

Abstract
Gangliosides are glycolipids in which an oligosaccharide headgroup containing one or more sialic acids is connected to a ceramide. Gangliosides reside in the outer leaflet of the plasma membrane and play a crucial role in various physiological processes such as cell signal transduction and neuronal differentiation by modulating structures and functions of membrane proteins. Because the detailed behavior of gangliosides and protein-ganglioside interactions are poorly known, we investigated the interactions between the gangliosides GM1 and GM3 and the proteins aquaporin (AQP1) and WALP23 using equilibrium molecular dynamics simulations and potential of mean force calculations at both coarse-grained (CG) and atomistic levels. In atomistic simulations, on the basis of the GROMOS force field, ganglioside aggregation appears to be a result of the balance between hydrogen bond interactions and steric hindrance of the headgroups. GM3 clusters are slightly larger and more ordered than GM1 clusters due to the smaller headgroup of GM3. The different structures of GM1 and GM3 clusters from atomistic simulations are not observed at the CG level based on the Martini model, implying a difference in driving forces for ganglioside interactions in atomistic and CG simulations. For protein-ganglioside interactions, in the atomistic simulations, GM1 lipids bind to specific sites on the AQP1 surface, whereas they are depleted from WALP23. In the CG simulations, the ganglioside binding sites on the AQP1 surface are similar, but ganglioside aggregation and protein-ganglioside interactions are more prevalent than in the atomistic simulations. Using the polarizable Martini water model, results were closer to the atomistic simulations. Although experimental data for validation is lacking, we proposed modified Martini parameters for gangliosides to more closely mimic the sizes and structures of ganglioside clusters observed at the atomistic level.
Introduction
Glycolipids are amphiphilic molecules composed of a pair of hydrophobic alkyl tails, anchoring the lipid to the bilayer, and a hydrophilic oligosaccharide headgroup. The headgroup resides on the bilayer surface and interacts with aqueous and plasma membrane constituents from the same cell or neighboring cells.1−5 Various species of glycolipids exist with different oligosaccharide headgroups, lipid backbones, and alkyl tails. In glycoglycerolipids, the headgroups are attached to glycerol, and in glycosphingolipids (GSLs), they are attached to ceramide. Gangliosides are GSLs with one or more sialic acids, hence one or more negative charges, in their oligosaccharide headgroups. In GSLs, the headgroups are connected to ceramide backbones via the 1-hydroxyl moiety of one of the sugars.6 Gangliosides are involved in many physiological processes,3,7,8 such as cell adhesion,9 pathogen recognition and viral infection,10,11 signal transduction,12 cell proliferation, and neuronal protection.8,13 The diversity of their biological functions is related to the diversity of their oligosaccharide headgroups.6 Two common GSLs are GM1 and GM3 (Figure 1), both with one sialic acid but different headgroup sizes. GM1 is enriched in the gray matter of the central nervous system and plays a key role in neurotrophic and neuroprotective processes8,14 as well as neurodegenerative diseases.15,16 GM3 is thought to regulate cell growth, adhesion, and motility.17 Abnormal lipid microdomains involving GM3 are associated with cancer development17 and insulin resistance in type 2 diabetes.18−20
Figure 1.
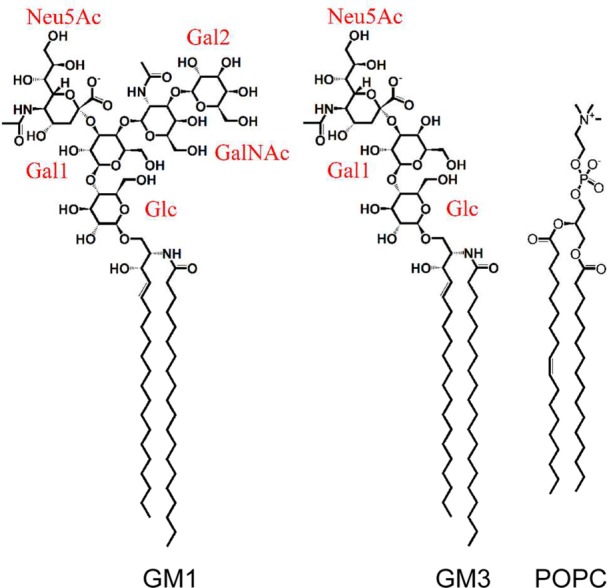
Structures of GM1, GM3, and POPC.
In bilayer environments, gangliosides segregate into microdomains (rafts) enriched in sphingomyelin, cholesterol, and gangliosides,21 and glycosynapses, which are formed by specific proteins and gangliosides but without cholesterol.22−24 These microdomains are highly dynamic, both spatially and temporally. Gangliosides can influence the lateral distributions of lipids in membranes.25 They can modulate the morphology of bilayers by inducing membrane curvature and are involved in membrane fusion and viral budding.26−29 Gangliosides specifically interact with and regulate the functions of various proteins, e.g., the epidermal growth factor receptor (EGFR)30 and insulin receptor.31 They also influence the lateral sorting of transmembrane lipid raft proteins.32
An important structural feature of gangliosides is their hydrogen bonding ability through their headgroups, which results in extensive ganglioside and protein-ganglioside interactions. However, the negative charges, steric hindrance, and hydration shell around the headgroups modulate these interactions. Other characteristics that contribute to ganglioside and protein-ganglioside interactions include the higher transition temperature due to long saturated tails, possible hydrogen bonding between the amide groups of the ceramide tails, and interdigitation between leaflets due to their long tails.24 Differences in ganglioside structures may result in significant differences between their interactions. GM1 and GM3 differ by two sugar units (GalNAc and Gal2, shown in Figure 1), but GM1 and GM3 clusters are separated from each other in membranes.33,34
Ganglioside and protein-ganglioside interactions have been investigated in model lipid mixtures both experimentally and with molecular dynamics (MD) simulations. The experiments have focused on detecting lipid domains containing gangliosides, as reviewed in refs (27 and 35), for example. The primary advantage of MD simulations is that they can resolve detailed lipid interactions.36−38 Manna et al.39 recently reviewed glycolipid MD simulations, highlighting the structural characterizations that affect lipid organizations in various environments. Hall et al. analyzed interactions in lipid mixtures of galactosylceramide (GalCer), a glycosphingolipid, and other lipid raft components.40,41 Sega et al. explored properties of GM3 bilayers by MD simulation.42 Patel et al.43,44 and Jedlovszky et al.45 investigated the conformations and distributions of GM1 in 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) bilayers. Mori et al. compared GM1 clusters in 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayers and lipid mixtures containing sphingomyelin and cholesterol.46
A notable part of the simulation studies has been conducted at the coarse-grained (CG) level using the Martini force field.47−49 In Martini, four heavy atoms are generally combined to form one CG bead. The force field is parametrized based on free energy landscapes of molecular interactions, particularly the partitioning free energies of molecules between oil- and water-like phases. Martini enables simulations of larger systems and/or longer time scales due to the reduced level of detail while still maintaining a reasonable degree of chemical accuracy. For instance, the plasma membrane simulations by Ingólfsson et al. found dynamic ganglioside clusters,50 whereas another simulation of complex lipid mixtures revealed correlation between bilayer curvature and GM3 clusters.51 Shorthouse et al.52 reviewed a coarse-grained model of GM3 and its application in protein-ganglioside interactions. Prasanna et al.53 investigated binding of GM1 to the serotonin 1A receptor. Jong et al. simulated sorting of proteins to liquid ordered (Lo) domains induced by gangliosides and lipid anchors.54 Basu et al.55 and Kociurzynski et al.56 simulated phase separation and transitions of lipid mixtures containing gangliosides.
Despite extensive studies of ganglioside-containing lipid mixtures, the strength of ganglioside and protein-ganglioside interactions is still poorly known (e.g, the size and stability of ganglioside clusters). In this study, we conducted both equilibrium MD simulations and free energy calculations of gangliosides, or gangliosides and proteins, in POPC bilayers both at the atomistic and CG levels to explore their interactions. We simulated GM1 and GM3 (Figure 1) as examples of gangliosides with different headgroup sizes and WALP23 and aquaporin (AQP1) as examples of membrane proteins. Our simulations provide detailed insight on the strength of ganglioside and protein-ganglioside interactions in a lipid bilayer environment and reveal how different types of ganglioside headgroups affect ganglioside aggregation.
Methods
System Setup
Both equilibrium simulations and PMF calculations were performed to investigate ganglioside and protein-ganglioside interactions. All simulations are summarized in Table 1.
Table 1. Summary of Simulations.
| system composition | simulation length (μs) | simulation levela | water modelb | electrostatic interactionsc | |
|---|---|---|---|---|---|
| Equilibrium Simulations | |||||
| GM1-POPC | upper leaflet: 24 GM1, 120 POPC | 6.5 | CG | W | shift |
| lower leaflet: 144 POPC | 6.5 | CG | PW | shift | |
| 6.5 | CG | PW | PME | ||
| 3 | AA | SPC | RF | ||
| GM3-POPC | upper leaflet: 24 GM3, 120 POPC | 6.5 | CG | W | shift |
| lower leaflet: 144 POPC | 6.5 | CG | PW | shift | |
| 6.5 | CG | PW | PME | ||
| 3 | AA | SPC | RF | ||
| GM1-GM3-POPC | upper leaflet: 12 GM1, 12 GM3, 120 POPC | 2 | AA | SPC | RF |
| lower leaflet: 144 POPC | |||||
| WALP23-GM1- POPC | protein: 1 WALP23 | 15 | CG | W | shift |
| upper leaflet: 23 GM1, 116 POPC | 15 | CG | PW | shift | |
| lower leaflet: 139 POPC | 15 | CG | PW | PME | |
| 2 | AA | SPC | RF | ||
| AQP1-GM1-POPC | protein: 1 AQP1 | 15 | CG | W | shift |
| upper leaflet: 41 GM1, 207 POPC | 15 | CG | PW | shift | |
| lower leaflet: 248 POPC | 15 | CG | PW | PME | |
| 2 | AA | SPC | RF | ||
| PMF Calculations | |||||
|---|---|---|---|---|---|
| GM1-GM1 | upper leaflet: 2 GM1, 254 POPC | 3.5×36 | CG | W | shift |
| lower leaflet: 256 POPC | 4.5×36 | CG | PW | shift | |
| 4.5×36 | CG | PW | PME | ||
| GM3-GM3 | upper leaflet: 2 GM1, 254 POPC | 3.5×36 | CG | W | shift |
| lower leaflet: 256 POPC | 4.5×36 | CG | PW | shift | |
| 4.5×36 | CG | PW | PME | ||
| GM3-WALP23 | protein: 1 WALP23 | 5.5×31 | CG | W | shift |
| upper leaflet: 1 GM3, 250 POPC | 5.5×31 | CG | PW | shift | |
| lower leaflet: 251 POPC | 5.5×31 | CG | PW | PME | |
| GM3-AQP1 | protein: 1 AQP1 | 1.5×23 | CG | W | shift |
| upper leaflet: 1 GM3, 290 POPC | 1.5×23 | CG | PW | shift | |
| lower leaflet: 291 POPC | 1.5×23 | CG | PW | PME | |
CG: coarse-grained simulation; AA: atomistic simulation.
