

Abstract
Identification of recurrent tumour‐specific chromosomal translocations and novel fusion oncogenes has important diagnostic, therapeutic and prognostic implications. Over the past decade, fluorescence in situ hybridization (FISH) analysis of tumour samples has been one of the most rapidly growing areas in genomic medicine and surgical pathology practice. Unlike traditional cytogenetics, FISH affords a rapid analysis of formalin‐fixed, paraffin‐embedded cells within a routine pathology practice workflow. As more diagnostic and treatment decisions are based on results of FISH, demand for the technology will become more widespread. Common FISH‐detected alterations are chromosome deletions, gains, translocations, amplifications and polysomy. These chromosome alterations may have diagnostic and therapeutic implications for many tumour types. Integrating genomic testing into cancer treatment decisions poses many technical challenges, but rapid progress is being made to overcome these challenges in precision medicine. FISH assessment of chromosomal changes relevant to differential diagnosis and cancer treatment decisions has become an important tool for the surgical pathologist. The aim of this review is to provide a theoretical and practical survey of FISH detected translocations with a focus on strategies for clinical application in surgical pathology practice.
Keywords: fluorescence in situ hybridization, molecular genetics/cytogenetics, targeted therapy, precision medicine, differential diagnosis
Introduction
In the late 1960s, in situ hybridization was first performed with radioisotope‐labelled probes followed by autoradiography 1, 2, 3. Fluorescence‐labelled probe technology started in the early 1980s with RNA probes that were directly labelled with fluorophores complementary to specific DNA sequences. Advances in fluorescence microscopy and digital imaging, as well as widespread availability of genomic and bioinformatic resources, have greatly improved the resolution, sensitivity, specificity, and accessibility of fluorescence in situ hybridization (FISH).
Interphase FISH, hereinafter simply referred to as FISH, is the usual clinical application of this diagnostic tool 4. FISH involves the use of fluorescence labelled fragments of DNA (probes) binding to interphase chromosomes of cytology specimens or paraffin embedded tissue sections. Common FISH‐detected alterations are chromosome deletions, gains, translocations, amplifications and polysomy. These chromosome alterations may have diagnostic and therapeutic implications for many tumour types 5, 6. Integrating precise genomic testing into cancer treatment decisions poses technical challenges, but rapid progress is being made in overcoming these difficulties. This review focuses on FISH based detection of translocations applicable to surgical pathology.
Chromosomal translocations: an overview
Chromosomal translocation refers to a chromosome rearrangement involving non‐homologous chromosome pairs. Translocations can be balanced, without net gain or loss of material, or unbalanced, with gain or loss of genetic loci. Translocation often creates fusion genes when two otherwise separated chromosome parts join 7, 8, 9. These fusion genes may behave as a hybrid chimeric oncogene, inducing tumorigenesis by gene overexpression, or they may interrupt an important control region, causing unresponsiveness to regulatory control factors. For example, receptor tyrosine kinase gain of function fusion oncogene is a mechanism whereby mesenchymal cells may be transformed into sarcomas. Constitutive activation of the insulin family kinase ALK results from unregulated dimerization of the fusion gene protein containing the active anaplastic lymphoma kinase (ALK) kinase domain. This mechanism underlies about half of inflammatory myofibroblastic tumours. Most Ewing sarcoma cases result from EWSR1‐FLI1 gene fusion deregulating the ETS transcription factor FLI1, leading to aberrant expression of Ewing sarcoma transforming genes. The pathognomonic fusion protein of dermatofibrosarcoma protuberans (DFSP) is PDGFB‐COL1A1. This amalgamation of the essential fibroblast collagen gene (COL1A1) with the angiogenic growth factor gene (PDGFB) leads to increased growth factor release. An overabundance of growth factor results in overstimulation of receptor tyrosine kinase PDGFR, activating not only endothelial cells but also fibroblasts and inflammatory cells 10.
Translocations may involve two or more different chromosomes or may exchange information between regions in the same chromosome. Intra‐chromosomal rearrangement may cause transposition of the involved loci with or without centromere involvement. Chromosome rearrangement, whether intra‐chromosomal or between chromosomes, invariably leads to a neoteric juxtaposition of previously unrelated gene elements into a new fusion gene that may or may not be transcribed. Fusion gene detection, therefore, is at the heart of FISH diagnostic testing.
Chromosomal translocation is detectable by several methods, including FISH, metaphase karyotyping, microarray comparative genomic hybridization, SNP‐array and next generation sequencing 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20. Most chromosomal translocations are detectable by FISH, either by split‐apart (Figure 1) or fusion probes (Figure 2). To be a reliable diagnostic tool, the probe sets must reproducibly show either an increase or a decrease in the optical distance between probe signals. Some translocations, especially those involving intra‐chromosomal inversion or deletion, have a relatively short physical distance between the fusion partners after translocation. These small distances cannot reliably be resolved at the light microscopic level. In this scenario, a PCR‐based method or RNA sequencing (RNA‐Seq) is favoured. RNA‐seq, a next generation sequencing technique, can detect both known and novel gene fusion events and allows RNA analysis through cDNA sequencing at a comprehensive genome‐wide scale. It can detect both cryptic intra‐chromosomal rearrangements and fusion products with uncharacterized fusion partners 14, 21, 22, 23, 24, 25, 26.
Figure 1.
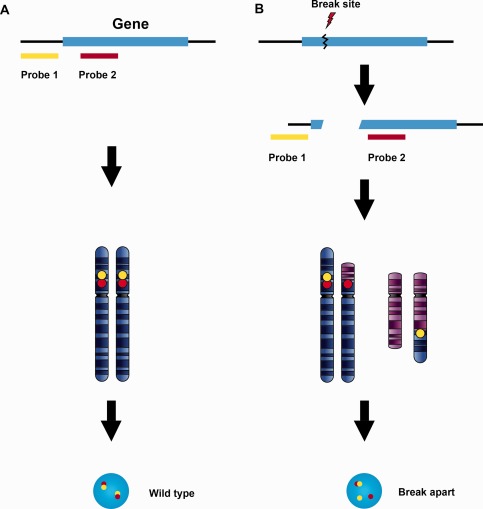
Break‐apart probe design for translocations with multiple fusion partners. A break‐apart probe set is composed of two probes specific for loci physically close to each other on the chromosome in their wild type configuration. The wild type signal pattern shows two pairs of closely approximated or fused signals (A). When translocation occurs involving a breakpoint between the two probe sites, the originally juxtaposed loci (fusion signals) split apart (B). The beauty of this design is that it detects chromosomal translocation regardless of the fusion partner involved. However, the break‐apart probe only identifies the breaking away of a gene fragment from its original location. It does not determine which chromosome receives the fragment or which genes may serve as fusion partners in the new location.
Figure 2.
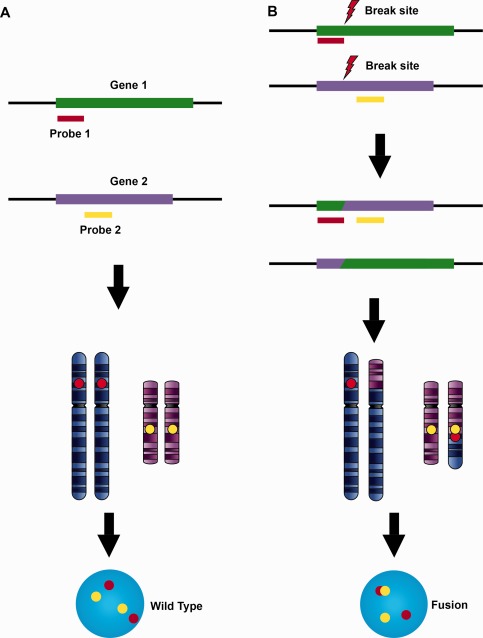
Fusion design for translocation between specific partners. A fusion probe set contains two locus‐specific probes labelled with different colours targeting the genes known to fuse in certain tumours or diseases. The fusion probes identify chromosomal loci far from each other either on the same chromosome or on different chromosomes. The wild type configuration shows two separated signals resulting from physically separated chromosome loci (A). When fusion occurs, the two probes move to a closely approximated position (fusion signal, B). This provides direct evidence of a gene fusion with definitive fusion partners identified.
Chromosomal translocations mainly associated with sarcomas
About 30% of sarcomas have well documented specific translocations, and the percentage continues to grow. With some exceptions, sarcoma translocations form fusion genes with the 5′ gene segment contributing a strong promoter and overexpression of the 3′ proto‐oncogene segment by transactivation. Recurrent gene fusions in soft tissue sarcomas tend to occur in tumours sharing similar morphological characteristics 27. Roughly half of the sarcoma‐associated fusion genes contain a member of the TET gene family, including FUS, Ewing sarcoma breakpoint region 1 (EWSR1) is the promoter and TAF15 is the proto‐oncogene partner. Most fusion genes are strongly associated with a particular tumour type, potentially making them ideal molecular diagnostic markers 10, 27, 28, 29. Some fusion proteins are actual or potential therapeutic targets, making the detection of fusion gene products valuable both for diagnostic and for therapeutic purposes.
More than 400 recurring translocations have been found in soft tissue sarcomas. The challenge now is to identify which of these recurring translocations truly have diagnostic, mechanistic, or therapeutic meaning for the associated sarcomas. Over 30 subtypes of mesenchymal tumour can be confirmed by FISH analysis based on tumour‐specific chimeric fusion sequences 28. FISH‐identifiable sarcomas include many common entities: Ewing sarcoma/primitive neuroectodermal tumour, synovial sarcoma, liposarcoma, round cell Ewing‐like sarcoma, pulmonary myxoid sarcoma, inflammatory myofibroblastic tumour and others (Table 1).
Table 1.
Chromosome alterations in mesenchymal and uncertain derivation solid tumours

|
Ewing sarcoma
Ewing sarcomas are composed of small round blue cells with highly malignant behaviour, usually affecting bone and extraosseous tissues of children and young adults. There is a specific balanced chromosomal rearrangement in most Ewing sarcomas, providing a valuable tool for diagnosis. The fusion partners of EWSR1 belong to several gene families, mainly encoding transcription factors. These proteins join the amino terminal of the EWSR1 gene with the carboxy terminal of an ETS gene family member. This fusion not only abrogates the RNA splicing function of EWSR1 but also removes essential trans‐activating control regions of the ETS gene partner. As many as 90% of the translocations involve the EWSR1 gene in the form of t(11;22)(q24;q12) translocation, which juxtaposes EWSR1 at 22q12 with the FLI1 gene on chromosome 11q24 30.
