar, next‐generation sequencing
Introduction
There is growing evidence that breast lesions characterized by adenosquamous proliferation (ASP), whether in the form of a radial sclerosing lesion (incorporating radial scar and complex sclerosing lesion; RSL), a sclerosing papilloma or similar lesions, are phenotypically similar to low‐grade adenosquamous carcinoma (LGASC) 1; their distinction is often subjective and is poorly defined in the literature. They may indeed form a continuous morphological spectrum with benign sclerosing lesions at one end and LGASC at the other, the latter typically being more extensive, and with prominent desmoplastic stroma 1, 2. RSLs have been shown to harbour areas that are clonal and neoplastic and clonal differences are seen within different areas of the same RSL 3. Macrodissection studies have shown PI3K/AKT/MTOR pathway mutations in over 63% of RSL, greater than twofold more frequent than in breast carcinoma 4. The PI3K/AKT/MTOR pathway has been termed a pro‐survival pathway because its activation is associated with cell survival, proliferation, migration and angiogenesis and as such it has a key role in normal human development. PI3K and PTEN have phosphorylating and dephosphorylating effects respectively on phosphatidylinositol. Phosphorylation by PI3K increases the activity of downstream kinases AKT and MTOR. PTEN thus has tumour suppressor activity. Inactivating germline mutations of PTEN are associated with hamartoma‐tumour syndromes. Its somatic loss is associated with various sporadic human cancers 5. Perhaps unsurprisingly, germline and mosaic (genetic or somatic) mutations in the PI3K/AKT/MTOR pathway are associated with overgrowth syndromes while somatic mutations are implicated in sporadic malignancies 6.
Phosphatidylinositol‐3‐kinase (PIK3CA) mutations are common in proliferative breast lesions 7 and indeed the majority component of RSL, particularly in late lesions, represents proliferative breast disease (PBD) such as usual ductal hyperplasia 8, 9. A similar lesion, which may form a spectrum with RSL, is infiltrative epitheliosis, an infrequently diagnosed breast lesion that comprises ASP in association with florid usual ductal hyperplasia in a non‐stellate configuration 10. Like RSL, infiltrative epitheliosis has been shown to harbour PIK3CA mutations, so is also thought to represent a clonal proliferation 11. The neoplastic risk of RSL, the best documented of these lesions, for common cancers may merely reside in the fact that they occur in the context of PBD, which has an inherent neoplastic risk that is essentially the same as that stated for RSL 12. It may in fact be the case that ASP arises in PBD due to this neoplastic risk, and that its subsequent growth and associated desmoplasia distorts the tissue within which it arises. The final histological diagnosis essentially depends upon whether this lesion resides centrally within a focus of PBD giving a stellate configuration (RSL), complicates a papilloma (sclerosing papilloma), arises intermixed with PBD, and/or papillomatosis at the nipple (syringomatous tumour), in a subareolar location (subareolar sclerosing duct hyperplasia) or in breast parenchyma (infiltrating epitheliosis). Although PBD itself is a risk factor for common cancers, ASP may be a direct precursor of low‐grade metaplastic carcinoma, particularly LGASC 1. Our aim was to determine whether microdissection specifically targeting the nidus and particularly the ASP of early RSL would reveal genetic mutations, isolated from that of background PBD. Four LGASCs, 13 early or cellular RSLs and four cases of PBD without ASP were assessed with a 50‐gene hot‐spot cancer panel using next generation (massive parallel) sequencing (NGS).