W: standard Martini water model; PW: polarizable Martini water model; SPC: simple point charge atomistic water model.
PME: particle mesh Ewald; RF: reaction field.
For equilibrium simulations, GM-POPC binary mixtures containing 17% GM1 or GM3 in the upper leaflet were simulated to investigate ganglioside interactions. We also simulated a GM1-GM3-POPC ternary mixture with 8.5% GM1 and 8.5% GM3 in the upper leaflet to illustrate the interactions between GM1 and GM3. Simulations of WALP23 (sequence: GWW(LA)8LWWA) and aquaporin 1 (AQP1) were conducted in binary mixtures with 17% GM1 in the upper leaflet to investigate protein-ganglioside interactions. Equilibrium simulations were conducted at both atomistic and CG levels. Free energy calculations were only conducted at the CG level. PMFs of the interaction between two GM1 lipids, two GM3 lipids, WALP23 and GM3, and AQP1 and GM3 in POPC bilayers were calculated as a function of distances in the x–y plane. For both the equilibrium simulations and PMF calculations, three parallel simulations were conducted at the CG level with different water models and parameters for electrostatic interactions (see below for detailed parameters).
The CG systems were built using the insane script.57 The CG model of GM lipid tail has the bead order AM1-AM2-T1A-C2A-C3A-C1B-C2B-C3B-C4B. The CG POPC lipid has the bead order NC3-PO4-GL1-GL2-C1A-C2A-D3A-C4A-C1B-C2B-C3B-C4B. WALP23 was built as an ideal helix and then equilibrated for 100 ns in a POPC bilayer before being used in the ganglioside simulations, whereas the AQP1 simulations were started from the crystal structure (PDB ID: 1J4N).58 Atomistic simulation systems were backmapped from equilibrated snapshots of the corresponding CG simulations.59 GM(d20:1/20:0) lipids were used in the atomistic simulations (i.e., the tail was constituted by a long-chain base of 20 carbons and an eicosyl fatty acid residue, as shown in Figure 1).
CG Simulation Details
CG simulations were performed with the Martini force field49 and the ganglioside lipid extension.48,50 Simulations were done with the GROMACS 4.6.5 package60,61 using an integration time step of 20 fs. Then, 0.15 M NaCl was added to mimic the physiological ionic strength. Temperature was maintained at 310 K using the v-rescale algorithm62 with a relaxation time of 2.0 ps. Semi-isotropic pressure coupling was applied with a reference pressure of 1 bar using the Parrinello–Rahman algorithm63,64 with a relaxation time of 12.0 ps; van der Waals interactions were turned off between 0.9 and 1.2 nm using the shift function in GROMACS. Three parallel simulations were conducted for each system with different Martini water models and parameters for electrostatic interactions. In the first set, called CG W, the standard Martini water49,65 was employed. The shift function in GROMACS was used to turn off the electrostatic interactions between 0 and 1.2 nm, and a dielectric constant of 15 was used. In the second set, called CG PW, polarizable Martini water was used with the same cutoff distance and a dielectric constant of 2.5.66 In the third and last set, called CG PW PME, polarizable Martini water and a dielectric constant of 2.5 were also used but additional long-range electrostatic interactions were included using the PME method.67,68 ElNeDyn approach was used to restrain the protein structures.69 All simulations and conditions are listed in Table 1.
Atomistic Simulation Details
Atomistic simulations were conducted with the GROMACS 4.0.7 package60,61 using an integration time step of 2 fs. Parameters for POPC and the lipid part of gangliosides are based on the GROMOS 53A6 united-atom force field,70 whereas parameters of ganglioside headgroups are based on the GROMOS hexopyranose force field.48,71,72 The systems were solvated in SPC water, and both counterions and 0.15 M NaCl were added. Temperature was maintained at 330 K by coupling the bilayer/protein and solution separately via the Berendsen thermostat algorithm73 with a relaxation time of 0.1 ps. A temperature of 330 K was used in atomistic simulations because we observed gel phase for gangliosides at 310 K. A 330 K simulation of the GM3-POPC mixture at the CG level was conducted as a control (see Discussion for details). Semi-isotropic pressure coupling with a relaxation time of 0.5 ps was applied to maintain the pressure at 1 bar.73 Nonbonded interactions within 0.9 nm were calculated every step based on a pair list that was updated every five steps. Interactions between 0.9 and 1.4 nm were calculated every five steps.74 Long-range electrostatic interactions were included using the reaction-field algorithm with a relative dielectric permittivity of 54.75 In most of the simulations, the proteins and lipids were free to move, but in the AQP1 simulations, position restraints of 10 kJ mol–1 nm–2 were applied on the protein backbone to maintain a backbone RMSD of ∼0.22 nm.
PMF Calculations
Umbrella sampling76 was employed to calculate the free energies of ganglioside and protein-ganglioside interactions at the CG level in POPC bilayers (Table 1). Starting structures for each window, spaced 1.0 Å, were taken from an initial 0.5 μs simulation in which one ganglioside molecule was pulled away from a second ganglioside or the protein in the plane of the bilayer. A force constant of 1000 kJ mol–1 nm–2 was used to control the distances between the centers of mass of gangliosides (or distances between ganglioside and protein) in the x–y plane. Twenty-three to thirty-six windows were used. MD simulations for each window were performed for 1.5–5.5 μs depending on the simulation system (see Table 1). The first 0.5 μs of each simulation was taken as equilibration, and the remaining parts were used to generate the PMF profiles based on the weighted histogram analysis method (WHAM).77 For the convergence to be verified, the trajectories were divided into blocks, and PMF profiles based on each block were calculated and compared. For GM3-AQP1 PMF, extensive sampling of GM3 around AQP1 is computationally too costly due to the large size of the protein. Therefore, PLUMED78 was used to control the orientation between GM3 and AQP1, so that GM3 approached AQP1 in one specific direction (see Figure S1) and a one-dimensional PMF profile as a function of the distance between them was obtained. GM3 was used to calculate the protein-ganglioside interactions instead of GM1 because the GM3 lipid has a smaller headgroup and the convergence of the PMF profile is faster. Given the simulation lengths required for the GM PMFs, accurate atomistic PMFs are currently not feasible with this protocol.
Modifications to the Martini Ganglioside Force Field
Because the aggregation of gangliosides in the CG simulations was significantly stronger than in the atomistic simulations (see section 1 of the results for details), we reconsidered the CG Martini GM1 and GM3 parameters. The structure and size of ganglioside clusters, and the radial distribution functions of gangliosides in the POPC bilayer at the atomistic level, were used as criteria. The atomistic to CG mapping (described in López et al.48) was kept unchanged, the bonded and nonbonded interactions were modified slightly (see section 3.1 of the results and the Supporting Information for detailed changes). For the bonded interactions, atomistic trajectories were converted to the CG level every 0.5 ns, and the distributions of the bonds, angles, and dihedrals were analyzed and used as references. Several new angles and dihedrals were introduced to better control ganglioside conformations at the CG level. For nonbonded interactions, different types of CG beads were assigned to the headgroups to reduce their interactions. Equilibrium simulations of GM1-POPC and GM3-POPC binary mixtures were conducted using the modified CG force field and compared with the previous parameters.
Data Analysis
The initial part of each simulation was excluded from the analyses. For the CG simulations of 6.5, 10, and 15 μs in length, the last 4, 7, and 12 μs were used for analysis, respectively, whereas for the atomistic simulations of 2 and 3 μs in length, the last 1 and 1.5 μs were used, respectively. The analysis of ganglioside cluster sizes was performed using the g_aggregate tool.79 Gangliosides were considered to be in the same cluster if the distance between their centers of mass in the x–y plane was within 1.5 nm. Radial distribution functions (RDFs) were calculated based on the distances between the centers of mass of lipids/ganglioside headgroups/tails and proteins in the x–y plane. RDF profiles were only calculated for the lipids in the upper leaflet. Hydrogen bonds were defined in the following way: the distance between the two heavy atoms is smaller than 3.5 Å, and the angle between donor, hydrogen atom, and acceptor is smaller than 150°. Order parameters of the lipid tails at the CG level were calculated based on the angle between the bonds of the lipid tails and the bilayer normal (approximated as the box z axis) using the g_ordercg tool.80
Results
First, the ganglioside-ganglioside interactions in simulations of mixed ganglioside-POPC bilayers using the original Martini force field and the GROMOS force field are compared. Sizes and structures of ganglioside clusters in the CG W, CG PW, CG PW PME, and atomistic simulations are described in detail. Next, proteins are included and the protein-ganglioside interactions at the CG and atomistic levels are examined. The distributions of GM lipids around WALP23 and AQP1 are presented. Lastly, modifications of the Martini ganglioside force field are conducted to mimic the ganglioside clustering in the atomistic POPC bilayer.
1. Interactions between Gangliosides
First, we analyze the ganglioside-POPC mixtures (Table 1). The bilayer properties (Table S1) and the density profiles of lipids (Figure S2) are calculated for the atomistic and three CG simulations. The polarizable Martini (PW) water and long-range electrostatic interactions only slightly affect bilayer properties. We focus here on ganglioside-ganglioside interactions and the formation of ganglioside clusters.