The fusion gene encodes a protein containing the upstream amino‐terminal domain of EWSR1 and the downstream carboxy‐terminal region of FLI1. This fusion protein uniquely functions in controlling cell growth and regulating the transcription of downstream genes responsible for uncontrolled cell division, and survival in the face of otherwise lethal additional mutations. The ERG translocation, t(21;22)(q22;12), resulting in EWSR1‐ERG fusion, represents only 10% of all Ewing sarcomas. The functional consequences of these variant fusion proteins are similar to those of the more common EWSR1‐FLI1 fusion. One study suggested a better outcome for patients with localized tumours expressing the most common t(11;22) transcript (EWSR1 exon 7 fused to FLI1 exon 6), but limited prognostic data is currently available for the less common fusion types 31.
Myxoid liposarcoma
The diagnosis of myxoid liposarcoma may be challenging by histology alone, especially on small biopsy tissues, since a variety of soft tissue tumours with myxoid change may mimic myxoid liposarcoma. Myxoid liposarcoma represents about 10% of all adult sarcomas and approximately 33% of all liposarcomas. Chromosomal translocations t(12,16)(q13;p11) and t(12;22)(q13;q12), rendering gene fusions of DNA‐damage inducible transcript 3 (DDIT3) with FUS or EWSR1, respectively, are characteristic of myxoid liposarcoma; one of these is identifiable in more than 95% of cases. Commonly used FISH probes are included in the Vysis DDIT3 dual colour, break‐apart rearrangement probe kit.
Synovial sarcoma
Synovial sarcoma is characterized by t(X;18)(p11;q11) translocation, which is present in virtually all tumours. The translocation fuses synovial sarcoma translocation gene (SS18) on chromosome 18 with either synovial sarcoma X breakpoint 1 (SSX1) (66%) or SSX2 (33%), located on chromosome Xp11 32, 33. The FISH test is highly specific and sensitive for synovial sarcoma. A characteristic t(X;18)(p11;q11) translocation or variants were found in 90% of synovial sarcomas. The t(X;18) translocation has not been found in other sarcomas.
Solitary fibrous tumour
Solitary fibrous tumour comprises a family of soft tissue lesions usually affecting adults, occurring at any site, and presumed to be of fibroblastic differentiation. The tumours were thought to derive from pericytes, but now there is evidence to support a fibroblastic or myofibroblastic origin. Recurrent fusions of the two genes, NGFI‐A–binding protein 2 (NAB2) and signal transducer and activator of transcription 6 (STAT6), both located near chromosomal region 12q13, have been identified in solitary fibrous tumours. The NAB2‐STAT6 fusion gene is derived from an inverted intra‐chromosomal fusion of the NAB2 and STAT6 genes on 12q13 34, 35. The fusion product contains the activation domain of STAT6 fused to the early growth response (EGR)‐binding domain of NAB2 [36]. Overexpression of the NAB2‐STAT6 fusion gene induces cell proliferation and activates expression of EGFR‐responsive genes. Chmielecki et al identified the NAB2‐STAT6 translocation by whole exome sequencing from 17 solitary fibrous tumours and matched blood 34. This fusion gene was confirmed in 29 of 55 (55%) solitary fibrous tumours by exome sequencing. Since the NAB2 and STAT6 genes are both located near 12q13, the break apart designed probe may detect some but not all of the NAB2‐STAT6 translocations. Mohajeri et al used six independent molecular genetic techniques directed at NAB2 or STAT6 rearrangement to identify NAB2–STAT6 fusion in 37 of 41 (90%) of the solitary fibrous tumours evaluated 37.
It should be noted that the NAB2‐STAT6 fusion is not consistently detected by FISH due to the very close proximity of the two genes 36 and, in this setting, reverse transcription polymerase chain reaction or RNA‐Seq, is the preferred method of detection. STAT6 immunostaining is highly sensitive and specific for detecting NAB2‐STAT6 fusion product and is now becoming standard practice.
Round cell Ewing‐like sarcoma
There is a small subset of sarcomas that clinically and histologically mimic Ewing sarcoma but fail to exhibit any of the cytogenetic abnormalities reported for these tumours. Recently cases of ‘Ewing‐like sarcoma’ have been found to have a recurrent chromosomal translocation, t(4;19)(q35;q13), resulting in fusion of capicua homologue (CIC) and double homeobox 4 (DUX4) 38. Fusion of the C‐terminal fragment of DUX4 with CIC enhances the transcriptional activity of CIC and deregulates expression of its downstream targets. CIC‐DUX4 fusion protein directly binds the ETS variant 5 (ETV5) promoter at a previously unrecognized site and upregulates expression of this oncogene. Four cases of CIC‐DUX4 sarcoma were identified using a combination of conventional cytogenetic, RT‐PCR, and FISH methods 39. FISH was positive for CIC‐DUX4 fusion in all four tumours. The distinctive histopathological features and rapid disease progression may warrant classification of CIC‐DUX4 sarcoma as a new translocation‐associated sarcoma.
Inflammatory myofibroblastic tumour
Inflammatory myofibroblastic tumour is a spindle cell neoplasm most frequently found in the urinary bladder but occasionally diagnosed at various body sites. Although these tumours usually behave in a benign fashion, they may be difficult to differentiate from sarcoma or sarcomatoid carcinoma. Roughly half of inflammatory myofibroblastic tumours have ALK rearrangements leading to elevated expression of ALK chimeric protein 40. The ALK translocations involve 2p23 with multiple fusion partners, including TPM3‐ALK, RANBP2‐ALK, TPM4‐ALK, EML4‐ALK, CLTC‐ALK, CARS‐ALK, ATIC‐ALK, SEC31A‐ALK and PPFIBP1‐ALK 41, 42. In each instance, the fusion gene overexpresses ALK due to promoter swapping. ALK rearrangements may prove useful for distinguishing inflammatory myofibroblastic tumour from morphologically similar neoplasms.
Therapy targeting tumour cells with ALK gene rearrangement induces partial tumour remission 43, 44, but, little is known about the pathogenesis of the 50% of inflammatory myofibroblastic tumours that lack ALK translocation.
Besides ALK, C‐ROS oncogene 1 (ROS1) and NTRK3 gene rearrangements, YWHAE–ROS1 and ETV6–NTRK3 are also found in ALK fusion negative inflammatory myofibroblastic tumours 45. Antonescu et al reported six (10%) ROS1 related translocations in a group of 62 cases. Most of the patients were children, and the tumours were located in the lung or abdomen 46. Two (3.2%) inflammatory myofibroblastic tumour cases showed TFG‐ROS1 fusions, t(3;6)(q12;q22).
Alveolar soft part sarcoma
Alveolar soft part sarcoma has a characteristic histopathology but a controversial histogenesis. Alveolar soft part sarcomas arising in many body sites have a diagnostic translocation. The rearrangement involves the alveolar soft part sarcoma locus (ASPSCR1) located on chromosome 17q25 and the transcription factor for the immunoglobulin heavy chain enhancer 3 (TFE3) gene, located on chromosome Xp11 47. The fusion protein is capable of inducing aberrant transcription of TFE3‐regulated genes and may confer resistance to cell‐cycle arrest signals and override apoptosis. Selvarajah et al investigated 17 alveolar soft part sarcomas from 11 patients by array comparative genomic hybridization and FISH. FISH identified the ASPSCR1‐TFE3 fusion in all cases 12.
The ASPSCRI‐TFE3 [der(17)t(X;17)(p11;q25)] translocation is found in 80% of alveolar soft part sarcomas. This translocation is also found in a distinctive subset of renal cell carcinoma (translocation renal cell carcinoma) which frequently has a papillary architecture.
Alveolar rhabdomyosarcoma
Alveolar rhabdomyosarcoma (ARMS) is a highly aggressive soft tissue sarcoma associated with translocations involving PAX3‐FOXO1 [t(2;13)(q35;q14)] or PAX7‐FOXO1 [t(1;13)(p36;q14)] reportedly accounting for 55–80% and 15–22% of ARMS, respectively 48. These translocations generate fusion proteins that function as transcriptional activators with oncogenic effects. FISH testing has higher sensitivity and specificity than RT‐PCR assay for these fusion transcripts 49. FISH analysis using the FOXO1split‐apart probe adds the ability to detect variant FOXO1 rearrangements not detectable by PCR.
Clear cell sarcoma of soft tissue
Clear cell sarcoma of soft tissue is a rare mesenchymal malignancy mainly occurring in young to middle‐aged adults of either sex, typically in the soft tissue of the lower extremities. The tumour is aggressive with a high frequency of local recurrence and distant metastasis. Almost all (90%) clear cell sarcomas are associated with EWSR1‐ATF1 translocation, t(12;22)(q13;q12), while a smaller subset of tumours (6%) bear an EWSR1‐CREB1 translocation 50. The specific translocation is unrelated to tumour prognosis. In a group of 33 clear cell sarcomas, RT‐PCR using RNA extracted from formalin‐fixed, paraffin‐embedded tissues demonstrated transcripts of the EWSR1‐ATF1 (31/33) or EWSR1‐CREB1 fusion gene (2/33) 51.
Chromosomal translocations in other solid tumours
A growing number of fusion oncogenes have been also associated with some common adult epithelial tumors, such as adenocarcinomas of the lung, prostate, colon, kidney, breast and other epithelial solid tumours (Table 2).
Table 2.