Materials and methods
Case selection
Archived cases from a 10‐year period were identified utilizing a SNOMED search to identify examples of LGASC as defined by Rosen 13 and sufficiently cellular early RSL to enable adequate DNA yield, using the accepted definition of Anderson and Battersby 8, 9. Four examples of LGASC were identified and of sufficient cellularity such that over 90% of the lesion comprised tumour cells within prominent desmoplastic stroma, with several examples showing perineural invasion (Figure 1). Over a hundred cases with RSL were examined from which 13 with cellular early RSL were selected, some of which were multiple and/or bilateral. ASP in several instances formed a discrete‐tumoural mass confined within the RSL (Figure 2), but in most instances they ramified between benign ducts, which often showed usual ductal hyperplasia, which made it difficult to obtain samples of ASP exclusive of background breast tissue while obtaining a sufficient yield of assessable DNA. While over 90% of the tissue targeted for microdissection was from the nidus of the RSLs, the ratio of ASP to nidus density ranged from 10% to greater than 90% because these are often paucicellular lesions. Four separate cases of benign proliferative disease (controls) without a sclerosing lesion were selected because the examples of RSL had insufficient uninvolved tissue to achieve a pure DNA yield. It is acknowledged this is less ideal than selecting control tissue from same subjects. However, gene mutations are well documented in proliferative breast tissue 7. Positive and negative controls (eg, for known PIK3CA mutations) were unavailable. The cases were anonymized prior to assessment. The study received institutional approval from the source laboratory. The cases were assessed with a cancer hot‐spot panel to detect single nucleotide variants (SNVs) and short insertions and deletions (indels). (Table 1)
Figure 1.
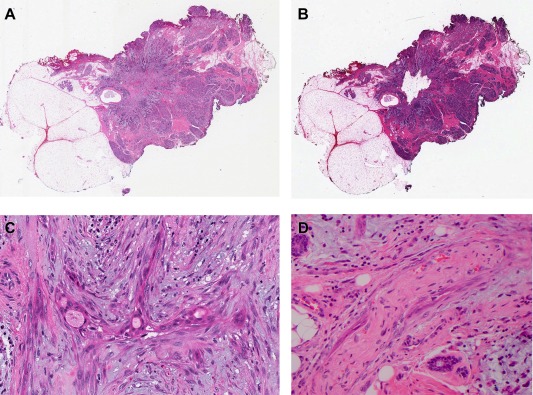
Sample A1: Low‐grade adenosquamous carcinoma. Displayed somatic mutation in PIK3CA: H1047L (H&E).
A. Whole slide scan – pre‐dissection.
B. Whole slide scan – post‐dissection.
C. Adenosquamous proliferation of LGASC (scan 28×).
D. Perineural invasion (scan 40×).
Figure 2.
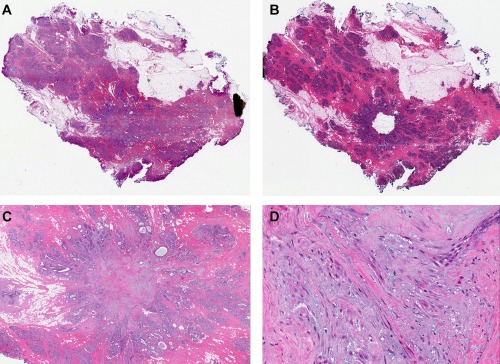
Sample B3: Early radial sclerosing lesion (RSL) with dense tumour‐like nidus. Displayed somatic mutation in PIK3CA: H1047R (H&E).
A. Whole slide scan – pre‐dissection.
B. Whole slide scan – post‐dissection.
C. Adenosquamous proliferation of early RSL (scan 1.7×).
D. Adenosquamous proliferation of early RSL (scan 29×).
Table 1.
Case and control samples selected for cancer hot‐spot sequencing
| Sample code | Diagnosis | Age (yr) | Laterality | Size (mm) | ASP nidus density (%) | Estimated sample purity (%) | DNA yield (ng/µL) |
|---|---|---|---|---|---|---|---|
| A1 | LGASC | 45 | Right | 25 | >90 | >90 | 31.2 |
| A2 | LGASC arising in IE? | 48 | Right | 20 | >90 | >90 | 42.8 |
| A3 | LGASC | 78 | Right | 17 | >90 | >90 | 18.5 |
| A4 | LGASC | 65 | Right | 13.5 | >90 | >90 | 17.2 |
| B1 | RSL x 2 (incorporated intraduct papilloma in one) | 41 | Left | <10 | 40 | >90 | 30.2 |
| B2 | RSL | 45 | Left | 5 | 10 | >90 | 33.4 |
| B3 | RSL | 45 | Right | Each <10 | >90 | >90 | 11 |
| B4 | RSL x 4 | 46 | Left | 2‐5 | 80 | >90 | 25 |
| B5 | RSL | 44 | Right | 12 | 30 | >90 | 15.8 |
| B6 | RSL | 48 | Left | 10 | 30 | >90 | 30.8 |
| B7 | RSL | 49 | Left | 10 | 40 | >90 | 28 |
| B8 | RSL (BRCA mutation, previous left carcinoma) | 38 | Bilateral:Right Left | <10<10 | 1050 | >90 | 23.2 |
| B9 | RSL x 5 | 45 | Left | Each <10 | 10 | >90 | 32 |
| B10 | RSL (adenomyoepithelial hyperplasia present) | 38 | Left | 12 | 30 | >90 | 19.5 |
| B11 | RSL | 40 | Bilateral:Right Left | ≤128 | 50 | >90 | 23.2 |
| B12 | RSL | 47 | Left | 7 | 35 | >90 | 37 |
| B13 | RSL | 24 | Right | <10 | 40 | >90 | 38.2 |
| C1 | UDH | 56 | N/A | N/A | >50 | >90 | 12.8 |
| C2 | UDH | 49 | N/A | N/A | >50 | >90 | 37.2 |
| C3 | UDH, CCC | 32 | N/A | N/A | >50 | >90 | 22 |
| C4 | AA | 53 | N/A | N/A | >50 | >90 | 26.4 |
Sample codes: A = LGASC, B = RSL, C = PBD controls.