1.1. Ganglioside Distribution in POPC Bilayers
The distribution of GM1 and GM3 is explored in POPC lipid simulations with 17% GM lipids in the upper leaflet (Figure 2). Under all of the tested conditions, the gangliosides cluster, but the degree of aggregation varies, as shown by the snapshots at the end of each simulation (Figure 2A). The averaged ganglioside cluster size and the number of clusters are calculated as a function of simulation time (Figure 2B and Table 2). GM1 aggregates most in the CG W simulations. Using PW decreases the cluster size significantly, whereas also including long-range electrostatic interactions (PME) has almost no additional effect. The average GM1 cluster sizes in the three CG simulations are 18, 5, and 4 lipids for the W, PW, and PW PME systems, respectively (Table 2). In the atomistic simulation, a large GM1 cluster exists at the beginning, as the initial conformation is backmapped from an equilibrated CG W snapshot. However, these large clusters quickly disassociate. After equilibration, more than 10 clusters are found over most of the simulation time with an average cluster size of only 2 lipids, indicating weaker GM1 interactions at the atomistic level (Figure 2B and Table 2). Simulations of GM3 reveal similar results, i.e., including PW weakens ganglioside interactions dramatically, and GM3 interactions at the atomistic level are the weakest among all of the four simulations. The GM3 cluster sizes in the three CG simulations (W, PW, and PW PME) and the atomistic simulation are 6, 3, 3, and 3 lipids, respectively (Table 2 and Figure 2B). GM3 clusters are somewhat smaller than GM1 at the CG level, whereas the opposite is observed at the atomistic level.
Figure 2.

Ganglioside interactions in the GM-POPC binary mixtures. (A) Snapshot of the upper leaflet at the end of the simulations. GM lipids and POPC are shown in red and gray, respectively. (B) Averaged sizes and numbers of ganglioside clusters as a function of simulation time. (C) PMF calculations of ganglioside interactions at the CG level. Abbreviations: CG, coarse-grained simulations; W, standard Martini water; PW, polarizable Martini water; PME, PME algorithm for long-range electrostatic interactions; AA, atomistic simulations.
Table 2. Average Size and Number of Ganglioside Clusters in POPC Bilayersa.
| lipid type | CG W | CG PW | CG PW PME | AA | CG W New FF | CG PW New FF | |
|---|---|---|---|---|---|---|---|
| GM1 | size | 18 ± 7 | 5 ± 2 | 4 ± 1 | 2 ± 1 | 3 ± 1 | 2 ± 1 |
| number | 2 ± 1 | 6 ± 2 | 6 ± 2 | 11 ± 2 | 10 ± 2 | 11 ± 2 | |
| GM3 | size | 6 ± 2 | 3 ± 1 | 3 ± 1 | 3 ± 1 | 4 ± 1 | 3 ± 1 |
| number | 5 ± 1 | 8 ± 2 | 8 ± 2 | 10 ± 2 | 7 ± 2 | 10 ± 2 | |
Abbreviations: CG W, CG simulations with standard Martini water; CG PW, CG simulations with polarizable water; CG PW PME, CG simulations with polarizable water in which PME was used for long-range electrostatic interactions; AA, atomistic simulations; New FF, simulations with reoptimized Martini ganglioside force field. Values are shown as average ± standard deviation.
The strength of ganglioside interactions revealed by three equilibrium simulations at the CG level is consistent with the PMF calculations: the association energy of GM1-GM1 is significantly larger than that of GM3-GM3 (in CG W simulations, the values are approximately −12 and −5.5 kJ/mol, respectively; Figure 2C). The calculated energies with the PW are roughly half of the values obtained with the standard Martini water (approximately −6.5 and −2.5 kJ/mol for GM1-GM1 and GM3-GM3, respectively).
1.2. Structures of Ganglioside Clusters
In addition to the cluster analyses, we calculated radial distribution function (RDF) profiles (Figure 3A and B; examples of convergence are shown in Figure S3) to investigate the structures of ganglioside clusters. RDFs for all CG simulations show peaks at ∼0.8 nm, but the magnitudes are decreased significantly by PW for both GM1 and GM3, which is consistent with the cluster sizes. However, in the atomistic simulations, the peak of the GM1-GM1 RDF is shifted to ∼1.6 nm and is much lower. For GM3, there is a strong peak at ∼0.6 nm and a second weak peak around 2.0 nm. Although the first peak of the GM3-GM3 RDF shows a magnitude comparable with that of CG PW, its width is much narrower, implying clusters with tighter packed structures.
Figure 3.

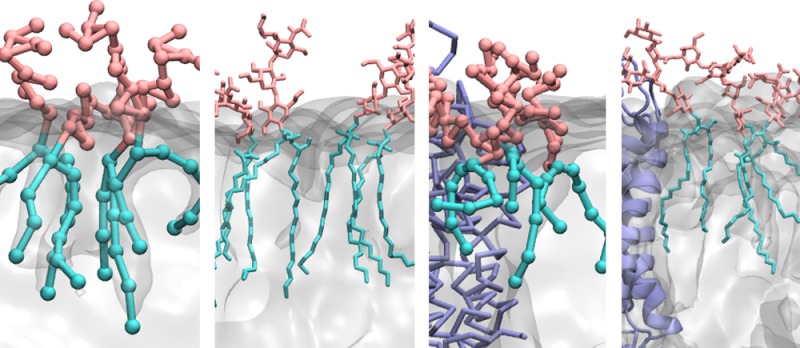
Structures of ganglioside clusters. (A) RDFs between the centers of mass of GM lipids and RDFs between the centers of mass of GM and POPC. (B) RDFs between the GM headgroups and between the GM tails. (C) GM1 and (D) GM3 clusters in the CG PW PME simulations. (E) GM1 and (F) GM3 clusters in the atomistic simulations. Gangliosides and POPC are shown in yellow and cyan, respectively. The ganglioside headgroups at the CG level are shown in pink. The sialic acid groups of gangliosides are shown in green. Hydrogen bonds are shown with dotted lines. Abbreviations: CG, coarse-grained simulations; W, standard Martini water; PW, polarizable Martini water; PME, PME algorithm for long-range electrostatic interactions; AA, atomistic simulations.
The RDFs of the headgroups and tails are plotted separately to further illustrate the ganglioside interactions (Figure 3B). Peaks are found for both headgroups and tails in all of the CG simulations. The headgroup peaks of GM1 are shifted to the right compared with GM3, which is consistent with the larger size of the GM1 headgroup. In the atomistic simulations of GM3, peaks are found for the RDFs of both the headgroups and tails at ∼0.6 nm, although the peak for tails is much weaker. However, for GM1, only the headgroup RDF shows a weak peak at ∼1.1 nm. Thus, at the atomistic level, both headgroups and tails interact with each other in GM3 clusters but only the headgroups aggregate in GM1 clusters. Minor differences in ganglioside cluster sizes and significant differences in the shapes of RDFs indicate different structures of ganglioside clusters.
The GM-POPC RDFs show peaks around 0.9 nm in the atomistic simulations, but the peaks are much weaker in the CG simulations (Figure 3A). In the atomistic simulations, these peaks are wider for GM1 than GM3 due to the larger spaces between GM1 lipid tails that may allow accommodation of POPC lipids.
Typical GM1 and GM3 clusters at both the CG and atomistic levels are shown in Figure 3C–F. Clusters in atomistic simulations consisting of two or three (Figure 3E and F) gangliosides are shown together with associated POPC molecules. In the GM1 clusters, Neu5Ac, and Gal2, GalNAc extend in different directions and form hydrogen bonds, whereas Gal, Glc, and ceramide groups are further away from each other (Figure 3E), consistent with the fact that peaks are only found for RDFs of headgroups but not for the RDFs of tails (Figure 3B). However, in the GM3 clusters, the headgroups are almost planar and tightly packed parallel to each other (Figure 3F). In this way, the ceramide groups are able to approach each other close enough to form hydrogen bonds (Figure 3F). The POPC lipid tails are inserted into the ganglioside clusters, whereas the phosphate groups form hydrogen bonds with the Glc and ceramide of the gangliosides, as shown in Figure 3E and F. The clusters in the CG simulations do not show distinctly different structures between GM1 and GM3 (Figure 3C and D), and both the headgroups and tails have extensive contacts with each other, which are consistent with the peaks in the RDFs in Figure 3B.
1.3. Hydrogen Bond Interactions Involving Gangliosides
Because the ganglioside headgroups can act as both hydrogen bond donors and acceptors, we analyzed intermolecular hydrogen bond interactions in the atomistic simulations; the results are shown in Table 3. Neu5Ac (the sialic acid) contributes most to the hydrogen bond interactions between gangliosides for both GM1 and GM3, which are consistent with Mori et al.’s simulations.46 In all of the atomistic simulations, hydrogen bonds involving Neu5Ac account for at least 55% of the total number of hydrogen bonds between the gangliosides. For GM3, these hydrogen bonds are mainly formed between Neu5Ac of different molecules, whereas for GM1, Gal2 also forms hydrogen bonds with Neu5Ac in addition to the hydrogen bonds between Neu5Ac groups. Although GM1 has a larger headgroup and more hydrogen bond donors and acceptors, the averaged intermolecular hydrogen bond numbers are smaller than for GM3.
