

Abstract
Cell division in Escherichia coli requires the recruitment of at least 10 essential proteins to the bacterial midcell. Recruitment of these proteins follows a largely linear dependency pathway in which depletion of one cell division protein leads to the absence from the division site of “downstream” proteins in the pathway. Analysis of events that underlie this pathway is complicated by the fact that a protein's ability to recruit “downstream” proteins is dependent on its own recruitment by “upstream” proteins. Hence, one cannot separate the individual contributions of various upstream proteins to any specific recruitment step. Here we present a method—premature targeting—for bypassing the normal localization requirements of a cell division protein and apply it to FtsQ, a protein recruited midway through the pathway. We fused FtsQ to the FtsZ-binding protein ZapA such that FtsQ was targeted to FtsZ rings independently of proteins FtsA and FtsK, which are normally required for FtsQ localization. Analysis of the resulting ZapA–FtsQ fusion suggests that FtsQ associates with a large complex of cell division proteins and that premature targeting of FtsQ can restore localization of this complex under conditions in which neither FtsQ nor the associated proteins would normally be localized.
Keywords: Cell division, divisome, FtsQ, ZapA, septal targeting
In Escherichia coli, cell division requires the coordinated constriction of the three layers of the Gram-negative cell envelope. Invagination is mediated by the septal ring, an assembly of proteins that localizes to the site of constriction and appears to remain at the site until separation of the two daughter cells. The septal ring is composed of multiple proteins, both essential and nonessential, which localize to the septum and/or are required for optimal cell division. The 10 known essential proteins are strictly required for cell division and localize to the constriction site in a defined hierarchical order (Fig. 1A; Buddelmeijer and Beckwith 2002). This hierarchy defines the septal recruitment “pathway,” in which a given protein requires all proteins upstream of it to localize to the division site, and is itself required for the localization of those that are farther downstream. The mode of localization to the septum within the pathway, however, is understood for only a few of the proteins, leaving open the questions of how the cell assembles the “divisome” into its final state and ultimately whether the order of assembly is critical to the cell division process.
Figure 1.

(A) The septal recruitment pathway for cell division proteins in E. coli. (B) Bacterial two-hybrid results summarized from several groups (Di Lallo et al. 2003; G. Karimova and D. Ladant, in prep.; M. Gonzalez and J. Beckwith, unpubl.). Cell division proteins are listed clockwise corresponding to their respective order in A. Lines indicate proposed interactions; circular arrows indicate proposed homodimerization. (C–E) Potential models for divisome assembly. (C) Linear assembly. (D) Cooperative binding by an upstream complex. (E) Sequential activities plus substrate recognition. (F) ZapA–FtsQ constructs used in this work. (Cyto) Cytoplasmic domain of FtsQ; (TM) transmembrane segment of FtsQ; (Peri) periplasmic domain of FtsQ. YFP and non-YFP constructs are identical except for the addition of YFP fused in frame to the N terminus of ZapA. (G) Proposed premature targeting of ZapA–FtsQ construct.
The assembly of the most upstream components of the septal ring is best understood. FtsZ is a widely conserved bacterial homolog of tubulin (Mukherjee et al. 1993; Lowe and Amos 1998) and was the first cell division protein shown to be localized to the bacterial midcell (Bi and Lutkenhaus 1991). FtsZ polymers form a ring or tight spiral at the bacterial midcell, which is thought to provide both a scaffold for assembly of subsequent septal ring components and possibly the force required for constriction (Romberg and Levin 2003). FtsA, a conserved member of the actin superfamily (Bork et al. 1992; van den Ent and Lowe 2000), and ZipA have both been shown to bind directly to the C terminus of FtsZ (Hale and de Boer 1997; Wang et al. 1997; Ma and Margolin 1999; Mosyak et al. 2000; Moy et al. 2000; Yan et al. 2000) and are thought to be required for stabilizing polymerized Z rings at the midcell (Pichoff and Lutkenhaus 2002). The molecular events that underlie recruitment among later proteins remain speculative.