AA: apocrine adenosis. CCC: columnar cell change. IE: Infiltrating epitheliosis. LGASC: low‐grade adenosquamous carcinoma. PBD: proliferative breast disease. RSL: radial sclerosing lesion. UDH: usual ductal hyperplasia. N/A: not available.
Microdissection and DNA extraction
Serial sections (8 μm) of formalin fixed paraffin embedded (FFPE) tissue from each case were similarly aligned on Superfrost™ Ultra Plus Adhesion Slides (Thermo Scientific). The first and sixth serial sections were stained with haematoxylin and eosin (H&E) (Figure 3). Using the first H&E section regions enriched with RSLs were imaged and selected using a P.A.L.M. DuoFlexCombi System (Zeiss). Reference points throughout sections one to six were used to map the RSL‐rich regions‐of‐interest onto each of these slides in turn. The laser on the P.A.L.M. DuoFlexCombi System was used to etch around the ASP foci on the unstained slides and the second H&E stained slide was used to confirm the regions of interest retained the enriched RSL morphology throughout the serial sections. In each case, the unstained cellular nidus, rich in ASP, were then precisely microdissected using a sterile needle under a stereo‐microscope. Genomic DNA was then purified using the GeneRead DNA FFPE Kit (Qiagen). Briefly, following deparafinization, proteinase K digestion was performed at 56°C for 1 hour. Formaldehyde modification of nucleic acids was reversed by incubation with buffer FTB and Uracil‐N‐glycosylase added to reverse cytosine deamination. Following RNase digestion, DNA was washed on the spin column, eluted and quantified using a Qubit® 3.0 Fluorometer. DNA were then stored at −20°C until sequence analysis.
Figure 3.
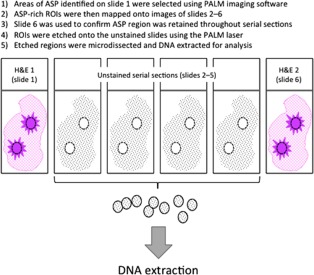
Schematic diagram of tissue selection for microdissection on PALM laser. ASP: adenosquamous proliferation. ROI: region of interest.
Hot‐spot panel with next generation (massive parallel) sequencing
We targeted a set of ∼2800 variants in known cancer genes present in the COSMIC database using an Ion Ampliseq Cancer hotspot v2 assay (Thermofisher Scientific) 14. The data were collected using TorrentSuite 4.2 and analyzed using Ion Reporter 4.6. Data were filtered to remove intronic, synonymous and common variants (based on 1000 genomes frequencies >1%). Overall, there was insufficient extracted DNA to allow confirmation of results by an orthogonal method (Sanger sequencing). However, there is well‐documented high concordance between NGS of whole‐exome and for cancer hot‐spot mutations, and Sanger sequencing. The latter is of use when there is poor gene coverage or there are novel variants detected by NGS 15, 16.