Table 3. Hydrogen Bond Interactions between Gangliosides in Atomistic Simulations.
| hydrogen
bonds between gangliosides | ||||||||
|---|---|---|---|---|---|---|---|---|
| simulation system | GM | Neu5Ac | Neu5Ac | Neu5Ac | Neu5Ac | Gal2 | Gal2 | Gal1 |
| GMa | Neu5Ac | Gal2 | GalNAc | Gal1 | Gal2 | GalNAc | Glc | |
| GM1-POPC | 1.97 ± 0.73 | 27.5% | 26.4% | 7.1% | 6.1% | 8.1% | 5.1% | <2% |
| GM3-POPC | 2.73 ± 1.62 | 65.5% | 30.2% | 3.4% | ||||
| hydrogen bonds between gangliosides and POPC | ||||||
|---|---|---|---|---|---|---|
| simulation system | GM | GM | Glc | Gal1 | Neu5Ac | Cerc |
| POPC | Phosb | Phos | Phos | Phos | Phos | |
| GM1-POPC | 2.73 ± 0.27 | 89.4% | 35.5% | 12.5% | 7.3% | 28.2% |
| GM3-POPC | 2.37 ± 0.28 | 90.7% | 34.2% | 14.8% | 8.9% | 32.5% |
Average numbers of hydrogen bonds between one molecule in the first group and all of the molecules in the second group and breakdown of the H-bonds in percentages due to different fragments. If two groups are the same, the hydrogen bond number between one molecule and all of the other molecules in the group is calculated. The nomenclature for the sugars is shown in Figure 1.
Phos: Phosphate group.
Cer: Ceramide.
Hydrogen bonds are also found between the linkage parts of gangliosides, in particular for GM3, which packed tightly to form clusters. The average number of hydrogen bonds between amide groups in the GM3 simulations is ∼1.6, which means there are one or two pairs of GM3 lipids forming this kind of hydrogen bond in each simulation frame (for GM1, this is only 0.4). These are notably common interactions when considering the ratio of ganglioside to POPC lipids in this system.
Extensive hydrogen bond interactions are also found between gangliosides and POPC lipids. Each ganglioside forms ∼2.5 hydrogen bonds with POPC lipids, and most of them are hydrogen bonds between the phosphate groups of the POPC and the Glc and ceramide backbones of gangliosides (Table 3). Hydrogen bonds between phosphate and Glc, and hydrogen bonds between phosphate and ceramide, each account for 28–35% of the total number of hydrogen bonds. The results are similar for GM1 and GM3.
1.4. Interactions between GM1 and GM3
For the interactions between GM1 and GM3 to be investigated, an atomistic MD simulation of a GM1-GM3-POPC ternary mixture was performed. Both GM1 and GM3 clusters are observed (Figure 4), as in the simulations of binary mixtures, as well as small mixed clusters of GM1 and GM3. Peaks are found for the GM3-GM3 RDFs for entire lipids, headgroups, and tails (Figure 4B–D) with comparable positions and magnitudes as in the binary mixtures (Figure 3A and B). For GM1-GM1, no clear peak is observed for the entire lipid RDF, but a weak peak for the headgroup RDF is found at ∼1.2 nm (Figure 4C) as in the binary mixtures (Figure 3B). These results do not reveal notable differences in structure and size of ganglioside clusters in binary and ternary mixtures for either GM1 or GM3. No strong interactions between GM1 and GM3 were found. None of the GM1-GM3 RDFs for lipids, headgroups, and tails show defined peaks (Figure 4B–D), suggesting limited GM1-GM3 clustering. GM1/GM3-POPC RDFs are shown in Figure 4E.
Figure 4.
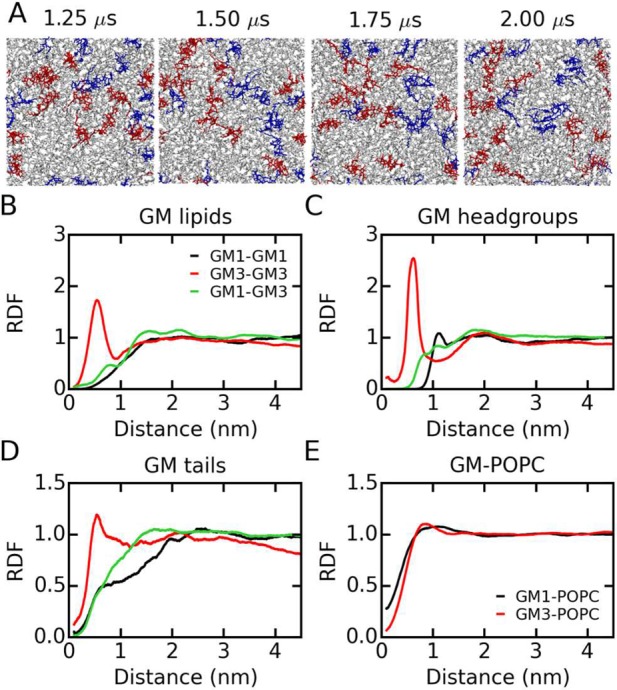
Ganglioside distributions in the atomistic GM1-GM3-POPC ternary mixture. (A) Snapshots of the simulation system. POPC lipids are shown in gray, and GM1 and GM3 are in red and blue, respectively. RDFs between the centers of mass of (B) GM lipids, (C) GM headgroups, (D) GM tails, and (E) GM and POPC lipids are shown.
We calculated the number of contacts between the gangliosides, averaged every 0.5 μs (Figure S4 and Table S2). The initial structure of the ternary mixture was created based on the last snapshot of the GM1-POPC simulation by changing some of the GM1 headgroups to GM3 headgroups; during the 2 μs simulation, the number of contacts between GM1 and GM3 decreased, whereas the GM3-GM3 contacts increased, and the number of GM1-GM1 contacts fluctuated. Our results are consistent with experiments conducted by Fujita et al.,33,34 which revealed separated GM1 and GM3 clusters, supporting the reliability of our atomistic simulations.
2. Protein-Ganglioside Interactions
2.1. Interactions between WALP23 and GM1
2D density maps (Figure 5A) and RDF profiles (Figure 5C) revealed that WALP23-GM1 interactions at the CG level are stronger than at the atomistic level. Using PW reduces the interactions dramatically, similarly to what we found in the lipid mixtures. Using PW PME makes the WALP23-GM1 interactions stronger than in the PW simulation without PME. In the simulation with PW (without PME), the RDF does not show peaks and is comparable to the corresponding RDF of the atomistic simulation (Figure 5C). In these two simulations (PW and AA), GM1 is depleted from WALP23. The GM1-protein RDF of the PW PME simulation shows a peak at ∼1.4 nm (Figure 5C). The 2D density maps are consistent with RDF profiles, as shown by the positions of high GM1 densities (Figure 5A). RDFs of the headgroups and tails in Figure 5D reveal that GM1 tail-WALP23 interactions are weaker than the GM1 headgroup-WALP23 interactions in all of the CG and atomistic simulations. The strength of GM-WALP23 interactions suggested by free energy calculations are consistent with the equilibrium simulations. The association energies of GM3-WALP23 are −2.5, −0.7, and −0.7 kJ/mol in the W, PW, and PW PME CG simulations, respectively (Figure 5B).
Figure 5.

Ganglioside-protein interactions in the GM1-POPC-protein simulations. (A) 2D density maps of GM1 lipids in the bilayer plane. The relative densities are shown. Structures of WALP23 and AQP1 are included to show their orientations during the density calculations. (B) PMF calculations of protein-ganglioside interactions at the CG level. (C) RDFs of GM1 and POPC lipids relative to the mass centers of the proteins. (D) RDFs of GM1 headgroups and tails relative to the mass centers of the proteins.
In the atomistic simulations, POPC is enriched around WALP23 compared with GM1 (see RDFs of GM1 and POPC lipids in Figure 5C). Figure 6A shows the snapshot at the end of the atomistic simulation. Most of the GM1 lipids are away from WALP23 except for one or two GM1 lipids (Figure 6A). The tails of that GM1 lipid do not come close to the peptide, but its headgroup tilts and interacts with the positively charged N-terminus via Neu5Ac (the sialic acid ,which has one negative charge) and GalNAc (Figure 6C).
Figure 6.

Interactions between GM1 lipids and WALP23 or AQP1 in atomistic simulations of GM1-POPC-protein systems. Snapshots of the simulation systems of (A) WALP23 and (B) AQP1 are shown. WALP23 is shown in cyan, and subunits 1–4 of AQP1 are shown in red, light blue, orange, and light green, respectively. POPC lipids and GM1 lipids are shown in gray and red, respectively. Additionally, in panel B, the two GM1 lipids, which are stably bound to AQP1 during the last 1 μs simulation, are in blue and the 7 GM1 lipids, which have interactions with AQP1 or the 2 stably bound GM1 lipids, are shown in magenta. Atomistic details of the interactions between GM1 and (C) WALP23 and (D) subunit 1 of AQP1. (E) Side view and (F) top view of the binding site of GM1 on subunit 1 of AQP1. Panels C–F are based on the last snapshots of the simulations. GM1 and protein residues are shown in yellow and cyan, and the backbones of WALP23 and AQP1 are shown in cyan and white, respectively.
2.2. Interactions between AQP1 and GM1
Gangliosides interact with AQP1 directly and indirectly by forming clusters with other gangliosides that contact AQP1 (see Figure 6B). Our CG simulations suggest that PW does not affect the strength of direct GM1-AQP1 interactions, as indicated by the high GM1 densities with comparable amplitudes at similar positions around the AQP1 in the CG W and PW simulations (Figure 5A). However, in the CG PW simulations, the regions with high relative GM1 densities (regions in blue and white) are smaller compared with those in the CG W simulations, which means the indirect GM1-AQP1 interactions are decreased because the GM1 clusters become smaller and less stable.
The same picture emerges from the RDFs in Figure 5C. The GM1-AQP1 RDFs in the CG simulations show at least two peaks. The first peak at ∼3.4 nm is comparable for all three CG simulations and corresponds to direct GM1-AQP1 interactions. However, the second peak at ∼3.8 nm is decreased significantly in CG PW simulations and corresponds to the GM1 lipids associated with the immediate GM1 shell around AQP1. The RDFs of GM1 headgroups in the CG simulations show the same trend (Figure 5D). The protein-GM1 RDF in the atomistic simulation (Figure 5C) shows two peaks at approximately 3.4 and 4.2 nm, respectively, but the amplitudes of these two peaks are much smaller than those in the CG simulations, indicating weaker AQP1-GM1 interactions at the atomistic level. The positions of the peaks of the GM1 tail RDFs are shifted to the right compared with the headgroups in all of the CG and atomistic simulations (Figure 5D).