Chromosome alterations in solid tumours of epithelial origin
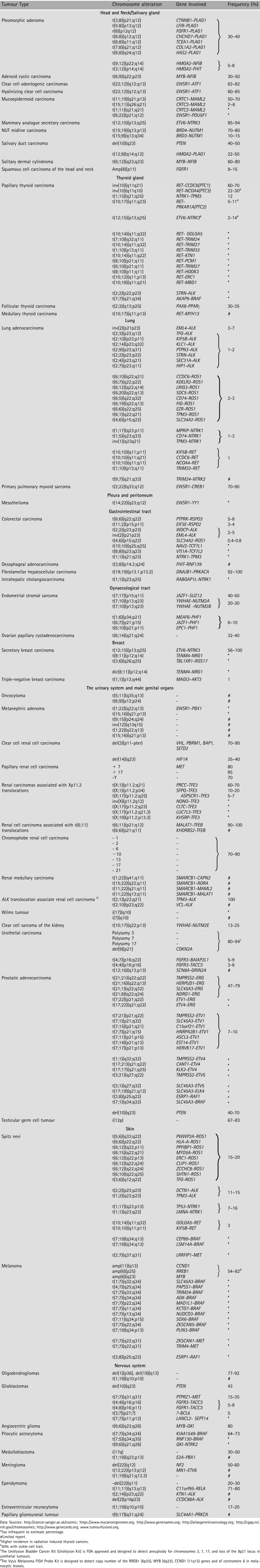
|
Chromosomal translocations mainly associated with lung cancer
EML4‐ALK translocation
ALK is a tyrosine kinase member of the insulin receptor superfamily normally expressed only in certain neurons of the developing central nervous system. Fusions of ALK with echinoderm microtubule‐associated protein‐like 4 (EML4), a protein involved in microtubule assembly, results in constitutive activation of the ALK kinase 52, 53. The ALK fusion oncogene was first identified in anaplastic large‐cell lymphoma, in which a t(2;5) chromosome rearrangement activates the ALK kinase by fusion with the nucleophosmin gene (NPM1) on chromosome 5. ALK fusions have been reported in non‐small cell lung carcinoma, breast cancer, colorectal cancer, renal cancer and other tumour types 41, 42, 54, 55. The common FISH test for ALK rearrangement uses dual colour labelled probes covering the ALK gene and 3′ flanking region of ALK (Figure 3). The year 2013 was the first time that the FDA simultaneously approved a novel anticancer drug (crizotinib, Pfizer) and its companion FISH detection kit (ALK FISH probe kit, Abbott Molecular), highlighting the critical role of FISH triage for guiding ALK‐targeted therapy 56, 57. A FDA approved Vysis ALK break apart FISH probe kit is recommended by the College of American Pathologists (CAP). In general, a sample is considered positive if >15% of cells are positive for ALK separation of the green and orange signals.
Figure 3.
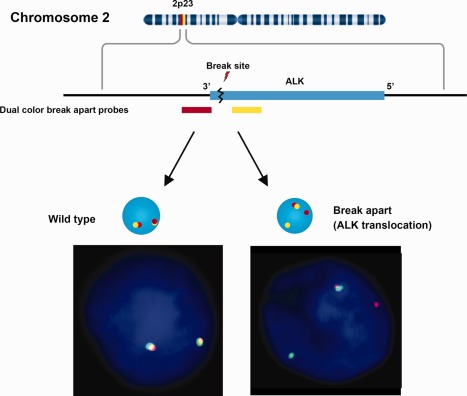
Signal patterns of ALK split‐apart probes. The commonly used ALK split‐apart FISH test uses dual colour labelled probes covering the ALK gene (yellow) and 3′ flanking region of ALK (red). When translocation occurs, the normal closely approximated signal pattern separates into distinct yellow and red signals (in commercial FISH ALK translocation kits Green and Red signals may be used). Since ALK translocations involve multiple fusion partners, a split‐apart design ensures that the probe set will uncover ALK translocations with most of the possible fusion partners.
ROS1 translocation
ROS1 is a receptor tyrosine kinase of the insulin receptor family. The ROS1 rearrangements lead to a constitutively activated fusion kinase, and are detected in 1.2–1.7% of lung adenocarcinoma cases 58, 59, 60. ROS1 activation stimulates other downstream signalling proteins including AKT1, MAPK1, MAPK3, IRS1 and PLCG2 59. Patients with ROS1 translocation are significantly younger than most patients with lung cancer and are more likely to be never‐smokers. Their cancers tend to be adenocarcinoma, and their tumours have a tendency towards higher grade. Several genes may serve as fusion partners for ROS1 including SDC4, SLC34A2, CD74, GOPC, and EZR, yet each of the ROS1 fusion products leads to constitutive ROS kinase activation 61. A dual probe break‐apart method is used to detect ROS1 rearrangement as with ALK rearrangement. Criteria similar to those used for ALK rearrangement screening are used to evaluate the ROS1 FISH test.
ROS1 fusions are also found in meningioma, cholangiocarcinoma and glioblastoma, but no systematic investigation of tyrosine kinase inhibitors has been published for ROS1‐fusion positive tumours of these primary sites 62, 63. The reported incidence of ROS1 translocation is also low in other tumour types: 8.7% (2/23) of cholangiocarcinomas, 0.5% (1/200) of ovarian carcinomas, 0.6% (3/495) of gastric adenocarcinomas, 0.8% (2/236) of colorectal carcinomas, 7.7% (2/26) of inflammatory myofibroblastic tumours, 2.9% (1/34) of angiosarcomas and 5% (1/20) of epithelioid haemangioendotheliomas 63, 64, 65, 66.
Rearranged during transfection translocation
Novel chromosomal translocations involving the rearranged during transfection (RET) tyrosine kinase gene were reported recently 61, 67. The RET gene belongs to the cadherin superfamily, and encodes a cell surface receptor tyrosine kinase that transduces signals for cell growth and differentiation 67, 68. Several RET rearrangements have been identified in non‐small cell lung carcinomas, including KIF5B‐RET, CCDC6‐RET, NCOA4‐RET, and TRIM33‐RET.
RET gene translocation occurs in approximately 1–2% of non‐small cell lung carcinomas and defines a clinically distinct subset of non‐small cell lung carcinomas. Patients with adenocarcinomas with RET fusion tend to be younger than most patients with lung cancer and they are more likely to be never‐smokers 69. In a study of 936 patients with non‐small cell lung carcinoma, the RET fusion gene was exclusively detected in 13 patients (11/633, 1.7% in adenocarcinoma; 2/24, 8.3% in adenosquamous cell carcinoma) 70. Of the 13 RET fusion‐positive patients, 9 (69%) had KIF5B‐RET, 3 (23%) had CCDC6‐RET, and 1 (7.6%) had NCOA4‐RET fusion. Their tumours tended to be poorly differentiated carcinomas that exhibited early lymph node metastasis. Drilon et al found RET fusions in 5 of 31 patients and showed that the RET kinase can be effectively inhibited by several small molecule inhibitors 71. Follow‐up imaging conducted after 4 and 12 weeks of therapy confirmed a partial response with a 66% decrease in measurable disease in the lungs and pleura.
EWSR1‐CREB1 translocation in primary pulmonary myxoid sarcomas
In the 2015 World Health Organization (WHO) Classification of Tumours of the Lung, Pleura, Thymus and Heart, pulmonary myxoid sarcoma with EWSR1‐CREB1 translocation was introduced as a new entity 72. Primary pulmonary myxoid sarcoma is rare, and arises most frequently in young females. The tumour consists of lobules with delicate, lacelike strands and cords of mildly atypical round or spindle cells in a prominent myxoid stroma. In addition to displaying a distinct morphology, it bears a EWSR1‐CREB1 translocation in 70% of cases 73. It should be noted that EWSR1‐CREB1 translocation is not unique for this entity; it can also be observed in clear cell sarcoma‐like tumour of the gastrointestinal tract and angiomatoid fibrous histiocytoma (Table 1)
Chromosomal translocations mainly associated with kidney cancer
TFE3 translocation
Renal cell carcinomas associated with Xp11.2 translocation are uncommon renal tumours that were recognized as a distinct entity in the 2016 World Health Organization classification of Tumours of the Urinary System and Male Genital Organs 74. These neoplasms comprise the majority of paediatric renal cell carcinomas and a smaller percentage of adult renal cell carcinomas. There are several different translocations involving chromosome Xp11.2, resulting in gene fusions of the TFE3 gene with various activating partners. At least five different fusion partners for TFE3 have been characterized, including ASPSCR1, PRCC, SFPQ, CLTC and NONO (Figure 4) 75, 76. Variant translocations with unknown fusion partners include t(X;3)(p11.2;q23) and t(X;10)(11.2;q23). Different gene fusions in Xp11.2 translocation renal cell carcinoma may be associated with different morphological features 77, 78.
Figure 4.
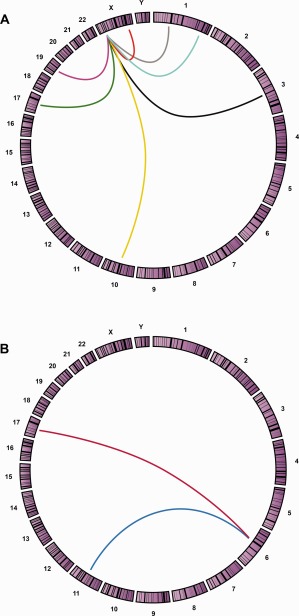
Genomic translocation relationships presented by Circos plot. Circos applies a circular ideogram displaying relationships between genomic intervals. The Circos plot is a ring composed of all chromosomes in proportionate size clockwise from 1 to Y. This graphic representation is used to visualize chromosomal translocations associated with a clinical entity. A specific translocation is shown with a line connecting the two chromosome partner loci involved in each translocation. The chromosomal translocations that may be involved in Xp11 translocation renal cell carcinomas are shown in A, including inv(X)(p11.2;q12) [NonO‐TFE3] (Red), t(X;1)(p11.2;p34) [PSF‐TEF3] (Gray), t(X;1)(p11.2;q21) [PRCC‐TFE3] (Blue), t(X;3)(p11.2;q23) [unknown‐TFE3] (Black), t(X;10)(p11.2;q23) [unknown‐TFE3] (Yellow), t(X;17)(p11.2;q23) [CLTC‐TFE3] (Magenta), t(X;17)(p11.2;q25) [ASPL‐TFE3] (Green), t(X;19)(p11.2;q13.1) [unknown‐TFE3] (Purple). Translocations involved in TFEB translocation renal cell carcinoma are shown in B, t(6;11)(p21;q13) [MALAT1‐TFEB] (Blue), t(6;17)(p21;q25) [ASPSCR1‐TFEB] (Red).