Interestingly, the almost strict linearity of recruitment observed in E. coli does not appear to be conserved in the Gram-positive bacteria Bacillus subtilis and Streptococcus pneumoniae (Errington et al. 2003; Morlot et al. 2004). In B. subtilis, localization of late cell division proteins is interdependent, suggesting they are assembled and recruited in a cooperative fashion (Errington et al. 2003; Morlot et al. 2004). Recent information derived from bacterial two-hybrid approaches suggests that the pathway in E. coli may in fact be more complex than originally thought. Indeed, it is suggested that many of the cell division proteins interact with at least one other cell division protein, and that several proteins interact with multiple proteins within the septal ring assembly (Fig. 1B; Di Lallo et al. 2003; G. Karimova and D. Ladant, in prep.). Interpretation of these two-hybrid results and understanding the ultimate role of these apparent interactions in divisome assembly are complicated as these interactions are not necessarily direct and should be confirmed by in vitro pull-down assays; however, if these two-hybrid interactions actually reflect the protein-binding events that direct assembly of these proteins at the septum, the significance of the linear assembly pathway and the molecular events responsible for the linearity seem even more mysterious.
The simplest model to explain recruitment in the E. coli system envisions a largely linear assembly via protein–protein interactions, such as cars in a train. In this model, each protein binds only to immediately adjacent proteins in the pathway, and interactions between adjacent proteins are independent of proteins localizing either farther upstream or downstream (Fig. 1C). Under this scenario, many of the positive signals seen by two-hybrid analysis could simply be a result of weak interactions or the close proximity of these cell division proteins when assembled into a complex rather than strong direct interactions. Alternatively, the two-hybrid data may be interpreted as indicating that recruitment involves cooperative binding of a recruited protein by a preassembled complex of upstream proteins. Under this model, only this complex of early proteins is able to provide the multiple required interactions that allow recruitment of the subsequent protein (Fig. 1D). Finally, one could envision that the linear pathway actually reflects a series of sequentially ordered septum modification events. In such a model, upstream proteins modify the septum (e.g., by modifying the cell wall murein), to create a localized signal at the septum that can be recognized by the next protein in the pathway (Fig. 1E). The assembly of high-molecular-weight penicillin-binding proteins (HMW PBPs) at the division site in S. pneumoniae (Morlot et al. 2004) seems to follow such a model.
All three models make specific predictions about the nature of recruitment events that occur between adjacent proteins in the pathway. In particular, the latter two models would predict that a protein must necessarily localize before the next protein in the pathway, as it must associate with other early proteins or perform some function at the septum before the next protein can be recruited. In the first model (linear assembly), however, the order of localization would not necessarily be important because recruitment would be simply a consequence of binding between two proteins. In this scenario, localization of either of a pair of interacting proteins would allow the recruitment of the other, and this interaction would be independent of upstream proteins in the pathway.
The linearity of the pathway, however, makes distinguishing among these models difficult. Since a protein within the pathway requires all proteins localizing upstream of it for its proper localization and a delocalized protein presumably cannot recruit the next protein in the pathway to the septum, it is difficult to identify which proteins are in fact strictly required for a given recruitment event. Hence, testing these predictions requires a method to divorce a given protein from its normal localization requirements, allowing recruitment by that protein to be analyzed in cells lacking upstream proteins. In this work we devised such a method and apply it to analyze the role of FtsQ in the assembly pathway.
FtsQ is a bitopic membrane protein with a short cytoplasmic sequence at its N terminus and a relatively large periplasmic sequence (Carson et al. 1991). It is in the middle of the localization pathway (Buddelmeijer and Beckwith 2002) and according to two-hybrid analysis (Di Lallo et al. 2003; G. Karimova and D. Ladant, in prep.) makes multiple contacts with other proteins in the pathway. These data raise the possibility that FtsQ could coordinate recruitment of many of the downstream proteins. Coimmune precipitations also indicate that FtsQ is likely part of a complex with downstream proteins FtsL and FtsB, although FtsQ localizes efficiently in their absence (Buddelmeijer and Beckwith 2004). Normally FtsQ requires FtsZ, FtsA, ZipA, and FtsK in order to localize to the septum, and its localization in turn is required for recruitment of downstream proteins such as FtsL and FtsI (Chen et al. 1999; Ghigo et al. 1999; Weiss et al. 1999; Chen and Beckwith 2001; Hale and de Boer 2002; Pichoff and Lutkenhaus 2002). If an early protein such as FtsA or FtsK is selectively depleted from cells, FtsQ is no longer localized to the septum and its ability to recruit proteins cannot be assayed. However, if FtsQ could somehow be localized “prematurely” to the septum in the absence of upstream proteins, one could analyze whether these upstream proteins contribute to the recruitment of downstream proteins by FtsQ or are simply required for the localization of FtsQ itself.