Results
Cases and controls were all female. The age at diagnosis of LGASC ranged from 45 to 78 (mean 59) years whereas for the 13 with early RSL it was 24–49 (mean 42) years. The age of the individuals with PBD ranged from 32 to 56 (mean 47) years. The 21 samples (4 LGASC, 13 RSL and 4 PBD) were assessed with a cancer hotspot‐sequencing panel, yielding between 11 and 81 variants per sample (average 21/sample). In most there were less than 30 variants, but one sample had nearly 81 variants, which were likely FFPE artefacts present at extremely low allelic frequency (61/81 variants with allele fraction < 0.05, and 62/81 C–T or A–G variants). Overall there were 448 candidate variants in 21 samples. The average depth of sequencing for variants was 988X (range 54–2000X), with 22 being below 500X. After filtering to remove intronic and synonymous coding region variants, as well as variants found in the thousand genomes project 17 at >1% (common inherited variants) there were 123 variants (average 5.8/sample). When working with DNA from FFPE tissue, there is a significant low level of artefactual mutation resulting in variants with an allele frequency of 1–10%. Unfortunately, there are also likely real variants present at low level. Hence the traditional approach is to exclude variants below 5% when looking for likely pathogenic variants, especially in tissues that have been selected to contain likely high mutation loads. When we excluded variants that were only present at a very low frequency in each sample (<5% allele frequency), 26 SNV remained (Table 2). Of these, 13 were in PIK3CA, of which 11 were variants affecting codon 1047, while the remaining 2 affected codon 545. Ten of the thirteen (77%) B samples (RSL) harboured mutations in PIK3CA of which eight (61.5%) had A/G (His1047Arg) mutations and two (15.5%) had G/A (Glu545Lys) mutations. In contrast, the A (LGASC) and C (PBD) samples had A/T (His1047Leu) mutations in one and two individuals respectively. The variants were generally present at 6–28% allelic frequency in each sample. There were a number of variants at high allelic frequencies (eg, JAK3 p.Val722Ile) which were deemed likely to be rare inherited heterozygous variants. Other variants of note were a PTEN variant present at low level in sample A4 (Arg558Cys) and an AKT variant present at low level in sample C2 (AKT1 Glu17Lys). The hot‐spot variants detected were not novel and included known tumour drivers. Hence, the chance of them being false positives was regarded to be zero. As stated above, Sanger confirmations are usually employed when a variant is novel.
Table 2.
Results of cancer hot‐spot sequencing
| Sample code | Gene | Locus | Type | Ref | Genotype | Variant Allele Frequency | Exon | Protein |
|---|---|---|---|---|---|---|---|---|
| A1 | PIK3CA | chr3:178952085 | SNV | A | A/T | 0.15 | 21 | p.His1047Leu |
| A2 | JAK3 | chr19:17945696 | SNV | C | C/T | 0.54 | 16 | p.Val722Ile |
| A3 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| A4 | PDGFRAPTENSTK11 | chr4:55141026chr10:89717676chr19:1220394 | SNVSNVSNV | GCG | G/AC/TG/A | 0.070.070.06 | 7124 | p.Arg234Gln p.Arg558Cys p.Gly163Ser |
| B1 | PIK3CARET | chr3:178952085chr10:43609955 | SNVSNV | AC | A/GC/T | 0.060.06 | 2111 | p.His1047Arg p.Thr636Met |
| B2 | PIK3CA | chr3:178952085 | SNV | A | A/G | 0.08 | 21 | p.His1047Arg |
| B3 | ATMJAK3PIK3CA | chr11:108138003chr19:17945696chr3:178952085 | SNVSNVSNV | TCA | T/CC/TA/G | 0.600.470.15 | 171621 | p.Phe858Leu p.Val722Ile p.His1047Arg |
| B4 | PIK3CA | chr3:178952085 | SNV | A | A/G | 0.08 | 21 | p.His1047Arg |
| B5 | PIK3CA | chr3:178952085 | SNV | A | A/G | 0.26 | 21 | p.His1047Arg |
| B6 | ATMPIK3CA | chr11:108155132chr3:178952085 | SNVSNV | GA | G/AA/G | 0.480.28 | 2621 | p.Ala1309Thr p.His1047Arg |
| B7 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| B8 | SMAD4PIK3CA | chr18:4860475chr3:178952085 | SNVSNV | AA | A/GA/G | 0.520.27 | 1221 | p.Ile525Val p.His1047Arg |
| B9 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| B10 | PIK3CA | chr3:178936091 | SNV | G | G/A | 0.19 | 10 | p.Glu545Lys |
| B11 | PIK3CA | chr3:178936091 | SNV | G | G/A | 0.16 | 10 | p.Glu545Lys |
| B12 | PIK3CASMARCB1 | chr3:178952085chr22:24134064 | SNVSNV | AC | A/GC/A | 0.260.06 | 212 | p.His1047Arg p.Thr72Lys |
| B13 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| C1 | PIK3CA | chr3:178952085 | SNV | A | A/T | 0.16 | 21 | p.His1047Leu |
| C2 | AKT1 | chr14:105246551 | SNV | C | C/T | 0.08 | 3 | p.Glu17Lys |
| C3 | SMARCB1 | chr22:24134064 | SNV | C | C/A | 0.06 | 2 | p.Thr72Lys |
| C4 | PIK3CA | chr3:178952085 | SNV | A | A/T | 0.17 | 21 | p.His1047Leu |
Sample codes: A1–A4 = LGASC, B1–B13 = RSL, C1–C4 = PBD controls.