Free energy calculations suggest little effects of PW on GM3-AQP1 direct interactions (the binding energies are −19, −24, and −19 kJ/mol in the W, PW, and PW PME CG simulations, respectively (Figure 5B)), consistent with the equilibrium simulations. These calculations do not give estimation of the real GM3-AQP1 binding free energy, as GM3 was restrained to the specific orientation of AQP1 (Figure S1). However, they provide reliable estimation of the effects of polarizable Martini water.
Despite quantitative and qualitative differences, the CG and atomistic simulations show similar overall binding sites for GM1 on the AQP1 surface, as indicated by the positions of high relative GM1 densities in the 2D density map plots (Figure 5A). Examples of the GM1-AQP1 interactions in the atomistic simulation are shown in Figure 6B. There are two GM1 lipids stably bound to AQP1 for the last 1 μs of the simulation, each forming ∼3 hydrogen bonds with AQP1 on average (blue lipids in Figure 6B). There are other GM1 lipids (magenta lipids in Figure 6B) interacting with AQP1 indirectly by forming clusters with the stably bound lipids. These lipids may also have transient direct contacts with AQP1 during some of the simulation time. Most interactions between GM1 and AQP1 occur at a specific site, as shown by the density maps in Figure 5A. The binding site of GM1 lipids on subunit 1 of AQP1 in the atomistic simulation (Figure 6D–F) is located in the vicinity of the M4 and M7 helices and the M4-M5 loop. Seven residues (Thr205, Asn207, Ser115, Leu121, Asp123, Asn124, and Ser124; Figure 6D) are involved in hydrogen bonds between GM1 and AQP1, each accounting for ∼10% of the total hydrogen bond numbers. Most of these residues have polar side chains, which can act as hydrogen bond donors and/or acceptors.
3. Modifications to the Martini Ganglioside Force Field
As shown in section 1, ganglioside-ganglioside interactions are more prevalent in the CG W simulation than in the atomistic simulation. Therefore, we modified the Martini ganglioside force field to reduce their self-interactions. The aim of the reparameterization was to better reproduce the sizes and structures of ganglioside clusters observed in the atomistic model. Specifically, some of the headgroup bead types were reassigned to reduce the attraction between gangliosides (see Figure 7 for the CG topology of gangliosides, and the mapping of atoms to CG beads). We also made minor modifications to the parameters used for bonded interactions in the headgroups and the linkage parts to improve the internal structure of the GM headgroup.
Figure 7.

Modifications to the Martini ganglioside force field. (A) The topology of GM1 and GM3 in the optimized force field. The names, types of CG beads, and the names of the sugars are labeled. (B) Mapping of atoms to CG beads for the GM1 headgroup. (C) The RDF profiles between the GM lipids, headgroups and tails, as well as the RDFs between GM lipids and POPC in the GM-POPC binary mixture simulations using the optimized force field.
3.1. Reoptimization of Bonded Interactions
The atomistic trajectories were converted to the CG level. The bond, angle, and dihedral distributions of the resulting GM lipid pseudo-CG trajectories were analyzed and taken as references to optimize the bonded interactions of CG GM lipids (Figures S5 and S6). The original and new parameters for the bonds, angles, and dihedrals are compared in Table S3.
Equilibrium values of the bonds between the AM1-AM2 and AM1-T1A beads were changed to be the same as the corresponding sphingomyelin parameters in the Martini force field (Figure 7 and Table S3) based on their distributions in the pseudo-CG trajectories. Constraints were applied to the bonds between the GM1-GM2 and GM1-GM3 beads of the Glc sugar in the optimized force field. More minor changes were introduced in some other bonds in the headgroups (Table S3).
Angles and dihedrals were modified to improve the orientation of the headgroups and the relative orientation of the sugars, as described in Table S3. The optimization improves the distributions of these angles and dihedrals but does not fit them perfectly to the atomistic simulations (Figures S5 and S6).
3.2. Reassignment of Ganglioside Headgroup Bead Types
For GM1, less “sticky” beads were assigned to Neu5Ac. Specifically, beads GM14 and GM15 were changed from type P1 to SP1, and bead GM17 from type P5 to P4. Bead GM6 of sugar Gal1 and GM7 of sugar GalNAc were changed from types Nda and P5 to SNda and P4, respectively. Sugars Glc and Gal2 were kept unchanged. The optimization of the GM1 lipid is based on the fact that hydrogen bond interactions mainly involve Gal2 and Neu5Ac. In the new GM1 force field, Gal2 and Neu5Ac are the most sticky sugars, whereas GalNAc is less sticky than Gal2 and Neu5Ac. Glc and Gal1 are not very sticky (Figure 7). The same changes were made to Neu5Ac in GM3; however, beads GM2 and GM5 were changed to type P1 to mimic the tightly and parallel packed ganglioside headgroups in GM3 clusters (Figure 7).
The ganglioside-POPC binary mixtures in section 1 were simulated again using these new Martini ganglioside parameters (newFF). The average cluster sizes of GM1 and GM3 in the CG W simulations were now 3 and 4, respectively, whereas using PW reduced the cluster sizes of GM1 and GM3 to 2 and 3, respectively (Table 2). The RDFs of ganglioside lipids, headgroups, and tails in Figure 7 also became closer to the atomistic results. The GM1-GM1 RDF is shifted to the right of the GM3-GM3 RDF in the simulations with the newFF. The RDFs of each sugar of the headgroups and the RDFs of specific CG beads in the CG simulations using the original and newFF are shown in Figures S7 and S8. The optimized parameters result in a significant reduction of ganglioside interactions.
The bilayer properties (Table S1) and lipid density profiles (Figure S9) of the newFF CG simulations were also calculated. The bilayer thicknesses are comparable with the atomistic simulations; however, the area per lipid (APL) still differs with the values in the atomistic simulations (Table S1). The diffusion coefficients of the GM lipids are larger than the corresponding values in the simulations with original Martini ganglioside force field due to weaker ganglioside clustering. However, the CG lipid diffusion coefficients are much larger than the atomistic values because of smoother interaction energy at the CG level. The chain order parameters of GM lipids are smaller than those of the atomistic simulations, suggesting the necessity of further optimizing the Martini ceramide building blocks (Table S1; see the Supporting Information for a detailed discussion). The density profiles of lipids are improved in the newFF CG simulations, particular for the density profiles of Gal sugar, which are shifted toward the solution (Figures S2 and S9). Although the new parameters did not exactly reproduce the bilayer properties at the atomistic level, they improved the ganglioside headgroup conformations and the strengths of clustering.
Discussion
Insights into Ganglioside-Ganglioside and Protein-Ganglioside Interactions at the Atomistic Level
Atomistic simulations revealed only weak ganglioside-ganglioside and protein-ganglioside interactions. GM1 and GM3 aggregate by two different strategies. GM3 clusters are more ordered and slightly larger than the GM1 clusters, and the GM3 lipids packed very tightly and almost parallel to each other (Figure 3). Sugars at the end of the headgroups (Neu5Ac and Gal2) contribute most to the hydrogen bond interactions, and the ceramide also makes contributions in the case of GM3 (Figure 3 and Table 3). This difference might be explained by the shapes and sizes of their headgroups. The two branches (Neu5Ac and GalNAc-Gal2, Figure 1) of the GM1 headgroup extend in different directions and contact each other, thereby preventing other parts of GM1 lipids (Glc, Gal1, and the ceramide tails) from approaching each other. The GM3 headgroups adjust their conformations to planar structures, resulting in more tightly packed parallel clusters. Simulations using the CHARMM force field also revealed weak clustering of GM1 (personal communication with Dr. Jeffery Klauda and Dr. Wonpil Im). For protein-ganglioside interactions, although GM1 lipids are depleted from WALP23, the headgroup tilted and interacted with the N-terminus of WALP23 during some of the simulation time (Figure 6A and C), whereas extensive hydrogen bonding was observed between GM1 and AQP1 (Figure 6B and D).
Saccharide and protein-saccharide interactions are crucial for molecular/cell recognization;81 however, investigating these interactions is challenging for both experiments82 and computations83 not only because of the diversity of the glycan structures84 but also because these interactions in solution are weak.81,83 Although atomistic simulations are the most detailed approach possible at the moment, sampling, in particular inside the bilayer environment, is a significant limitation. In addition, anomeric and exoanomeric effects85 and CH-π interactions86 are difficult to model and may affect the reliability of the atomistic simulations of saccharides.
Effects of Polarizable Martini Water (PW) on Ganglioside-Ganglioside and Protein-Ganglioside Interactions at the CG Level
In the CG simulations, both ganglioside (Figures 2 and 3 and Table 2) and protein-ganglioside interactions (Figure 5) are stronger than in the atomistic simulations. The ganglioside clusters of GM1 and GM3 did not show apparent differences at the CG level.
Previous simulations indicate that the partitioning free energies of charged amino acid residues at the POPC/water interface87 and the association energies of charged amino acid pairs88 are improved significantly by PW. Phase transition of bilayers containing GM1 also appear to be modeled better by PW.56 We found that PW reduced ganglioside and ganglioside-WALP23 interactions in equilibrium simulations and quantified the effects of PW and PW PME by PMF calculations (Figure 2C and Figure 5B). We included PW PME simulations, although this is not standard practice with Martini, to compare our results to those of Kociurzynski et al.56 In simulations of GM1, GM3, and WALP23-GM1, the PW roughly reduces the binding free energies by half. The PW increased GM1-AQP1 binding free energies slightly. However, PME did not significantly affect ganglioside and protein-ganglioside interactions. The changes of GM1-GM1 and GM3-GM3 PMFs when using PW are different, although these lipids have the same charge.
In the protein-ganglioside simulations, PW does not affect the direct GM1-AQP1 interactions significantly (Figure 5). This might be explained by the positions of the charged residues of AQP1. For each subunit of AQP1, there are only 2–3 charged residues located on the surface of the protein, whereas the others are buried in the protein and do not interact with the GM1 headgroups (see Figure S10). Thus, the interactions between GM1 and AQP1 are dominated by LJ interactions between the CG beads, and the changes in electrostatic interactions induced by the PW have a minor effect.