In this paper, we describe a technique for premature targeting of FtsQ to the nascent Z ring by fusing it to the protein ZapA. ZapA is a small cytoplasmic protein that appears to be a nonessential component of the septal ring. It localizes to FtsZ rings in both E. coli and B. subtilis, and purified ZapA protein can bind to FtsZ in vitro and promote bundling of FtsZ filaments (Gueiros-Filho and Losick 2002; Anderson et al. 2004). These data suggest that ZapA binds directly to FtsZ in vivo independently of other cell division proteins. Therefore, we predict that FtsQ, if fused to ZapA (diagrammed in Fig. 1F), should localize in the absence of FtsA, ZipA, and FtsK (Fig. 1G). The fact that ZapA is nonessential in both B. subtilis and E. coli (Gueiros-Filho and Losick 2002; Johnson et al. 2004) indicates that it is not required for localization of other cell division proteins and hence is unlikely to be responsible for any observed recruitment by a ZapA fusion protein.
The results presented here demonstrate that FtsQ when fused to ZapA is prematurely targeted to midcell. Our studies on the functioning of the prematurely targeted FtsQ in the assembly pathway provide new insights into this pathway and help to distinguish between various models for how proteins are assembled at the nascent division site.
Results
Based on the known properties of the ZapA protein, we reasoned that fusion of full-length E. coli ZapA to the N terminus of FtsQ would result in the localization of FtsQ to the sites of FtsZ rings independently of FtsA, ZipA, and FtsK, three proteins normally required for FtsQ localization. Critical to this experiment, however, is the confirmation that, in E. coli, ZapA is part of the septal ring and localizes to FtsZ rings independently of other cell division proteins.
Positioning ZapA in the E. coli septal recruitment pathway
In order to determine whether localization of ZapA is dependent on FtsZ and not other downstream proteins, we introduced the plasmid pNG53, which expresses a YFP–ZapA fusion under IPTG control, into strains carrying either the ftsA12(Ts) or ftsZ84(Ts) temperature-sensitive mutations to create NWG128 and NWG129. Strains carrying either mutation grow and divide normally at 30°C, but fail to divide when grown at 42°C, resulting in the formation of filamentous, undivided cells. A GFP fusion to a cell division protein such as FtsQ that depends on both FtsZ and FtsA for localization fails to localize in cells carrying these mutations at 42°C, but localizes normally at 30°C (Fig. 2; Chen et al. 1999). The YFP–ZapA fusion protein, although failing to localize in ftsZ84 cells grown at the restrictive temperature, localizes efficiently in ftsA12 cells (Fig. 2). These results indicate that, in E. coli, ZapA localizes to the septal ring and its localization is dependent on FtsZ, independent of FtsA, and presumably independent of proteins further downstream such as FtsK and FtsQ, all of which require FtsA for their normal localization.
Figure 2.

Localization of YFP–ZapA requires FtsZ but not FtsA. YFP–ZapA and GFP–FtsQ localize in ftsZ84(Ts) or ftsA12(Ts) cells grown at 30°C (permissive temperature). When grown at 42°C (restrictive temperature), YFP–ZapA localizes in cells carrying the ftsA12(Ts) allele but not in those containing ftsZ84(Ts). GFP–FtsQ does not localize in either background at 42°C. The strains used are JOE95, JOE97, NWG128, and NWG129.
Construction and localization of a ZapA–FtsQ fusion
To test the ability of ZapA to localize FtsQ prematurely, we constructed the plasmid pNG107, which expresses a YFP–ZapA–FtsQ fusion (Fig. 1F) at low levels from an IPTG-inducible PTrc promoter carrying –10 and –35 down mutations (Weiss et al. 1999). The YFP–ZapA–FtsQ fusion expressed from pNG107 localized to the midcell in wild-type cells and was able to complement a strain depleted for FtsQ (data not shown). To confirm that YFP–ZapA–FtsQ localization was independent of FtsA and FtsK, pNG107 was introduced into a strain carrying the ftsA12(Ts) mutation or into the FtsK depletion strain NWG326. The latter strain carries a chloramphenicol replacement of the ftsK gene complemented by a low-copy plasmid expressing FtsK under an arabinose-inducible PBAD promoter. When grown in arabinose, FtsK is expressed and the cells grow and divide normally. Upon switching to glucose, ftsK expression is repressed, FtsK protein is depleted, and cells become unable to divide, resulting in filamentous growth (Chen and Beckwith 2001). Both strains also carry an FtsZ–CFP fusion construct under IPTG control integrated into the chromosome (Chen and Beckwith 2001). This allowed us to confirm that the observed localization of YFP signal overlapped the sites of FtsZ rings as visualized by CFP. To confirm that our cells were properly depleted, isogenic strains expressing YFP–FtsQ from pJC64 were always grown in parallel for comparison.