Genotype codes: A: adenine. C: cytosine. G: guanine. T: thymine. SNV: single nucleotide variant.
Discussion
RSL is an apparently self‐limited benign sclerosing lesion, categorized into early and late forms 8, 9. The phenotypic similarity of ASP in benign sclerosing lesions to the characteristic neoplastic ducts of LGASC suggests that ASP represents a form of early neoplasia 1, 2. Clonality has been demonstrated within RSL 3, 4 and more recently in infiltrating epitheliosis 11, lesions that contain ASP. We sought to determine if selective microdissection of the ASP‐rich nidus of early RSLs, attempting to avoid sampling background benign PBD, would reveal evidence of specific mutations and show evidence of clonality. PIK3CA mutations were present in 13 samples. Ten (77%) of the RSLs harboured mutations in PIK3CA of which eight (61.5%) had H1047R mutations and two (15.5%) had E545K mutations, while one LGASC and two PBD (usual ductal hyperplasia and apocrine adenosis) samples had H1047L mutations. One LGASC showed a PTEN (Arg558Cys) mutation. Our results support the notion that ASP in the setting of RSL is a clonal proliferation, but which appears to involute more often, creating a paucicellular elastotic ‘late’ RSL 1. However, because ASP infrequently formed a pure mass, there was a variable content of normal breast tissue and PBD, which is difficult to avoid because the ASP arises within PBD. Our results suggest that the presence of PIK3CA mutations in RSL may not be only attributable to the common association of usual ductal hyperplasia, which has recently been suggested to be an early clonal neoplastic proliferation [23], and that ASP may be a clonal lesion in its own right. Its relationship to LGASC remains unclear in view of the small sample size and the fact that our RSL and LGASC samples were unmatched because of the nature of the lesions in the cases selected.
The PI3Ka protein is a lipid kinase involved in cellular processes vital for cancer progression, such as cell growth, proliferation, motility, survival and metabolism. Dysregulation of PI3Ka signaling is therefore one of the most frequent oncogenic events. Gain of function somatic mutations within the gene encoding PIK3CA on chromosome 3q26.3 are frequently observed in a variety of human tumors, including in up to one third of breast carcinomas. One of the commonest and most frequently studied mutations is an amino acid substitution at position 1047 from a histidine (H) to an arginine (R) H1047R (His1047Arg) 18, which results in a gain of protein function. In mouse models, over expression of PIK3CAH1047R has been shown to induce mammary cells to de‐differentiate into a multipotent stem cell state and to then differentiate into either luminal, basal or dual cell lineages. This induced cell plasticity results in multi‐lineage tumours 19, 20. Other common gain of function mutations in PIK3CA are found at codons 542 and 545.