The TFE3 break‐apart FISH assay has proven useful for detecting TFE3 gene fusions in Xp11.2 translocation renal cell carcinoma, and the FISH assay, in comparison with immunohistochemical evaluation, is less often adversely affected by technical and fixation issues. Rao et al analysed 24 poorly differentiated renal cancers with break‐apart TFE3 FISH 79. Seventeen cases showed TFE3 rearrangement associated with Xp11.2 translocation by FISH, including seven previously unclassified renal cell tumours, supporting the diagnostic value and clinical application of FISH for enigmatic renal tumours. On the basis of commercially available and widely tested break‐apart FISH assays, the TFE3 split‐signal pattern was considered positive when ≥10% of the tumour nuclei showed separation of the two coloured signals by more than one signal diameter (Figure 5 ).
Figure 5.
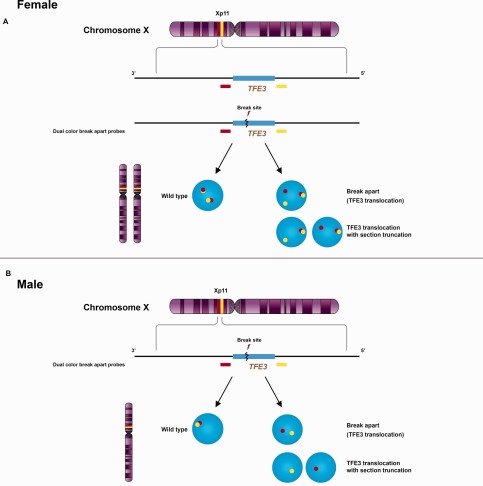
Signal patterns of TFE3 split‐apart probes. Renal cell carcinomas associated with Xp11.2 translocation involve several different translocations resulting in gene fusions of the TFE3 gene with various fusion partners. The break‐apart FISH assay uses probes both upstream (yellow) and downstream (red) to TFE3 showing different signal patterns in female (A) and male (B) patients. A positive result in a female patient shows a fused or closely approximated normal yellow‐red signal pair (uninvolved X chromosome) and either a pair of split‐apart signals or a single yellow or red signal due to section truncation artifact. Because males have only one X‐chromosome, a positive result in a male patient consists either of a pair of split‐apart yellow and red signals, or of a single yellow or red signal due to section truncation.
Transcription factor EB translocation
The t(6;11) translocation renal cell carcinoma is rare and usually bears a translocation between transcription factor EB (TFEB) and MALAT1 on chromosome 11q12, but rarely TFEB may partner with KHDRBS2 on chromosome 6q11 (Figure 4B) 80. Renal cell carcinomas with t(6;11)(p21;q12) are characterized by translocation involving TFEB. Identification of either TFEB protein overexpression or t(6;11) translocation is useful for the diagnosis of TFEB renal cell carcinoma. Cytogenetic karyotypic analysis and RT‐PCR are also common methodologies for identifying this translocation. Unfortunately, these methods are limited by the need for fresh viable tumour cells for culture or flash frozen tissue to quantitatively assay TFEB mRNA. More recently, a break‐apart FISH assay to detect TFEB gene translocation has been validated on formalin‐fixed paraffin‐embedded tissues, and appears to be superior to the TFEB IHC assay. Rao et al reported seven patients with TFEB renal cell carcinoma confirmed diagnostically by using FISH technology. All seven cases were TFEB positive by both fusion and split‐apart probe designs 81.
SMARCB1/SMARCB1 genetic alterations and ALK translocation
Renal medullary carcinoma is a rare, aggressive tumour with a poor clinical outcome. It belongs to a SMARCB1‐deficient tumour family 82. Inactivation of tumour suppressor gene SMARCB1 is the hallmark of this tumour. Cheng et al studied a total of 19 renal cancers that included 5 renal medullary carcinomas, 2 paediatric rhabdoid tumours of kidney, 10 high grade renal cell carcinomas and 2 urothelial carcinomas 83. All renal medullary carcinomas were from African American patients with sickle‐cell trait who presented with extensive extrarenal metastases at the time of diagnosis. All five renal medullary carcinomas and two renal rhabdoid tumours showed complete loss of SMARCB1 expression. Another study by Calderaro et al found recurrent alterations involving chromosome 22q11 in each of four renal medullary carcinomas associated with sickle‐cell trait. Each of these showed hemizygous SMARCB1 deletion 84. RNA sequencing further identified fusion transcripts involving SMARCB1 in these tumours. The transcripts resulted from balanced translocations disrupting SMARCB1 and fusing it to various partners including CAPN2, RORA, MAML2 and MALAT1.
ALK translocation was also reported in renal medullary carcinomas from young patients with sickle‐cell trait to be associated with VCL‐ALK translocation 85. The incidence of ALK rearrangement associated with all renal cell carcinomas is low, with an overall frequency of <1%. In a study including 534 adult renal cell carcinoma patients, ALK rearrangements occurred in two papillary renal cell carcinomas 86. More recently, Kusano et al reported two cases of RCC harbouring a novel STRN‐ALK fusion 87. In both cases, the patients were in their 30s at the time of nephrectomy.
Chromosomal translocations mainly associated with prostate cancer
A significant event in prostate carcinogenesis involves gene fusion between members of the E‐twenty‐six transforming factor (ETS) family of genes, including ERG, and the transmembrane protease, serine 2 (TMPRSS2, 21q22.3) gene 88, 89, 90, 91. The genes involved are the androgen‐regulated gene TMPRSS2 and ETS transcription factor family members, including ERG (21q22.2), ETV1 (7p21.2), or ETV4 (17q21). TMPRSS2‐ERG gene fusion has been identified in about 50% of prostate tumours (ranging from 27% to 79%). These fusions appear to represent a specific early event in prostatic carcinogenesis that can be observed in high grade prostatic intra‐epithelial neoplasia 90. The TMPRSS2‐ERG rearrangement can be identified by both dual colour split apart probe or by tricolour probes (Figure 6) 92, 93, 94.
Figure 6.
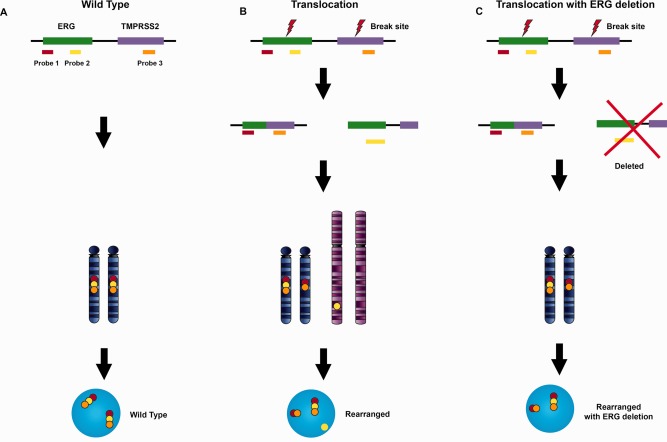
Signal patterns of TMPRSS2‐ERG translocation by tri‐colour split‐apart probes. The TMPRSS2‐ERG tri‐colour fused probe uses three different colour probes recognizing the up‐stream end of TMPRSS2 (orange), and both up‐ and down‐ stream ends of ERG (yellow, red), respectively. The probes hybridizing to the wild type TMPRSS2/ERG loci show the normal co‐localized red‐yellow‐orange pattern (A). TMPRSS2/ERG loci rearrangement via breaks in both ERG and TMPRSS2 genes leads to a red‐orange fusion of ERG down‐stream signal to the TMPRSS2 signal. The ERG up‐stream signal may either split from the down‐stream sequence as in (B), or be deleted entirely as shown with the signal pattern shown in (C).
Chromosomal translocations mainly associated with breast cancer
A rare variant of breast cancer, secretory breast cancer, has a characteristic recurrent translocation between ETS variant 6 (ETV6) and neurotrophic tyrosine kinase type 3 (NTRK3), t(12;15)(p13;q25) 95, 96. This translocation appears to be the initial hit required for formation of secretory breast cancer. Retroviral transfer of ETV6‐NTRK3 into murine mammary epithelial cells resulted in transformed cells that readily formed tumours in nude mice.
The biological consequence of this translocation is expression of a chimeric tyrosine kinase protein with potent transforming capability. The fusion protein strongly activates the MAPK1 and the PIK3CA/AKT1 pathways 96. The occurrence of the ETV6‐NTRK3 translocation and consequential expression of the oncogene is an initiating event in the genesis of secretory breast cancer 97. The ETV6‐NTRK3 translocation is demonstrated either by break‐apart or by fusion FISH design. Positive signal patterns have been seen in most secretory breast carcinoma tumour cells using either assay strategy.
Chromosomal translocations mainly associated with colorectal cancer
The first recurrent fusion gene in colorectal cancer was reported in 2011 98. Bass et al correlated the genomes of nine colorectal cancers with paired non‐neoplastic tissue controls. Recurrent VTI1A‐TCF7L2 fusion was identified in 3% of colorectal cancers 98. Their findings indicated that functionally important fusion events also occur in colorectal cancers, but the clinical impact of these is yet to be elucidated. RNA‐sequencing studies reported by Seshagiri et al identified multiple fusion transcripts in colorectal cancer, including recurrent gene fusions involving R‐spondin family members EIF3E‐RSPO2 and PTPRK‐RSPO3 that occurred with a cumulative frequency of 10% in colorectal cancers 99. Other translocations reported in recent years include EML4‐ALK, C2orf44‐ALK, SLC34A2‐ROS1 and NAV2‐TCF7L1 66.
Chromosomal translocations mainly associated with thyroid cancer
More than 10 rearrangements involving RET have been reported; all of the rearrangements are formed by fusion of the RET tyrosine kinase domain with a fusion partner gene 100, 101, 102, 103. RET rearrangements have been identified almost exclusively in papillary thyroid carcinomas; most frequent were RET‐CCDC6 (60%), RET‐NCOA4 (20%) and RET‐ PRKAR1A (5–12%) 67, 68, 104. The fusion genes code for constitutively activated proteins driving uncontrolled proliferation of follicular cells.
Kelly et al reported that STRN‐ALK fusion occurs in a subset of patients with highly aggressive types of thyroid cancer and provide initial evidence suggesting that it may represent a therapeutic target for these patients. STRN‐ALK fusion protein leads to constitutive activation of ALK kinase 105.
Follicular thyroid carcinoma accounts for approximately 20% of all thyroid malignancies. The t(2;3)(q13;p25) translocation, fusion of the thyroid transcription factor, paired box gene 8 (PAX8) with peroxisome proliferator activated receptor γ (PPARG) is most commonly observed (30%) in follicular thyroid carcinoma 106. The fusion product acts as an oncogene by accelerating cellular growth, down‐regulating apoptosis, promoting anchorage‐independence, and causing loss of contact inhibition. It is noteworthy that rearrangement of these genes has also been identified in benign thyroid adenomas with a break‐apart frequency between 3% and 6% 107, 108.