As expected, both YFP–FtsQ and YFP–ZapA–FtsQ appeared as rings overlapping the CFP–FtsZ signal at the bacterial midcell (Fig. 3) when expressed in ftsA12(Ts) cells grown at 30°C or cells complemented for FtsK by growth in arabinose. Upon depletion for FtsA at 42°C or for FtsK by growth in glucose, YFP–ZapA–FtsQ remained colocalized with CFP–FtsZ, forming regularly spaced rings along the filamentous cell, whereas the control, YFP–FtsQ, was no longer localized. Western blots indicated that the lack of YFP–FtsQ localization was not due to lower amounts of protein (data not shown). Since the YFP is fused to the N terminus of the ZapA–FtsQ fusion, it is possible that the septal signal we see is due to a breakdown product consisting of YFP–ZapA rather than the full-length fusion; however, Western blot analysis (data not shown) in combination with the results described below indicate that the majority of full-length fusion protein is recruited to the septum under these conditions. These results suggest that the ZapA moiety is effectively recruiting the ZapA–FtsQ fusion to the septum independently of FtsA and FtsK, both of which are normally required for FtsQ localization.
Figure 3.
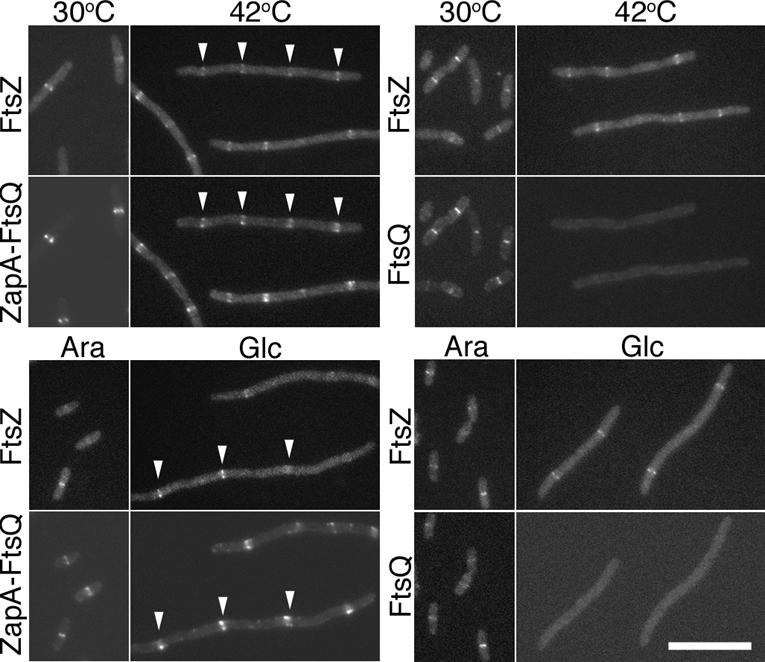
YFP–ZapA–FtsQ colocalizes with FtsZ rings in cells depleted for FtsA or FtsK. (Top panel) Localization of CFP–FtsZ (FtsZ) with either YFP–ZapA–FtsQ (ZapA–FtsQ) or YFP–FtsQ (FtsQ) in ftsA12(Ts) cells at permissive (30°C) or restrictive (42°C) temperatures. (Bottom panel) Localization of the same in ΔftsK cells when FtsK expression from a complementing plasmid is induced with arabinose (Ara) or repressed with glucose (Glc). Examples of colocalization of YFP–ZapA–FtsQ with FtsZ–CFP in cells depleted for FtsK or FtsA are indicated by white arrowheads. Bar, 10 μm. The strains used are NWG327, NWG328, NWG373, and NWG374.