Ganglioside Molecular Dynamics
Because atomistic ganglioside simulations at 310 K resulted in the gangliosides entering a gel phase, we performed our simulations at 330 K to maintain the bilayer at the liquid disordered phase; this has the added benefit that lipid diffusion and simulation convergence are faster. A CG W simulation of the GM3-POPC mixture at 330 K revealed similar GM3 cluster sizes and numbers with the simulation at 310 K (Figure S11A compared to Figures 2B and 3A and B), indicating that the differences of ganglioside clustering at the CG and AA levels are not the result of different temperatures.
In vivo, gangliosides have high phase transition temperatures because of their saturated tails and are probably segregated from lipids with lower phase transition temperatures24 to form highly ordered lipid microdomains such as lipid rafts with sphingomyelin and cholesterol. The temperature we used in our atomistic simulation prevents phase separation and reduces the effects of different phase transition temperatures of POPC and gangliosides on ganglioside segregation; therefore, interactions between the headgroups play the most important role in ganglioside aggregation in our simulations. The ganglioside clustering is probably decreased compared to the situation in vivo due to the high temperature we used. To evaluate the effects of both headgroup interactions and phase separation on ganglioside aggregation, additional simulations in different lipid environments and at lower temperatures using enhanced sampling techniques might be useful, although in atomistic simulations sampling remains a serious challenge.
One may expect a strain between leaflets as we only included GM lipids in one leaflet and the APLs of GM1, GM3, and POPC are not identical. However, in the CG simulations (using the original Martini ganglioside force field) of pure POPC, symmetrical GM1-POPC, and GM3-POPC bilayers, the averaged APLs are 64.2 ± 0.8, 64.7 ± 0.8, and 64.1 ± 0.8 Å2, respectively, suggesting only minor differences between the APLs of the GM lipids and POPC. Therefore, we ignore the effects of bilayer asymmetry on ganglioside clustering and bilayer properties.
In the CG simulations, the gangliosides tails we used have 3–4 CG tail beads (and one linker bead) each and correspond to GM (d18:1/18:0) lipids at the atomistic level according to the four to one mapping Martini philosophy. However, the GM lipids with longer tails (d20:1/d20:0) were used in our atomistic simulations. To address the inconsistency of the ganglioside tails at the CG and atomistic levels, we conducted control CG simulations using GM lipids with longer tails (AM1-AM2-T1A-C2A-C3A-C4A-C1B-C2B-C3B-C4B-C5B) at 330 K. We found that the lengths of the tails did not influence ganglioside clustering at the CG level in both simulations using the original and modified Martini ganglioside force fields (Figures S11B and C compared to Figures 2B and 3A and B).
Modifications to the Martini Ganglioside Force Field
We used the extent of ganglioside aggregation and the internal structures of the gangliosides as reference data to modify the Martini ganglioside force field. The detailed atomistic structures of ganglioside clusters were also considered in the reoptimization, but we do not aim to reproduce the different structures of GM1 and GM3 clusters exactly at the CG level. Ganglioside clustering is reduced significantly by the modified force field. This is achieved by assigning new types for the CG beads used for the ganglioside headgroups, suggesting that attraction of the headgroups is an important driving force for ganglioside clustering at the CG level. The parameters of the amide linkage at the CG level are changed to be the same as those used for Martini sphingomyelin for consistency.
We would like to note that different types of CG beads are used for the same building blocks of GM1 and GM3 lipids. Specifically, in GM3, a P1 type is used for bead GM2 in the Glc sugar and GM5 in the Gal1 sugar, but the corresponding types are SP1 in GM1. This is not consistent with the Martini building block philosophy but is needed in this case. Because the two branches of the GM1 headgroup point in different directions (Neu5Ac and GalNAc-Gal2; see Figure 1), Glc and Gal1 may not be able to approach each other closely, and the interactions between them are shielded. However, in GM3, all three sugars packed tightly in the clusters. The interactions involving Glc and Gal1 are much stronger than in GM1. Therefore, we used different types of beads for Glc and Gal1 to mimic the cluster sizes and structures in the atomistic simulations. Slightly different dihedrals are used for GM1 and GM3 due to the different orientations of the sugars of their headgroups (Table S3).
It has been reported that membrane incorporation restricts the motion of ganglioside headgroups.89 The Glc sugar was buried in the interfacial region of the phosphatidylcholine bilayers, whereas the other sugars were extended fully into solution.40,90 The density profiles of the sugars in our atomistic and CG newFF simulations reproduced these experimental results (Figures S2 and S9). However, the GM clustering behavior in our simulations should ultimately be subjected to more rigorous testing, including experimental data that is currently lacking. The new GM parameters are available on the Martini Web site (cgmartini.nl) and in the CHARMM-GUI Martini maker.91
Conclusions
In atomistic simulations, the sugars at the end of the headgroups (Neu5Ac and Gal2) contribute most of the hydrogen bonds between GM headgroups. GM1 and GM3 pack differently in their respective clusters due to the different headgroup geometry with GM3 lipids packing more tightly, leading to GM3 clusters being more stable and a little larger than GM1 clusters. In atomistic simulations, GM1 binds stably to specific positions of AQP1 by hydrogen bonding between polar residues of the protein and the GM1 headgroups. The GM1 lipids are depleted from WALP23.
The ganglioside and protein-ganglioside interactions in the CG Martini simulations are more prevalent than in the atomistic simulations. Using the polarizable Martini water decreases ganglioside and protein-ganglioside interactions in most of the conditions. We proposed an optimized Martini force field for gangliosides that reproduces the strengths of ganglioside aggregations at the atomistic level.
Acknowledgments
This work is supported by the Natural Sciences and Engineering Research Council (Canada). D.P.T. is an Alberta Innovates Health Solutions (AIHS) Scientist and Alberta Innovates Technology Futures Strategic Chair in (Bio)Molecular Simulation. S.J.M. is supported by an ERC Advanced grant. R.X.G. is supported by fellowships from AIHS and the Canadian Institutes for Health Research (funding reference number: MFE-140949). Calculations were carried out on Compute Canada facilities. We thank Jeff Klauda, Wonpil Im, and Soohyung Park for sharing data on the clustering behavior of GM lipids with the CHARMM force field. The authors would like to dedicate this paper to Klaus Schulten on the occasion of his 70th birthday and in recognition of his seminal contributions to, among many other topics, membrane biophysics.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.6b07142.
Comparison of bilayer properties, modifications to the nonbonded parameters of the Martini ganglioside force field, data of bilayer properties, averaged lipid contact numbers in the GM1-GM3-POPC ternary mixture simulation, comparison of original and modified parameters of Martini ganglioside force field, orientation restraint of GM3 around AQP1 for PMF calculations, density profiles of lipids along bilayer normal in the AA and the CG simulations with original Martini ganglioside force field, convergence of RDF profiles, lipid contact numbers as a function of simulation time in the GM1-GM3-POPC ternary mixture simulations, distributions of angles and dihedrals of the ganglioside headgroups, RDFs between each sugar and specific beads of ganglioside headgroups, density profiles of lipids along bilayer normal in the CG simulations with modified Martini ganglioside force field, positions of AQP1 charged residues, GM3 clustering at 330 K, atomistic topology files of GM1 and GM3, and modified CG topology files of GM1 and GM3 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bush C. A.; Martin-Pastor M.; Imbery A. Structure and Conformation of Complex Carbohydrates of Glycoproteins, Glycolipids, and Bacterial Polysaccharides. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 269–293. 10.1146/annurev.biophys.28.1.269. [DOI] [PubMed] [Google Scholar]
- Carter H.; Johnson P.; Weber E. Glycolipids. Annu. Rev. Biochem. 1965, 34, 109–142. 10.1146/annurev.bi.34.070165.000545. [DOI] [PubMed] [Google Scholar]
- Malhotra R. Membrane Glycolipids: Functional Heterogeneity: A Review. Biochem. Anal. Biochem. 2012, 1, 108. 10.4172/2161-1009.1000108. [DOI] [Google Scholar]
- Shaw N. Bacterial Glycolipids. Bacteriol Rev. 1970, 34, 365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeley C. C.; Siddiqui B.. Chemistry of Mammalian Glycolipids. In The Glycoconjugates; Horowitz M. I., Pigman W., Eds.; Academic Press: New York, 1977. [Google Scholar]