Chromosomal translocations mainly associated with salivary gland tumours
Approximately 20% of salivary gland tumours harbour chromosomal translocation 109. There are four major recurrent translocations in malignant salivary gland tumours: the MYB‐NFIB fusion in adenoid cystic carcinoma, the CRTC1‐MAML2 fusion in mucoepidermoid carcinoma, the ETV6‐NTRK3 fusion in mammary analogue secretory carcinoma (MASC), and the EWSR1‐ATF1 fusion in hyalinizing clear cell carcinoma (HCCC) (Table 2) 96, 97, 110, 111. The identification of recurrent tumour‐specific chromosomal translocations and novel fusion oncogenes has diagnostic, therapeutic, and prognostic implications.
Pleomorphic adenoma
Pleomorphic adenoma is a benign salivary gland tumour most often arising in the parotid gland. The tumour presents with a highly specific and recurrent pattern of chromosome abnormalities in 70% of cases. There are four cytogenetic categories of pleomorphic adenoma. These involve 8q12 (39%), 12q13‐15 (8%), other recurring translocations (23%), and cytogenetically unchanged cases (30%). Of cases with rearrangements, the 8q12 rearrangement [t(3;8)(p21;q12)], represents almost half of the diagnostic translocations found.
Mucoepidermoid carcinoma
Mucoepidermoid carcinoma represents a distinct type of polymorphous tumour, containing three cellular elements in varying proportions: squamous cells, mucus‐secreting cells, and ‘intermediate’ cells. The CRTC1‐MAML2 fusion is present in 55% of mucoepidermoid carcinomas. The fusion oncogene derived from chromosomal translocation that binds cyclic‐AMP response element protein related transcription co‐factor 1 (CRTC1) with mastermind‐like 2 NOTCH signalling coactivator (MAML2), t(11;19)(q12;p13) is aetiological for some mucoepidermoid carcinomas. When present, this marker confers a favourable survival outcome when compared with fusion‐negative tumours 112.
Adenoid cystic carcinoma
Recurrent t(6;9)(q22–23;p23–24) translocation in adenoid cystic carcinoma results in a fusion of the MYB proto‐oncogene with transcription factor gene NFIB 113. MYB is a leucine zipper transcription factor at 6q22‐24 that participates in the regulation of cell proliferation, apoptosis, and differentiation 114. West et al used FISH to investigate MYB translocation in 37 of these tumours. MYB‐NFIB translocation was present in 49% of adenoid cystic carcinomas but not in other salivary gland tumours or non‐salivary gland neoplasms 115. A subset of about 20% of adenoid cyst carcinomas lacks any detectable MYB gene fusion 116, 117. The high frequency of this fusion in adenoid cystic carcinoma indicates that the transcript may play a developmental role in a significant subset of these tumours. Therapies directed against MYB‐NFIB transcriptional targets may improve the prognosis for this chemoresistant neoplasm.
Mammary analogue secretory carcinoma
MASC is a recently recognized salivary gland tumour that is histologically, immunohistochemically, and genetically similar to secretory carcinoma of the breast 118. MASC harbours a t(12;15) (p13;q25) translocation resulting in fusion of an E‐twenty‐six family member (ETV6) and the neurotrophic tyrosine kinase receptor type 3 (NTRK3) gene 119. Histologically, conventional MASC displays a lobulated growth pattern of a tumour composed of microcystic, tubular, and solid structures. Between epithelial structures, one finds abundant eosinophilic homogenous or bubbly secretions. Majewska et al investigated seven MASC by FISH, and the ETV6‐NTRK3 rearrangement was identified in six cases 120. For FISH analysis, a dual‐colour break‐apart probe for the ETV6 gene exhibits a ‘split’ fluorescent signal in nuclei where the ETV6 gene participates in translocation. MASC behaves as a low‐grade carcinoma with 15–20% recurrence, lymph node metastases in 15–20% of cases, and occasional mortality 118, 121. ETV6 FISH is useful to diagnose difficult cases and to exclude the tumours that morphologically mimic MASC.
Hyalinizing clear cell carcinoma
HCCC is a recently described minor salivary gland tumour composed of glycogen‐rich clear cells with hyalinized or myxoid stroma. Gene fusion between Ewing sarcoma breakpoint region 1 (EWSR1) and the activating transcription factor 1 (ATF1) gene is a relatively consistent finding in HCCC 122. However, EWSR1‐ATF1 translocation could also be found in other types of tumour including angiomatoid fibrous histiocytoma, angiosarcoma of the parotid gland, clear cell sarcoma of soft tissue, and clear cell sarcoma‐like tumour from the GI tract 51, 123, 124, 125, 126. In one study, the ATF1 gene translocation was found in 82% of HCCC cases, but in none of the other tumours in the differential diagnosis 109.
Chromosomal translocations mainly associated with Spitz nevi and melanoma
The genetic underpinnings of Spitz nevi are poorly understood, and alterations in melanoma‐associated oncogenes are typically absent. Spitzoid neoplasms harbour kinase fusions of ROS1 (17%), NTRK1 (16%), ALK (10%), BRAF (5%) and RET (3%) in a mutually exclusive pattern 127. The chimeric proteins are constitutively activated, which stimulate oncogenic signalling and initiate the tumorigenesis. In a study of 140 spitzoid neoplasms, Wiesner et al demonstrated that kinase fusions were present in 18 of 30 Spitz nevi (60%) and within 6 of 8 atypical Spitz tumours (75%). The kinase gene fusions involved ROS1 (26%, 16/73), ALK (11%, 8/75), NTRK1 (11%, 8/75), BRAF (5%, 4/75) and RET (3%, 2/75). FISH can be used as an ancillary tool in the diagnosis of ambiguous melanocyte neoplasms, since Spitz nevi are benign and do not require wide excision 127.
Molecular alterations commonly seen in melanomas are typically absent from Spitz nevi and Spitz nevi contain translocations are not usually found in melanoma 91, 128, 129, 130. Translocations involving BRAF and MET genes have been recently reported in melanoma patients (Table 2). BRAF fusion genes characteristically activate the MAPK pathway with transformation abilities 131, 132. This activation of the MAPK pathway renders the tumours potentially sensitive to MEK inhibition.
MET is a high‐affinity tyrosine kinase receptor with functions in angiogenesis, cellular motility, growth and invasion 133. Yeh et al analysed 1202 equivocal pigmented skin lesions representing spitzoid melanoma, Spitz nevi, conventional nevi, deep penetrating nevus‐like lesions and blue nevi. The patients' tumours were tested by comparative genomic hybridization. MET translocations were found in six melanocytic tumours resulting in the following fusion genes: TRIM4‐MET, ZKSCAN1‐MET, PPFIBP1‐MET LRRFIP1‐MET, EPS15‐MET and DCTN1‐MET 134. MET fusion genes are driven by the promoter of the partner gene and express MET without control.
Chromosomal translocations mainly associated with tumours of the central nervous system
The WHO 2016 classification of tumours of the central nervous system emphasizes the importance of molecular classification 135. More than 10 translocations have been reported in glioblastomas and gene fusions occur in approximately 30–50% of glioblastoma patients (Table 2) 136, 137, 138, 139. PTPRZ1‐MET translocation was detected in 15% of glioblastomas in independent cohorts, rendering it the most frequently recurring transcript in glioblastoma. Recently, Bao et al investigated 272 gliomas by RNA seq and identified 67 in‐frame fusion transcripts, including three recurrent fusion transcripts: FGFR3‐TACC3, RNF213‐SLC26A11 and PTPRZ1‐MET 139. Fourteen other rare fusion transcripts containing sequences of genes involved in the canonical glioblastoma signalling pathways were also found.
Co‐deletion of 1p/19q has been observed in up to 70% of oligodendrogliomas and 50% of mixed oligoastrocytomas 140, 141, 142, 143. Deletions of 1p and 19q have been associated with prolonged survival in patients with oligodendrogliomas and mixed oligoastrocytomas. Jenkins et al hypothesized that the majority of 1p and 19q deletions in gliomas were derived from a translocation t(1;19)(q10;p10), which has a prevalence of 81% in all 1p/19q deletion cases 144.
KIAA1549‐BRAF translocations are characteristic of pilocytic astrocytoma 138, 145. The translocation was found by whole‐genome sequencing of 96 pilocytic astrocytomas 145. These new molecular revelations suggested that BRAF targeted therapy may be applicable in pilocytic astrocytomas since the majority of tumours harbour KIAA1549‐BRAF fusions (Table 2).
Ependymoma arises from the ependymal cells of the brain and spinal cord. Surgery and irradiation are the major treatments for this disease since chemotherapy is ineffective in most patients 146. Parker et al showed that more than two‐thirds of supratentorial ependymomas contained chromosomal translocations between avian reticuloendotheliosis viral oncogene homologue A (RELA) and chromosome 11 open reading frame 95(C11orf95) 147. The resulting fusion protein activates NF‐κB target genes, transforming the cells 148. This finding is significant not only for understanding the biology of the tumour but also affords potential for an effective treatment.
Conclusions
Over the past decade, FISH analysis of neoplasms has become one of the most rapidly growing areas in genomic medicine and surgical pathology practice. As more diagnostic and treatment algorithms incorporate the results of FISH, demand for the technology will become more widespread. Common FISH‐detected alterations are chromosome deletions, gains, translocations, amplifications and polysomy. These chromosome alterations may have diagnostic and therapeutic implications for many tumour types. Integrating genomic testing into cancer treatment decisions poses many technical challenges, but rapid progress is being made in overcoming these precision medicine challenges. Next generation sequencing platforms allow detection of fusion genes at both the transcriptional and genomic levels. The rapidly emerging RNA sequencing (RNA‐seq) technology has empowered an increasing pace of fusion gene identification. RNA‐seq is superior to current molecular test methods in its capacity to detect gene mutations simultaneously, the precise mapping of break‐point and joint sequences, and the discovery of cryptic fusions and fusion genes with unknown partners or with short physical distance between the fusion partners.