Studies of early neoplasia in breast have shown that single nucleotide variations (mutations) are common, that aneuploidy may also be present and that these changes may be detected in a patient's early neoplasia and associated cancer indicating a common ancestral clone. However, although activating PIK3CA mutations are commonly detected in early neoplasia and are common in breast cancer, they do not necessarily correlate with progression to invasive carcinoma or play a prominent role in promoting the progression to cancer at an early stage 21. So, while PIK3CA mutations may provide a growth advantage in early neoplasia they may not be present in any ensuing carcinoma, which might explain why they were more common in RSL than in LGASC in this series. PIK3CA mutations are present in up to 50% of benign proliferative breast lesions (usual ductal hyperplasia and columnar cell change) 7, in 22% and 42% of intraduct papillomata with and without hyperplasia 22, and in nearly 64% of macrodissected radial scars 4. It has recently been proposed that due to evidence of i) activating mutations in the PI3K/AKT/MTOR pathway, ii) loss of heterozygosity and iii) its mass forming proliferative nature, usual ductal hyperplasia is best regarded as an early neoplastic intraductal proliferation 23. Indeed, PIK3CA mutations are common in infiltrating epitheliosis, a lesion which contains mass forming usual ductal hyperplasia 11. Papillomata without cytological atypia have an up to two fold increased risk of subsequent breast cancer, similar to that seen with usual ductal hyperplasia/proliferative breast disease. RSL have a similar risk of subsequent carcinoma but this risk may be attributable to coexistent PBD including usual ductal hyperplasia because RSLs arise in that setting 12. The frequency of PIK3CA mutations in macrodissected RSL could therefore potentially reflect a high content of PBD. Infiltrating epitheliosisis also a lesion characterized by a high content of usual ductal hyperplasia (epitheliosis) along with scleroelastic stromal changes containing tubules, strands and cords of spindle cells with variable squamous differentiation (adenosquamous proliferation/ASP), which resembles a miniature LGASC 10. In a recent study of infiltrating epitheliosis, all eight cases showed mutations in components of the PI3K pathway with seven cases harbouring PIK3CA mutations representing six hotspot H1047R mutations and one E542K mutation, and the remaining case harbouring a PIK3R1 (L380del) small deletion. In one case the same PIK3CA (H1047R) and SF3B1 (K700E) mutations were identified in infiltrating epitheliosis, ductal carcinoma in situ (DCIS) and LGASC, suggesting that the lesions were clonally related and that the infiltrating epitheliosis and DCIS may have represented a ‘substrate for the development of the LGASC’. Unfortunately, none of our LGASC cases had an adjacent RSL to enable such comparison. The authors also suggested that infiltrating epitheliosis may form a spectrum with RSL 11.
In summary, our data support the notion that RSL may represent a form of early neoplasia. The detection of recurrent PIK3CA mutations in these cases implies clonality. The mutation drives cellular proliferation and provides a survival advantage. The factors that determine whether ASP involutes or persists (in the setting of a RSL), and possibly even progresses to other forms of neoplasia, remain unknown and would indeed be an interesting new line of research.
Author contributions
MW and TO conceived the study. MW selected the cases for assessment. TO performed the microdissection, macrodissection and DNA extraction. RA performed the genetic testing and analyses. MW wrote the first draft of the manuscript, which was subsequently expanded and edited by all authors.
Acknowledgements
Special thanks are owed to Rosemary Clay and Samantha Graham who retrieved slides and blocks from file, Megan Chandler who cut sections for microdissection, Hedley Coleman for slide scanning and Marie Steyn and Lucia Simone who obtained the articles for literature review, and to Grant Taggart and Warick Delprado for their support of the project. The authors also acknowledge the support received from the University of Sydney's Bosch Institute Molecular Biology and Advanced Microscopy facilities and the expert help of Facility staff, especially Donna Lai, Sheng Hua and Louise Cole.
No conflicts of interest were declared.