- Kolter T. Ganglioside Biochemistry. ISRN Biochem. 2012, 2012, 506160. 10.5402/2012/506160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledeen R. W.; Wu G. The Multi-Tasked Life of GM1 Ganglioside, a True Factotum of Nature. Trends Biochem. Sci. 2015, 40, 407–418. 10.1016/j.tibs.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Schengrund C.-L. Gangliosides: Glycosphingolipids Essential for Normal Neural Development and Function. Trends Biochem. Sci. 2015, 40, 397–406. 10.1016/j.tibs.2015.03.007. [DOI] [PubMed] [Google Scholar]
- Hakomori S.-i. Cell Adhesion/Recognition and Signal Transduction through Glycosphingolipid Microdomain. Glycoconjugate J. 2000, 17, 143–151. 10.1023/A:1026524820177. [DOI] [PubMed] [Google Scholar]
- Gout E.; Garlatti V.; Smith D. F.; Lacroix M.; Dumestre-Pérard C.; Lunardi T.; Martin L.; Cesbron J.-Y.; Arlaud G. J.; Gaboriaud C. Carbohydrate Recognition Properties of Human Ficolins Glycan Array Screening Reveals the Sialic Acid Binding Specificity of M-Ficolin. J. Biol. Chem. 2010, 285, 6612–6622. 10.1074/jbc.M109.065854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo-Useros N.; Lorizate M.; Puertas M. C.; Rodriguez-Plata M. T.; Zangger N.; Erikson E.; Pino M.; Erkizia I.; Glass B.; Clotet B. Siglec-1 is a Novel Dendritic Cell Receptor that Mediates Hiv-1 Trans-Infection through Recognition of Viral Membrane Gangliosides. PLoS Biol. 2012, 10, e1001448. 10.1371/journal.pbio.1001448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremer E.; Hakomori S.-I.; Bowen-Pope D.; Raines E.; Ross R. Ganglioside-Mediated Modulation of Cell Growth, Growth Factor Binding, and Receptor Phosphorylation. J. Biol. Chem. 1984, 259, 6818–6825. [PubMed] [Google Scholar]
- Schnaar R. L.; Gerardy-Schahn R.; Hildebrandt H. Sialic Acids in the Brain: Gangliosides and Polysialic Acid in Nervous System Development, Stability, Disease, and Regeneration. Physiol. Rev. 2014, 94, 461–518. 10.1152/physrev.00033.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aureli M.; Mauri L.; Ciampa M. G.; Prinetti A.; Toffano G.; Secchieri C.; Sonnino S. GM1 Ganglioside: Past Studies and Future Potential. Mol. Neurobiol. 2016, 53, 1824–1842. 10.1007/s12035-015-9136-z. [DOI] [PubMed] [Google Scholar]
- Hirano-Sakamaki W.; Sugiyama E.; Hayasaka T.; Ravid R.; Setou M.; Taki T. Alzheimer’s Disease is Associated with Disordered Localization of Ganglioside GM1Molecular Species in the Human Dentate Gyrus. FEBS Lett. 2015, 589, 3611–3616. 10.1016/j.febslet.2015.09.033. [DOI] [PubMed] [Google Scholar]
- Yanagisawa K. GM1 Ganglioside and Alzheimer’s Disease. Glycoconjugate J. 2015, 32, 87–91. 10.1007/s10719-015-9579-5. [DOI] [PubMed] [Google Scholar]
- Hakomori S.-i. Glycosynaptic Microdomains Controlling Tumor Cell Phenotype through Alteration of Cell Growth, Adhesion, and Motility. FEBS Lett. 2010, 584, 1901–1906. 10.1016/j.febslet.2009.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inokuchi J.-i. Membrane Microdomains and Insulin Resistance. FEBS Lett. 2010, 584, 1864–1871. 10.1016/j.febslet.2009.10.012. [DOI] [PubMed] [Google Scholar]
- Inokuchi J.-i. Physiopathological Function of Hematoside (GM3 Ganglioside). Proc. Jpn. Acad., Ser. B 2011, 87, 179–198. 10.2183/pjab.87.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inokuchi J.-i. GM3 and Diabetes. Glycoconjugate J. 2014, 31, 193–197. 10.1007/s10719-013-9516-4. [DOI] [PubMed] [Google Scholar]
- Lingwood D.; Simons K. Lipid Rafts as a Membrane-Organizing Principle. Science 2010, 327, 46–50. 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- Hakomori S.-i. The Glycosynapse. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 225–232. 10.1073/pnas.012540899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todeschini A. R.; Hakomori S.-i. Functional Role of Glycosphingolipids and Gangliosides in Control of Cell Adhesion, Motility, and Growth, through Glycosynaptic Microdomains. Biochim. Biophys. Acta, Gen. Subj. 2008, 1780, 421–433. 10.1016/j.bbagen.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerlund B.; Slotte J. P. How the Molecular Features of Glycosphingolipids Affect Domain Formation in Fluid Membranes. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 194–201. 10.1016/j.bbamem.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Lingwood D.; Ries J.; Schwille P.; Simons K. Plasma Membranes are Poised for Activation of Raft Phase Coalescence at Physiological Temperature. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 10005–10010. 10.1073/pnas.0804374105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantù L.; Del Favero E.; Sonnino S.; Prinetti A. Gangliosides and the Multiscale Modulation of Membrane Structure. Chem. Phys. Lipids 2011, 164, 796–810. 10.1016/j.chemphyslip.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Cantù L.; Corti M.; Brocca P.; Del Favero E. Structural Aspects of Ganglioside-Containing Membranes. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 202–208. 10.1016/j.bbamem.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Cantù L.; Del Favero E.; Brocca P.; Corti M. Multilevel Structuring of Ganglioside-Containing Aggregates: From Simple Micelles to Complex Biomimetic Membranes. Adv. Colloid Interface Sci. 2014, 205, 177–186. 10.1016/j.cis.2013.10.016. [DOI] [PubMed] [Google Scholar]
- Prinetti A.; Loberto N.; Chigorno V.; Sonnino S. Glycosphingolipid Behaviour in Complex Membranes. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 184–193. 10.1016/j.bbamem.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Yoon S.-J.; Nakayama K.-i.; Hikita T.; Handa K.; Hakomori S. -i. Epidermal Growth Factor Receptor Tyrosine Kinase is Modulated by GM3 Interaction with N-Linked GlcNAc Termini of the Receptor. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 18987–18991. 10.1073/pnas.0609281103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabayama K.; Sato T.; Saito K.; Loberto N.; Prinetti A.; Sonnino S.; Kinjo M.; Igarashi Y.; Inokuchi J.-i. Dissociation of the Insulin Receptor and Caveolin-1 Complex by Ganglioside GM3 in the State of Insulin Resistance. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 13678–13683. 10.1073/pnas.0703650104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S.; Mayor S. The GPI-Anchor and Protein Sorting. Cell. Mol. Life Sci. 2001, 58, 1969–1987. 10.1007/PL00000831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita A.; Cheng J.; Fujimoto T. Segregation of GM1 and GM3 Clusters in the Cell Membrane Depends on the Intact Actin Cytoskeleton. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2009, 1791, 388–396. 10.1016/j.bbalip.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Fujita A.; Cheng J.; Hirakawa M.; Furukawa K.; Kusunoki S.; Fujimoto T. Gangliosides GM1 and GM3 in the Living Cell Membrane Form Clusters Susceptible to Cholesterol Depletion and Chilling. Mol. Biol. Cell 2007, 18, 2112–2122. 10.1091/mbc.E07-01-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta G.; Surolia A. Glycosphingolipids in Microdomain Formation and Their Spatial Organization. FEBS Lett. 2010, 584, 1634–1641. 10.1016/j.febslet.2009.11.070. [DOI] [PubMed] [Google Scholar]
- Ingólfsson H. I.; Arnarez C.; Periole X.; Marrink S. J. Computational ‘Microscopy’of Cellular Membranes. J. Cell Sci. 2016, 129, 257–268. 10.1242/jcs.176040. [DOI] [PubMed] [Google Scholar]
- Khakbaz P.; Klauda J. B. Probing the Importance of Lipid Diversity in Cell Membranes Via Molecular Simulation. Chem. Phys. Lipids 2015, 192, 12–22. 10.1016/j.chemphyslip.2015.08.003. [DOI] [PubMed] [Google Scholar]
- Lorent J. H.; Levental I. Structural Determinants of Protein Partitioning into Ordered Membrane Domains and Lipid Rafts. Chem. Phys. Lipids 2015, 192, 23–32. 10.1016/j.chemphyslip.2015.07.022. [DOI] [PubMed] [Google Scholar]
- Manna M.; Róg T.; Vattulainen I. The Challenges of Understanding Glycolipid Functions: An Open Outlook Based on Molecular Simulations. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2014, 1841, 1130–1145. 10.1016/j.bbalip.2013.12.016. [DOI] [PubMed] [Google Scholar]
- Hall A.; Róg T.; Karttunen M.; Vattulainen I. Role of Glycolipids in Lipid Rafts: A View through Atomistic Molecular Dynamics Simulations with Galactosylceramide. J. Phys. Chem. B 2010, 114, 7797–7807. 10.1021/jp912175d. [DOI] [PubMed] [Google Scholar]
- Hall A.; Róg T.; Vattulainen I. Effect of Galactosylceramide on the Dynamics of Cholesterol-Rich Lipid Membranes. J. Phys. Chem. B 2011, 115, 14424–14434. 10.1021/jp203234n. [DOI] [PubMed] [Google Scholar]
- Sega M.; Vallauri R.; Brocca P.; Melchionna S. Molecular Dynamics Simulation of a GM3 Ganglioside Bilayer. J. Phys. Chem. B 2004, 108, 20322–20330. 10.1021/jp046898m. [DOI] [Google Scholar]
- Patel R. Y.; Balaji P. V. Characterization of the Conformational and Orientational Dynamics of Ganglioside GM1 in a Dipalmitoylphosphatidylcholine Bilayer by Molecular Dynamics Simulations. Biochim. Biophys. Acta, Biomembr. 2007, 1768, 1628–1640. 10.1016/j.bbamem.2007.02.020. [DOI] [PubMed] [Google Scholar]
- Patel R. Y.; Balaji P. V. Characterization of Symmetric and Asymmetric Lipid Bilayers Composed of Varying Concentrations of Ganglioside GM1 and DPPC. J. Phys. Chem. B 2008, 112, 3346–3356. 10.1021/jp075975l. [DOI] [PubMed] [Google Scholar]
- Jedlovszky P.; Sega M.; Vallauri R. GM1 Ganglioside Embedded in a Hydrated DOPC Membrane: A Molecular Dynamics Simulation Study. J. Phys. Chem. B 2009, 113, 4876–4886. 10.1021/jp808199p. [DOI] [PubMed] [Google Scholar]