FISH assessment of chromosomal changes is a continuously evolving technology. Its role in clarifying cancer diagnoses and its contributions to the decisions involved in choice of cancer therapy will become ever more important to surgical pathologists and the clinicians and patients they serve.
Author contributions
LC and SZ were involved in conception and design of the paper. All the authors (LC, SZ, LW, GTM and DDD) participated in data acquisition and analysis, and writing the article. All the authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank Natasha Gibson for excellent editorial assistance.
No conflicts of interest were declared.
References
- 1. Buongiorno‐Nardelli M, Amaldi F. Autoradiographic detection of molecular hybrids between RNA and DNA in tissue sections. Nature 1970; 225 : 946–948. [DOI] [PubMed] [Google Scholar]
- 2. John HA, Birnstiel ML, Jones KW. RNA‐DNA hybrids at the cytological level. Nature 1969; 223 : 582–587. [DOI] [PubMed] [Google Scholar]
- 3. Gall JG, Pardue ML. Formation and detection of RNA‐DNA hybrid molecules in cytological preparations. Proc Natl Acad Sci U S A 1969; 63 : 378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Katsanis SH, Katsanis N. Molecular genetic testing and the future of clinical genomics. Nat Rev Genet 2013; 14 : 415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cheng L, Zhang DY, Eble JN. Molecular Genetic Pathology (2nd edn) Springer: New York, NY, 2013. [Google Scholar]
- 6. Cheng L, Eble JN. Molecular Surgical Pathology (1st edn). Springer: New York, NY, 2013. [Google Scholar]
- 7. Mitelman F, Johansson B, Mertens F. Fusion genes and rearranged genes as a linear function of chromosome aberrations in cancer. Nat Genet 2004; 36 : 331–334. [DOI] [PubMed] [Google Scholar]
- 8. Lin C, Yang L, Tanasa B, et al Nuclear receptor‐induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 2009; 139 : 1069–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Doroshow JH, Kummar S. Translational research in oncology–10 years of progress and future prospects. Nat Rev Clin Oncol 2014; 11 : 649–662. [DOI] [PubMed] [Google Scholar]
- 10. Mertens F, Johansson B, Fioretos T, et al The emerging complexity of gene fusions in cancer. Nat Rev Cancer 2015; 15 : 371–381. [DOI] [PubMed] [Google Scholar]
- 11. Martin CL, Warburton D. Detection of chromosomal aberrations in clinical practice: from karyotype to genome sequence. Annu Rev Genomics Hum Genet 2015; 16 : 309–326. [DOI] [PubMed] [Google Scholar]
- 12. Selvarajah S, Pyne S, Chen E, et al High‐resolution array CGH and gene expression profiling of alveolar soft part sarcoma. Clin Cancer Res 2014; 20 : 1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Friedman AA, Letai A, Fisher DE, et al Precision medicine for cancer with next‐generation functional diagnostics. Nat Rev Cancer 2015; 15 : 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kumar‐Sinha C, Kalyana‐Sundaram S, Chinnaiyan AM. Landscape of gene fusions in epithelial cancers: seq and ye shall find. Genome Med 2015; 7 : 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheng L, Zhang S, MacLennan GT, et al Laser‐assisted microdissection in translational research: theory, technical considerations, and future applications. Appl Immunohistochem Mol Morphol 2013; 21 : 31–47. [DOI] [PubMed] [Google Scholar]
- 16. Roukos V, Misteli T. The biogenesis of chromosome translocations. Nat Cell Biol 2014; 16 : 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bunting SF, Nussenzweig A. End‐joining, translocations and cancer. Nat Rev Cancer 2013; 13 : 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vorsanova SG, Yurov YB, Iourov IY. Human interphase chromosomes: a review of available molecular cytogenetic technologies. Mol Cytogenet 2010; 3: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Speicher MR, Carter NP. The new cytogenetics: blurring the boundaries with molecular biology. Nat Rev Genet 2005; 6 : 782–792. [DOI] [PubMed] [Google Scholar]
- 20. Cui C, Shu W, Li P. Fluorescence in situ hybridization: cell‐based genetic diagnostic and research applications. Front Cell Dev Biol 2016; 4 : 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Byron SA, Van Keuren‐Jensen KR, Engelthaler DM, et al Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nat Rev Genet 2016; 17 : 257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next‐generation sequencing technologies. Nat Rev Genet 2016; 17 : 333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martin JA, Wang Z. Next‐generation transcriptome assembly. Nat Rev Genet 2011; 12 : 671–682. [DOI] [PubMed] [Google Scholar]
- 24. Yoshihara K, Wang Q, Torres‐Garcia W, et al The landscape and therapeutic relevance of cancer‐associated transcript fusions. Oncogene 2015; 34 : 4845–4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Latysheva NS, Babu MM. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Res 2016; 44 : 4487–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet 2011; 12 : 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mertens F, Antonescu CR, Mitelman F. Gene fusions in soft tissue tumors: recurrent and overlapping pathogenetic themes. Genes Chromosomes Cancer 2016; 55 : 291–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fletcher CDM, Bridge JA, Hogendoorn PCW, F Mertens (Eds). WHO/IARC Classification of Tumours, 4th Edition, Volume 5. IARC Press, Lyon, 2015. [Google Scholar]
- 29. Fletcher CD. The evolving classification of soft tissue tumours ‐ an update based on the new 2013 WHO classification. Histopathology 2014; 64 : 2–11. [DOI] [PubMed] [Google Scholar]
- 30. Thway K, Fisher C. Tumors with EWSR1‐CREB1 and EWSR1‐ATF1 fusions. The current status. Am J Surg Pathol 2012; 36 : e1–e11. [DOI] [PubMed] [Google Scholar]
- 31. Grunewald TG, Bernard V, Gilardi‐Hebenstreit P, et al Chimeric EWSR1‐FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat Genet 2015; 47 : 1073–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nielsen TO, Poulin NM, Ladanyi M. Synovial sarcoma: recent discoveries as a roadmap to new avenues for therapy. Cancer Discov 2015; 5 : 124–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Svejstrup JQ. Synovial sarcoma mechanisms: a series of unfortunate events. Cell 2013; 153 : 11–12. [DOI] [PubMed] [Google Scholar]
- 34. Chmielecki J, Crago AM, Rosenberg M, et al Whole‐exome sequencing identifies a recurrent NAB2‐STAT6 fusion in solitary fibrous tumors. Nat Genet 2013; 45 : 131–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Robinson DR, Wu YM, Kalyana‐Sundaram S, et al Identification of recurrent NAB2‐STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat Genet 2013; 45 : 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kouba E, Simper NB, Chen S, et al Solitary fibrous tumour of the genitourinary tract: a clinicopathological study of 11 cases and their association with the NAB2‐STAT6 fusion gene. J Clin Pathol 2016. Oct 31. pii: jclinpath-2016-204088. doi: 10.1136/jclinpath-2016-204088. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 37. Mohajeri A, Tayebwa J, Collin A, et al Comprehensive genetic analysis identifies a pathognomonic NAB2/STAT6 fusion gene, nonrandom secondary genomic imbalances, and a characteristic gene expression profile in solitary fibrous tumor. Genes Chromosomes Cancer 2013; 52 : 873–886. [DOI] [PubMed] [Google Scholar]
- 38. Gambarotti M, Benini S, Gamberi G, et al CIC‐DUX4 Fusion‐positive round cell sarcomas of soft tissue and bone: a single institution morphologic and molecular analysis of 7 cases. Histopathology 2016; 69 : 624–634. [DOI] [PubMed] [Google Scholar]
- 39. Choi EY, Thomas DG, McHugh JB, et al Undifferentiated small round cell sarcoma with t(4;19)(q35;q13.1) CIC‐DUX4 fusion: a novel highly aggressive soft tissue tumor with distinctive histopathology. Am J Surg Pathol 2013; 37 : 1379–1386. [DOI] [PubMed] [Google Scholar]
- 40. Choi E, Williamson SR, Montironi R, et al Inflammatory myofibroblastic tumour of the urinary bladder: the role of immunoglobulin G4 and the comparison of two immunohistochemical antibodies and fluorescence in‐situ hybridization for the detection of anaplastic lymphoma kinase alterations. Histopathology 2015; 67 : 20–38. [DOI] [PubMed] [Google Scholar]
- 41. Mano H. ALKoma: a cancer subtype with a shared target. Cancer Discov 2012; 2 : 495–502. [DOI] [PubMed] [Google Scholar]
- 42. Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer 2013; 13 : 685–700. [DOI] [PubMed] [Google Scholar]
- 43. Butrynski JE, D'adamo DR, Hornick JL, et al Crizotinib in ALK‐rearranged inflammatory myofibroblastic tumor. N Engl J Med 2010; 363 : 1727–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gaudichon J, Jeanne‐Pasquier C, Deparis M, et al Complete and repeated response of a metastatic ALK‐rearranged inflammatory myofibroblastic tumor to Crizotinib in a teenage girl. J Pediatr Hematol Oncol 2016; 38 : 308–311. [DOI] [PubMed] [Google Scholar]
- 45. Yamamoto H, Yoshida A, Taguchi K, et al ALK, ROS1 and NTRK3 gene rearrangements in inflammatory myofibroblastic tumours. Histopathology 2016; 69 : 72–83. [DOI] [PubMed] [Google Scholar]
- 46. Antonescu CR, Suurmeijer AJ, Zhang L, et al Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 gene fusions and rare novel RET rearrangement. Am J Surg Pathol 2015; 39 : 957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hodge JC, Pearce KE, Wang X, et al Molecular cytogenetic analysis for TFE3 rearrangement in Xp11.2 renal cell carcinoma and alveolar soft part sarcoma: validation and clinical experience with 75 cases. Mod Pathol 2014; 27 : 113–127. [DOI] [PubMed] [Google Scholar]
- 48. Brown RE, Buryanek J, Katz AM, et al Alveolar rhabdomyosarcoma: morphoproteomics and personalized tumor graft testing further define the biology of PAX3‐FKHR(FOXO1) subtype and provide targeted therapeutic options. Oncotarget 2016; 7 : 46263–46272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thway K, Rockcliffe S, Gonzalez D, et al Utility of sarcoma‐specific fusion gene analysis in paraffin‐embedded material for routine diagnosis at a specialist centre. J Clin Pathol 2010; 63 : 508–512. [DOI] [PubMed] [Google Scholar]