References
- 1. Wilsher MJ. Adenosquamous proliferation of the breast and low‐grade adenosquamous carcinoma. A common precursor of an uncommon cancer? Pathology 2014; 46: 402–410. [DOI] [PubMed] [Google Scholar]
- 2. Wilsher MJ, Snook K. Bilateral, multicentric low‐grade adenosquamous carcinomas of the breast, each arising within a separate radial scar/complex sclerosing lesion. Pathology 2014; 46: 85–88. [DOI] [PubMed] [Google Scholar]
- 3. Iqbal M, Shoker BS, Foster CS, et al Molecular and genetic abnormalities in radial scar. Hum Pathol 2002; 33: 715–722. [DOI] [PubMed] [Google Scholar]
- 4. Wolters KL, Ang D, Warrick A, et al Frequent PIK3CA mutations in radial scars. Diagn Mol Pathol 2013; 22: 210–214. [DOI] [PubMed] [Google Scholar]
- 5. Blumenthal GM, Dennis PA. PTEN hamartoma tumor syndromes. Eur J Hum Genet 2008; 16: 1289–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee JH, Huynh M, Silhavy JL, et al De novo somatic mutations in components of the PI3K‐AKT3‐mTOR pathway cause hemimegalencephaly. Nat Genet 2012; 44: 941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ang DC, Warrick AL, Shilling A, et al Frequent phosphatidylinositol‐3‐kinase mutations in proliferative breast lesions. Mod Pathol 2014; 27: 740–750. [DOI] [PubMed] [Google Scholar]
- 8. Anderson TJ, Battersby S. Radial scars of benign and malignant breasts: comparative features and significance. J Pathol 1985; 147: 23–32. [DOI] [PubMed] [Google Scholar]
- 9. Battersby S, Anderson TJ. Myofibroblast activity of radial scars. J Pathol 1985; 147: 33–40. [DOI] [PubMed] [Google Scholar]
- 10. Eusebi V, Millis RR. Epitheliosis, infiltrating epitheliosis, and radial scar. Semin Diagn Pathol 2010; 27: 5–12. [DOI] [PubMed] [Google Scholar]
- 11. Eberle CA, Piscuoglio S, Rakha EA, et al Infiltrating epitheliosis of the breast: characterization of histologic features, immunophenotype and genomic profile. Histopathology 2016; 68: 1030–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sanders ME, Page DL, Simpson JF, et al Interdependence of radial scar and proliferative disease with respect to invasive breast carcinoma risk in patients with benign breast biopsies. Cancer 2006; 106: 1453–1461. [DOI] [PubMed] [Google Scholar]
- 13. Rosen PP, Ernsberger D. Low‐grade adenosquamous carcinoma. A variant of metaplastic mammary carcinoma. Am J Surg Pathol 1987; 11: 351–358. [DOI] [PubMed] [Google Scholar]
- 14. RR Singh, KP Patel, MJ Routbort, et al Clinical validation of a next‐generation sequencing screen for mutational hotspots in 46 cancer‐related genes. J Mol Diagn 2013; 15: 607–622. [DOI] [PubMed] [Google Scholar]
- 15. Simen BB, Yin L, Goswami CP, et al Validation of a next‐generation–sequencing cancer panel for use in the clinical laboratory. Arch Pathol Lab Med 2015; 139: 508–517. [DOI] [PubMed] [Google Scholar]
- 16. Hamilton A, Tétreault M, Dyment DA, et al Concordance between whole‐exome sequencing and clinical Sanger sequencing: implications for patient care. Mol Genet Genomic Med 2016; 4: 504–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abecasis GR, Auton A, Brooks LD, et al An integrated map of genetic variation from 1,092 human genomes. 1000 Genomes Project Consortium. Nature 2012; 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gkeka P, Evangelidis T, Pavlaki M, et al Investigating the structure and dynamics of the PIK3CA wild‐type and H1047R oncogenic mutant. PLoS Comput Biol 2014; 10: e1003895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koren S, Reavie L, Couto JP, et al PIK3CA(H1047R) induces multipotency and multi‐lineage mammary tumours. Nature 2015; 525: 114–118. [DOI] [PubMed] [Google Scholar]
- 20. Van Keymeulen A, Lee MY, Ousset M, et al Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 2015; 525: 119–123. [DOI] [PubMed] [Google Scholar]
- 21. Brunner AL, Li J, Guo X, et al A shared transcriptional program in early breast neoplasias despite genetic and clinical distinctions. Genome Biol 2014; 15: R71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Troxell ML, Levine J, Beadling C, et al High prevalence of PIK3CA/AKT pathway mutations in papillary neoplasms of the breast. Mod Pathol 2010; 23: 27–37. [DOI] [PubMed] [Google Scholar]
- 23. Jahn SW, Kashofer K, Thüringer A, et al Mutation profiling of usual ductal hyperplasia of the breast reveals activating mutations predominantly at different levels of the PI3K/AKT/mTOR pathway. Am J Pathol 2016; 186: 15–23. [DOI] [PubMed] [Google Scholar]