- Mori K.; Mahmood M. I.; Neya S.; Matsuzaki K.; Hoshino T. Formation of GM1 Ganglioside Clusters on the Lipid Membrane Containing Sphingomyeline and Cholesterol. J. Phys. Chem. B 2012, 116, 5111–5121. 10.1021/jp207881k. [DOI] [PubMed] [Google Scholar]
- López C. A.; Rzepiela A. J.; De Vries A. H.; Dijkhuizen L.; Hünenberger P. H.; Marrink S. J. Martini Coarse-Grained Force Field: Extension to Carbohydrates. J. Chem. Theory Comput. 2009, 5, 3195–3210. 10.1021/ct900313w. [DOI] [PubMed] [Google Scholar]
- López C. s. A.; Sovova Z.; van Eerden F. J.; de Vries A. H.; Marrink S. J. Martini Force Field Parameters for Glycolipids. J. Chem. Theory Comput. 2013, 9, 1694–1708. 10.1021/ct3009655. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; Risselada H. J.; Yefimov S.; Tieleman D. P.; De Vries A. H. The Martini Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- Ingólfsson H. I.; Melo M. N.; van Eerden F. J.; Arnarez C. m.; Lopez C. A.; Wassenaar T. A.; Periole X.; de Vries A. H.; Tieleman D. P.; Marrink S. J. Lipid Organization of the Plasma Membrane. J. Am. Chem. Soc. 2014, 136, 14554–14559. 10.1021/ja507832e. [DOI] [PubMed] [Google Scholar]
- Koldsø H.; Shorthouse D.; Hélie J.; Sansom M. S. Lipid Clustering Correlates with Membrane Curvature as Revealed by Molecular Simulations of Complex Lipid Bilayers. PLoS Comput. Biol. 2014, 10, e1003911. 10.1371/journal.pcbi.1003911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorthouse D.; Hedger G.; Koldsø H.; Sansom M. S. Molecular Simulations of Glycolipids: Towards Mammalian Cell Membrane Models. Biochimie 2016, 120, 105–109. 10.1016/j.biochi.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanna X.; Jafurulla M.; Sengupta D.; Chattopadhyay A. The Ganglioside GM1 Interacts with the Serotonin 1A Receptor via the Sphingolipid Binding Domain. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2818–2826. 10.1016/j.bbamem.2016.08.009. [DOI] [PubMed] [Google Scholar]
- de Jong D. H.; Lopez C. A.; Marrink S. J. Molecular View on Protein Sorting into Liquid-Ordered Membrane Domains Mediated by Gangliosides and Lipid Anchors. Faraday Discuss. 2013, 161, 347–363. 10.1039/C2FD20086D. [DOI] [PubMed] [Google Scholar]
- Basu I.; Mukhopadhyay C. In Silico Phase Separation in the Presence of GM1 in Ternary and Quaternary Lipid Bilayers. Phys. Chem. Chem. Phys. 2015, 17, 17130–17139. 10.1039/C5CP01970B. [DOI] [PubMed] [Google Scholar]
- Kociurzynski R.; Pannuzzo M.; Böckmann R. A. Phase Transition of Glycolipid Membranes Studied by Coarse-Grained Simulations. Langmuir 2015, 31, 9379–9387. 10.1021/acs.langmuir.5b01617. [DOI] [PubMed] [Google Scholar]
- Wassenaar T. A.; Ingólfsson H. I.; Böckmann R. A.; Tieleman D. P.; Marrink S. J. Computational Lipidomics with Insane: A Versatile Tool for Generating Custom Membranes for Molecular Simulations. J. Chem. Theory Comput. 2015, 11, 2144–2155. 10.1021/acs.jctc.5b00209. [DOI] [PubMed] [Google Scholar]
- Sui H.; Han B.-G.; Lee J. K.; Walian P.; Jap B. K. Structural Basis of Water-Specific Transport through the AQP1 Water Channel. Nature 2001, 414, 872–878. 10.1038/414872a. [DOI] [PubMed] [Google Scholar]
- Wassenaar T. A.; Pluhackova K.; Böckmann R. A.; Marrink S. J.; Tieleman D. P. Going Backward: A Flexible Geometric Approach to Reverse Transformation from Coarse Grained to Atomistic Models. J. Chem. Theory Comput. 2014, 10, 676–690. 10.1021/ct400617g. [DOI] [PubMed] [Google Scholar]
- Pronk S.; Páll S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; van der Spoel D.; et al. GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; Van Der Spoel D.; Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Parrinello M.; Rahman A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. 10.1063/1.328693. [DOI] [Google Scholar]
- Nosé S. A Molecular Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 1984, 52, 255–268. 10.1080/00268978400101201. [DOI] [Google Scholar]
- Marrink S. J.; De Vries A. H.; Mark A. E. Coarse Grained Model for Semiquantitative Lipid Simulations. J. Phys. Chem. B 2004, 108, 750–760. 10.1021/jp036508g. [DOI] [Google Scholar]
- Yesylevskyy S. O.; Schäfer L. V.; Sengupta D.; Marrink S. J. Polarizable Water Model for the Coarse-Grained Martini Force Field. PLoS Comput. Biol. 2010, 6, e1000810. 10.1371/journal.pcbi.1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle Mesh Ewald: An N Log (N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Periole X.; Cavalli M.; Marrink S.-J.; Ceruso M. A. Combining an Elastic Network with a Coarse-Grained Molecular Force Field: Structure, Dynamics, and Intermolecular Recognition. J. Chem. Theory Comput. 2009, 5, 2531–2543. 10.1021/ct9002114. [DOI] [PubMed] [Google Scholar]
- Oostenbrink C.; Villa A.; Mark A. E.; Van Gunsteren W. F. A Biomolecular Force Field Based on the Free Enthalpy of Hydration and Solvation: The GROMOS Force-Field Parameter Sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- Pol-Fachin L.; Rusu V. H.; Verli H.; Lins R. D. GROMOS 53A6GLYC, an Improved GROMOS Force Field for Hexopyranose-Based Carbohydrates. J. Chem. Theory Comput. 2012, 8, 4681–4690. 10.1021/ct300479h. [DOI] [PubMed] [Google Scholar]
- Lins R. D.; Hünenberger P. H. A New GROMOS Force Field for Hexopyranose-Based Carbohydrates. J. Comput. Chem. 2005, 26, 1400–1412. 10.1002/jcc.20275. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J.; Postma J. v.; van Gunsteren W. F.; DiNola A.; Haak J. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- van Gunsteren W. F.; Berendsen H. J. Computer Simulation of Molecular Dynamics: Methodology, Applications, and Perspectives in Chemistry. Angew. Chem., Int. Ed. Engl. 1990, 29, 992–1023. 10.1002/anie.199009921. [DOI] [Google Scholar]
- Tironi I. G.; Sperb R.; Smith P. E.; van Gunsteren W. F. A Generalized Reaction Field Method for Molecular Dynamics Simulations. J. Chem. Phys. 1995, 102, 5451–5459. 10.1063/1.469273. [DOI] [Google Scholar]
- Torrie G. M.; Valleau J. P. Nonphysical Sampling Distributions in Monte Carlo Free-Energy Estimation: Umbrella Sampling. J. Comput. Phys. 1977, 23, 187–199. 10.1016/0021-9991(77)90121-8. [DOI] [Google Scholar]
- Kumar S.; Rosenberg J. M.; Bouzida D.; Swendsen R. H.; Kollman P. A. The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method. J. Comput. Chem. 1992, 13, 1011–1021. 10.1002/jcc.540130812. [DOI] [Google Scholar]
- Bonomi M.; Branduardi D.; Bussi G.; Camilloni C.; Provasi D.; Raiteri P.; Donadio D.; Marinelli F.; Pietrucci F.; Broglia R. A. Plumed: A Portable Plugin for Free-Energy Calculations with Molecular Dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. 10.1016/j.cpc.2009.05.011. [DOI] [Google Scholar]
- Barnoud J.; Rossi G.; Monticelli L. Lipid Membranes as Solvents for Carbon Nanoparticles. Phys. Rev. Lett. 2014, 112, 068102. 10.1103/PhysRevLett.112.068102. [DOI] [PubMed] [Google Scholar]
- Castillo N.; Monticelli L.; Barnoud J.; Tieleman D. P. Free Energy of WALP23 Dimer Association in DMPC, DPPC, and DOPC Bilayers. Chem. Phys. Lipids 2013, 169, 95–105. 10.1016/j.chemphyslip.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Cohen M. Notable Aspects of Glycan-Protein Interactions. Biomolecules 2015, 5, 2056–2072. 10.3390/biom5032056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings R. D.; Pierce J. M. The Challenge and Promise of Glycomics. Chem. Biol. 2014, 21, 1–15. 10.1016/j.chembiol.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez S.; Tvaroska I. Carbohydrate–Protein Interactions: Molecular Modeling Insights. Adv. Carbohydr. Chem. Biochem. 2014, 71, 12–136. 10.1016/B978-0-12-800128-8.00001-7. [DOI] [PubMed] [Google Scholar]
- Krasnova L.; Wong C.-H. Understanding the Chemistry and Biology of Glycosylation with Glycan Synthesis. Annu. Rev. Biochem. 2016, 85, 599–630. 10.1146/annurev-biochem-060614-034420. [DOI] [PubMed] [Google Scholar]
- Foley B. L.; Tessier M. B.; Woods R. J. Carbohydrate Force Fields. WIREs Comput. Mol. Sci. 2012, 2, 652–697. 10.1002/wcms.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson K. L.; Bartlett G. J.; Diehl R. C.; Agirre J.; Gallagher T.; Kiessling L. L.; Woolfson D. N. Carbohydrate–Aromatic Interactions in Proteins. J. Am. Chem. Soc. 2015, 137, 15152–15160. 10.1021/jacs.5b08424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G.; Tieleman D. P. Using the Wimley–White Hydrophobicity Scale as a Direct Quantitative Test of Force Fields: The Martini Coarse-Grained Model. J. Chem. Theory Comput. 2011, 7, 2316–2324. 10.1021/ct2002623. [DOI] [PubMed] [Google Scholar]
- de Jong D. H.; Periole X.; Marrink S. J. Dimerization of Amino Acid Side Chains: Lessons from the Comparison of Different Force Fields. J. Chem. Theory Comput. 2012, 8, 1003–1014. 10.1021/ct200599d. [DOI] [PubMed] [Google Scholar]
- Jarrell H.; Singh D.; Grant C. W. Oligosaccharide Order in a Membrane-Incorporated Complex Glycosphingolipid. Biochim. Biophys. Acta, Biomembr. 1992, 1103, 331–334. 10.1016/0005-2736(92)90105-U. [DOI] [PubMed] [Google Scholar]
- McIntosh T. J.; Simon S. A. Long-and Short-Range Interactions between Phospholipid/Ganglioside GM1 Bilayers. Biochemistry 1994, 33, 10477–10486. 10.1021/bi00200a032. [DOI] [PubMed] [Google Scholar]
- Qi Y.; Ingólfsson H. I.; Cheng X.; Lee J.; Marrink S. J.; Im W. CHARMM-GUI Martini Maker for Coarse-Grained Simulations with the Martini Force Field. J. Chem. Theory Comput. 2015, 11, 4486–4494. 10.1021/acs.jctc.5b00513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.