- 50. Wang WL, Mayordomo E, Zhang W, et al Detection and characterization of EWSR1/ATF1 and EWSR1/CREB1 chimeric transcripts in clear cell sarcoma (melanoma of soft parts). Mod Pathol 2009; 22 : 1201–1209. [DOI] [PubMed] [Google Scholar]
- 51. Hisaoka M, Ishida T, Kuo TT, et al Clear cell sarcoma of soft tissue: a clinicopathologic, immunohistochemical, and molecular analysis of 33 cases. Am J Surg Pathol 2008; 32 : 452–460. [DOI] [PubMed] [Google Scholar]
- 52. Shaw AT, Engelman JA. ALK in lung cancer: past, present, and future. J Clin Oncol 2013; 31 : 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cheng L, Alexander RE, Maclennan GT, et al Molecular pathology of lung cancer: key to personalized medicine. Mod Pathol 2012; 25 : 347–369. [DOI] [PubMed] [Google Scholar]
- 54. Swanton C, Govindan R. Clinical implications of genomic discoveries in lung cancer. N Engl J Med 2016; 374 : 1864–1873. [DOI] [PubMed] [Google Scholar]
- 55. Hirsch FR, Scagliotti GV, Mulshine JL, et al Lung cancer: current therapies and new targeted treatments. Lancet 2017; 389: 299–311. [DOI] [PubMed] [Google Scholar]
- 56. Alkan A, Koksoy EB, Utkan G. First‐line crizotinib in ALK‐positive lung cancer. N Engl J Med 2015; 372 : 781–782. [DOI] [PubMed] [Google Scholar]
- 57. Shen L, Ji HF. Ceritinib in ALK‐rearranged non‐small‐cell lung cancer. N Engl J Med 2014; 370 : 2537. [DOI] [PubMed] [Google Scholar]
- 58. Morton MJ, Zhang S, Lopez‐Beltran A, et al Telomere shortening and chromosomal abnormalities in intestinal metaplasia of the urinary bladder. Clin Cancer Res 2007; 13 : 6232–6236. [DOI] [PubMed] [Google Scholar]
- 59. Bergethon K, Shaw AT, Ou SH, et al ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol 2012; 30 : 863–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Uguen A, De Braekeleer M. ROS1 fusions in cancer: a review. Future Oncol 2016; 12 : 1911–1928. [DOI] [PubMed] [Google Scholar]
- 61. Takeuchi K, Soda M, Togashi Y, et al RET, ROS1 and ALK fusions in lung cancer. Nat Med 2012; 18 : 378–381. [DOI] [PubMed] [Google Scholar]
- 62. Charest A, Lane K, McMahon K, et al Fusion of FIG to the receptor tyrosine kinase ROS in a glioblastoma with an interstitial del(6)(q21q21). Genes Chromosomes Cancer 2003; 37 : 58–71. [DOI] [PubMed] [Google Scholar]
- 63. Davies KD, Doebele RC. Molecular pathways: ROS1 fusion proteins in cancer. Clin Cancer Res 2013; 19 : 4040–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gu TL, Deng X, Huang F, et al Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS One 2011; 6 : e15640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lee J, Lee SE, Kang SY, et al Identification of ROS1 rearrangement in gastric adenocarcinoma. Cancer 2013; 119 : 1627–1635. [DOI] [PubMed] [Google Scholar]
- 66. Aisner DL, Nguyen TT, Paskulin DD, et al ROS1 and ALK fusions in colorectal cancer, with evidence of intratumoral heterogeneity for molecular drivers. Mol Cancer Res 2014; 12 : 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mulligan LM. RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer 2014; 14 : 173–186. [DOI] [PubMed] [Google Scholar]
- 68. Shaw AT, Hsu PP, Awad MM, et al Tyrosine kinase gene rearrangements in epithelial malignancies. Nat Rev Cancer 2013; 13 : 772–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Vargas AJ, Harris CC. Biomarker development in the precision medicine era: lung cancer as a case study. Nat Rev Cancer 2016; 16 : 525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang R, Hu H, Pan Y, et al RET fusions define a unique molecular and clinicopathologic subtype of non‐small‐cell lung cancer. J Clin Oncol 2012; 30 : 4352–4359. [DOI] [PubMed] [Google Scholar]
- 71. Drilon A, Wang L, Hasanovic A, et al Response to Cabozantinib in patients with RET fusion‐positive lung adenocarcinomas. Cancer Discov 2013; 3 : 630–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Travis WD, Brambilla E, Burke Allen P., Marx Alexander, Nicholson Andrew G. (Eds). World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of the Lung, Pleura, Thymus and Heart 4th Edn. IARC Press, Lyon, 2015. [DOI] [PubMed] [Google Scholar]
- 73. Thway K, Nicholson AG, Lawson K, et al Primary pulmonary myxoid sarcoma with EWSR1‐CREB1 fusion: a new tumor entity. Am J Surg Pathol 2011; 35 : 1722–1732. [DOI] [PubMed] [Google Scholar]
- 74. Moch H, Humphrey PA, Ulbright TM, et al WHO Classification of Tumors of the Uronary System and Male Genital Organ (4th edn). International Agency for Research on Cancer (IARC) Press: Lyon, France, 2016. [Google Scholar]
- 75. Smith NE, Illei PB, Allaf M, et al t(6;11) renal cell carcinoma (RCC): expanded immunohistochemical profile emphasizing novel RCC markers and report of 10 new genetically confirmed cases. Am J Surg Pathol 2014; 38 : 604–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kauffman EC, Ricketts CJ, Rais‐Bahrami S, et al Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat Rev Urol 2014; 11 : 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Argani P. MiT family translocation renal cell carcinoma. Semin Diagn Pathol 2015; 32 : 103–113. [DOI] [PubMed] [Google Scholar]
- 78. Argani P, Zhong M, Reuter VE, et al TFE3‐fusion variant analysis defines specific clinicopathologic associations among Xp11 translocation cancers. Am J Surg Pathol 2016; 40 : 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rao Q, Williamson SR, Zhang S, et al TFE3 greak‐apart FISH has a higher sensitivity for Xp11.2 translocation‐associated renal cell carcinoma compared with TFE3 or cathepsin K immunohistochemical staining alone: expanding the morphologic spectrum. Am J Surg Pathol 2013; 37 : 804–815. [DOI] [PubMed] [Google Scholar]
- 80. Malouf GG, Su X, Yao H, et al Next‐generation sequencing of translocation renal cell carcinoma reveals novel RNA splicing partners and frequent mutations of chromatin‐remodeling genes. Clin Cancer Res 2014; 20 : 4129–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rao Q, Liu B, Cheng L, et al Renal cell carcinomas with t(6;11)(p21;q12): a clinicopathologic study emphasizing unusual morphology, novel alpha‐TFEB gene fusion point, immunobiomarkers, and ultrastructural features, as well as detection of the gene fusion by fluorescence in situ hybridization. Am J Surg Pathol 2012; 36 : 1327–1338. [DOI] [PubMed] [Google Scholar]
- 82. Lopez‐Beltran A, Cheng L, Raspollini MR, et al SMARCB1/INI1 genetic alterations in renal medullary carcinomas. Eur Urol 2016; 69 : 1062–1064. [DOI] [PubMed] [Google Scholar]
- 83. Cheng JX, Tretiakova M, Gong C, et al Renal medullary carcinoma: rhabdoid features and the absence of INI1 expression as markers of aggressive behavior. Mod Pathol 2008; 21 : 647–652. [DOI] [PubMed] [Google Scholar]
- 84. Calderaro J, Masliah‐Planchon J, Richer W, et al Balanced translocations disrupting SMARCB1 are hallmark recurrent genetic alterations in renal medullary carcinomas. Eur Urol 2016; 69 : 1055–1061. [DOI] [PubMed] [Google Scholar]
- 85. Marino‐Enriquez A, Ou WB, Weldon CB, et al ALK rearrangement in sickle cell trait‐associated renal medullary carcinoma. Genes Chromosomes Cancer 2011; 50 : 146–153. [DOI] [PubMed] [Google Scholar]
- 86. Sukov WR, Hodge JC, Lohse CM, et al ALK alterations in adult renal cell carcinoma: frequency, clinicopathologic features and outcome in a large series of consecutively treated patients. Mod Pathol 2012; 25 : 1516–1525. [DOI] [PubMed] [Google Scholar]
- 87. Kusano H, Togashi Y, Akiba J, et al Two cases of renal cell carcinoma harboring a novel STRN‐ALK fusion gene. Am J Surg Pathol 2016; 40 : 761–769. [DOI] [PubMed] [Google Scholar]
- 88. Tomlins SA, Rhodes DR, Perner S, et al Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005; 310 : 644–648. [DOI] [PubMed] [Google Scholar]
- 89. Tomlins SA, Laxman B, Dhanasekaran SM, et al Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 2007; 448 : 595–599. [DOI] [PubMed] [Google Scholar]
- 90. Tomlins SA, Bjartell A, Chinnaiyan AM, et al ETS gene fusions in prostate cancer: from discovery to daily clinical practice. Eur Urol 2009; 56 : 275–286. [DOI] [PubMed] [Google Scholar]
- 91. Palanisamy N, Ateeq B, Kalyana‐Sundaram S, et al Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med 2010; 16 : 793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yoshimoto M, Joshua AM, Chilton‐Macneill S, et al Three‐color FISH analysis of TMPRSS2/ERG fusions in prostate cancer indicates that genomic microdeletion of chromosome 21 is associated with rearrangement. Neoplasia 2006; 8 : 465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Fisher KW, Zhang S, Wang M, et al TMPRSS2‐ERG gene fusion is rare compared to PTEN deletions in stage T1a prostate cancer. Mol Carcinog 2017; 56: 814–820. [DOI] [PubMed] [Google Scholar]
- 94. Williamson SR, Zhang S, Yao JL, et al ERG‐TMPRSS2 rearrangement is shared by concurrent prostatic adenocarcinoma and prostatic small cell carcinoma and absent in small cell carcinoma of the urinary bladder: evidence supporting monoclonal origin. Mod Pathol 2011; 24 : 1120–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Li Z, Tognon CE, Godinho FJ, et al ETV6‐NTRK3 fusion oncogene initiates breast cancer from committed mammary progenitors via activation of AP1 complex. Cancer Cell 2007; 12 : 542–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Del Castillo M, Chibon F, Arnould L, et al Secretory breast carcinoma: a histopathologic and genomic spectrum characterized by a joint specific ETV6‐NTRK3 gene fusion. Am J Surg Pathol 2015; 39 : 1458–1467. [DOI] [PubMed] [Google Scholar]
- 97. Tognon C, Knezevich SR, Huntsman D, et al Expression of the ETV6‐NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell 2002; 2 : 367–376. [DOI] [PubMed] [Google Scholar]
- 98. Bass AJ, Lawrence MS, Brace LE, et al Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A‐TCF7L2 fusion. Nat Genet 2011; 43 : 964–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Seshagiri S, Stawiski EW, Durinck S, et al Recurrent R‐spondin fusions in colon cancer. Nature 2012; 488 : 660–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer 2013; 13 : 184–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Cabanillas ME, McFadden DG, Durante C. Thyroid cancer. Lancet 2016; 388 : 2783–2795. [DOI] [PubMed] [Google Scholar]
- 102. Cancer Genome Atlas Research Network . Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014; 159 : 676–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Nikiforov YE, Nikiforova MN. Molecular genetics and diagnosis of thyroid cancer. Nat Rev Endocrinol 2011; 7 : 569–580. [DOI] [PubMed] [Google Scholar]
- 104. Romei C, Ciampi R, Elisei R. A comprehensive overview of the role of the RET proto‐oncogene in thyroid carcinoma. Nat Rev Endocrinol 2016; 12 : 192–202. [DOI] [PubMed] [Google Scholar]
- 105. Kelly LM, Barila G, Liu P, et al Identification of the transforming STRN‐ALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer. Proc Natl Acad Sci U S A 2014; 111 : 4233–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Raman P, Koenig RJ. Pax‐8‐PPAR‐gamma fusion protein in thyroid carcinoma. Nat Rev Endocrinol 2014; 10 : 616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Caria P, Frau DV, Dettori T, et al Optimizing detection of RET and PPARg rearrangements in thyroid neoplastic cells using a home‐brew tetracolor probe. Cancer Cytopathol 2014; 122 : 377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Sharifah NA, Zakaria Z, Chia WK. FISH analysis using PPAR gamma‐specific probes for detection of PAX8‐PPAR gamma translocation in follicular thyroid neoplasms. Methods Mol Biol 2013; 952 : 187–196. [DOI] [PubMed] [Google Scholar]
- 109. Weinreb I. Translocation‐associated salivary gland tumors: a review and update. Adv Anat Pathol 2013; 20 : 367–377. [DOI] [PubMed] [Google Scholar]
- 110. Amelio AL, Fallahi M, Schaub FX, et al CRTC1/MAML2 gain‐of‐function interactions with MYC create a gene signature predictive of cancers with CREB‐MYC involvement. Proc Natl Acad Sci U S A 2014; 111 : E3260–E3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Tonon G, Modi S, Wu L, et al t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a notch signaling pathway. Nat Genet 2003; 33 : 208–213. [DOI] [PubMed] [Google Scholar]
- 112. Anzick SL, Chen WD, Park Y, et al Unfavorable prognosis of CRTC1‐MAML2 positive mucoepidermoid tumors with CDKN2A deletions. Genes Chromosomes Cancer 2010; 49 : 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wysocki PT, Izumchenko E, Meir J, et al Adenoid cystic carcinoma: emerging role of translocations and gene fusions. Oncotarget 2016; 7 : 66239–66254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ramsay RG, Gonda TJ. MYB function in normal and cancer cells. Nat Rev Cancer 2008; 8 : 523–534. [DOI] [PubMed] [Google Scholar]
- 115. West RB, Kong C, Clarke N, et al MYB expression and translocation in adenoid cystic carcinomas and other salivary gland tumors with clinicopathologic correlation. Am J Surg Pathol 2011; 35 : 92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Stenman G, Persson F, Andersson MK. Diagnostic and therapeutic implications of new molecular biomarkers in salivary gland cancers. Oral Oncol 2014; 50 : 683–690. [DOI] [PubMed] [Google Scholar]
- 117. North JP, Garrido MC, Kolaitis NA, et al Fluorescence in situ hybridization as an ancillary tool in the diagnosis of ambiguous melanocytic neoplasms: a review of 804 cases. Am J Surg Pathol 2014; 38 : 824–831. [DOI] [PubMed] [Google Scholar]
- 118. Skalova A, Vanecek T, Sima R, et al Mammary analogue secretory carcinoma of salivary glands, containing the ETV6‐NTRK3 fusion gene: a hitherto undescribed salivary gland tumor entity. Am J Surg Pathol 2010; 34 : 599–608. [DOI] [PubMed] [Google Scholar]
- 119. Skalova A, Weinreb I, Hyrcza M, et al Clear cell myoepithelial carcinoma of salivary glands showing EWSR1 rearrangement: molecular analysis of 94 salivary gland carcinomas with prominent clear cell component. Am J Surg Pathol 2015; 39 : 338–348. [DOI] [PubMed] [Google Scholar]
- 120. Majewska H, Skalova A, Stodulski D, et al Mammary analogue secretory carcinoma of salivary glands: a new entity associated with ETV6 gene rearrangement. Virchows Arch 2015; 466 : 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Connor A, Perez‐Ordonez B, Shago M, et al Mammary analog secretory carcinoma of salivary gland origin with the ETV6 gene rearrangement by FISH: expanded morphologic and immunohistochemical spectrum of a recently described entity. Am J Surg Pathol 2012; 36 : 27–34. [DOI] [PubMed] [Google Scholar]
- 122. Antonescu CR, Katabi N, Zhang L, et al EWSR1‐ATF1 fusion is a novel and consistent finding in hyalinizing clear‐cell carcinoma of salivary gland. Genes Chromosomes Cancer 2011; 50 : 559–570. [DOI] [PubMed] [Google Scholar]
- 123. Kao YC, Lan J, Tai HC, et al Angiomatoid fibrous histiocytoma: clinicopathological and molecular characterisation with emphasis on variant histomorphology. J Clin Pathol 2014; 67 : 210–215. [DOI] [PubMed] [Google Scholar]
- 124. Gru AA, Becker N, Pfeifer JD. Angiosarcoma of the parotid gland with a t(12;22) translocation creating a EWSR1‐ATF1 fusion: a diagnostic dilemma. J Clin Pathol 2013; 66 : 452–454. [DOI] [PubMed] [Google Scholar]
- 125. Hallor KH, Mertens F, Jin Y, et al Fusion of the EWSR1 and ATF1 genes without expression of the MITF‐M transcript in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer 2005; 44 : 97–102. [DOI] [PubMed] [Google Scholar]
- 126. Wang J, Thway K. Clear cell sarcoma‐like tumor of the gastrointestinal tract: an evolving entity. Arch Pathol Lab Med 2015; 139 : 407–412. [DOI] [PubMed] [Google Scholar]
- 127. Wiesner T, He J, Yelensky R, et al Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun 2014; 5 : 3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Bradish JR, Cheng L. Molecular pathology of malignant melanoma: changing the clinical practice paradigm toward a personalized approach. Hum Pathol 2014; 45 : 1315–1326. [DOI] [PubMed] [Google Scholar]
- 129. Cancer Genome Atlas Research Network . Genomic classification of cutaneous melanoma. Cell 2015; 161 : 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Shain AH, Bastian BC. From melanocytes to melanomas. Nat Rev Cancer 2016; 16 : 345–358. [DOI] [PubMed] [Google Scholar]
- 131. Hutchinson KE, Lipson D, Stephens PJ, et al BRAF fusions define a distinct molecular subset of melanomas with potential sensitivity to MEK inhibition. Clin Cancer Res 2013; 19 : 6696–6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Ross JS, Wang K, Chmielecki J, et al The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer 2016; 138 : 881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Bottaro DP, Rubin JS, Faletto DL, et al Identification of the hepatocyte growth factor receptor as the c‐met proto‐oncogene product. Science 1991; 251 : 802–804. [DOI] [PubMed] [Google Scholar]
- 134. Yeh I, Botton T, Talevich E, et al Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat Commun 2015; 6 : 7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Louis DN, Ohgaki H, Wiestler OD, WK Cavenee (Eds). WHO Classification of Tumours of the Central Nervous System. WHO/IARC Classification of Tumours, 4th Edition Revised, Volume 1. IARC Press, Lyon: 2016. [Google Scholar]
- 136. Shah N, Lankerovich M, Lee H, et al Exploration of the gene fusion landscape of glioblastoma using transcriptome sequencing and copy number data. BMC Genomics 2013; 14 : 818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Frattini V, Trifonov V, Chan JM, et al The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet 2013; 45 : 1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Ceccarelli M, Barthel FP, Malta TM, et al Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016; 164 : 550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Bao ZS, Chen HM, Yang MY, et al RNA‐seq of 272 gliomas revealed a novel, recurrent PTPRZ1‐MET fusion transcript in secondary glioblastomas. Genome Res 2014; 24 : 1765–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Eckel‐Passow JE, Lachance DH, Molinaro AM, et al Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med 2015; 372 : 2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Mur P, Mollejo M, Hernandez‐Iglesias T, et al Molecular classification defines 4 prognostically distinct glioma groups irrespective of diagnosis and grade. J Neuropathol Exp Neurol 2015; 74 : 241–249. [DOI] [PubMed] [Google Scholar]
- 142. Wesseling P, van den Bent M, Perry A. Oligodendroglioma: pathology, molecular mechanisms and markers. Acta Neuropathol 2015; 129 : 809–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Rodriguez FJ, Vizcaino MA, Lin MT. Recent advances on the molecular pathology of glial neoplasms in children and adults. J Mol Diagn 2016; 18 : 620–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Jenkins RB, Blair H, Ballman KV, et al A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 2006; 66 : 9852–9861. [DOI] [PubMed] [Google Scholar]
- 145. Jones DT, Hutter B, Jager N, et al Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 2013; 45 : 927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Merchant TE, Li C, Xiong X, et al Conformal radiotherapy after surgery for paediatric ependymoma: a prospective study. Lancet Oncol 2009; 10 : 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Parker M, Mohankumar KM, Punchihewa C, et al C11orf95‐RELA fusions drive oncogenic NF‐kappaB signalling in ependymoma. Nature 2014; 506 : 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Versteeg R. Cancer: tumours outside the mutation box. Nature 2014; 506 : 438–439. [DOI] [PubMed] [Google Scholar]