Recruitment of FtsL
Previous results indicated that FtsQ, FtsL, and FtsB exist in a stable complex in the cell and that the FtsL–FtsB complex, presumably including FtsQ, can form in the absence of FtsK, a condition in which none of the three proteins are normally localized to the septum (Buddelmeijer and Beckwith 2004). In the previous section we showed that fusion to ZapA results in the localization of FtsQ to the septum independently of FtsK. Therefore, if FtsQ, FtsL, and FtsB are part of an FtsK-independent complex, FtsL and FtsB should be recruited to the septum by the ZapA–FtsQ fusion protein in cells depleted for FtsK. To test this prediction, an IPTG-inducible construct expressing the ZapA–FtsQ fusion (construct diagrammed in Fig. 1F) was integrated into the chromosome at the λ att site using λ InCh and expression confirmed by Western blot analysis (data not shown). This construct was then introduced into an FtsK depletion strain expressing a GFP–FtsL fusion integrated into the chromosome at the φ80 att site as described previously (Chen and Beckwith 2001). In order to ensure that our recruitment was specific to the ZapA–FtsQ fusion protein, we also introduced identical chromosomal constructs expressing either ZapA or FtsQ alone into isogenic FtsK depletion strains for comparison.
As predicted, when the ZapA–FtsQ protein is expressed in the FtsK depletion strain described above, GFP–FtsL localizes efficiently to regularly spaced rings when compared to control cells expressing either ZapA or FtsQ alone (Fig. 4A–C; Table 1, rows 1–3). The ring spacing (an inverse measure of ring frequency) for GFP–FtsL in the ZapA–FtsQ-expressing cells of 9.5 μm/ring (Table 1, row 1) is very similar to the published spacing of 8.8 μm/ring for FtsZ localization in FtsK-depleted cells and 13 μm/ring for FtsL localization in FtsN-depleted cells (Chen and Beckwith 2001). This indicates that ZapA–FtsQ is recruiting GFP–FtsL at a near maximal expected level if it were to recruit GFP–FtsL to every available Z ring. In contrast, expression of FtsQ or ZapA alone from an identical chromosomal construct in isogenic FtsK-depleted cells resulted in significantly reduced levels of localization of GFP–FtsL (Fig. 4B,C; Table 1, rows 2–3). Thus, ZapA–FtsQ appears able to direct the recruitment of FtsL to FtsZ rings in the absence of FtsK, and this recruitment is not due to the individual effects of expressing either ZapA or FtsQ alone.
Figure 4.

ZapA–FtsQ restores localization of FtsL in cells depleted for FtsK, FtsA, or both. Localization of GFP–FtsL is shown in cells depleted for the indicated cell division protein(s) and expressing, as indicated, ZapA–FtsQ (A,D,G), wild-type FtsQ (B,E,H), or ZapA (C,F) from the λatt site. Representative images are shown. A quantitative summary of results for all cells analyzed is presented in Table 1, rows 1–8. Bar, 10 μm.
Table 1.
Quantitative summary of localization data
| GFP fusion | FtsA | FtsKa | ZapA—FtsQ or FtsQ(wt) | Strain | Cells | Avg cell length (μm) | Fraction of cells w/rings | Rings/cell | Ring spacingb (μm/ring) | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | GFP—FtsL | + | - | ZapA—FtsQ | NWG252 | 65 | 22.1 | 0.95 | 2.3 | 9.5 |
| 2 | + | - | FtsQ | NWG390 | 44 | 28.6 | 0.39 | 0.5 | 52.4 | |
| 3 | + | - | ZapA | NWG448 | 46 | 27.3 | 0.61 | 0.9 | 29.9 | |
| 4 | - | + | ZapA—FtsQ | NWG341 | 38 | 40.5 | 1.00 | 3.1 | 13.3 | |
| 5 | - | + | FtsQ | NWG418 | 36 | 41.6 | 0.56 | 0.9 | 48.3 | |
| 6 | - | + | ZapA | NWG447 | 20 | 33.0 | 0.8 | 1.2 | 28.7 | |
| 7 | - | - | ZapA—FtsQ | NWG259 | 77 | 49.3 | 1.00 | 4.2 | 11.8 | |
| 8 | - | - | FtsQ | NWG411 | 47 | 47.9 | 0.36 | 0.4 | 112.6 | |
| 9 | GFP—FtsI | + | - | ZapA—FtsQ | NWG277 | 53 | 25.1 | 0.94 | 2.3 | 10.8 |
| 10 | + | - | FtsQ | NWG394 | 54 | 24.4 | 0.65 | 0.9 | 28.0 | |
| 11 | - | + | ZapA—FtsQ | NWG340 | 18 | 39.1 | 1.00 | 2.9 | 13.5 | |
| 12 | - | + | FtsQ | NWG417 | 32 | 45.0 | 0.69 | 0.8 | 55.4 | |
| 13 | - | - | ZapA—FtsQ | NWG299 | 30 | 51.2 | 1.00 | 3.5 | 14.5 | |
| 14 | - | - | FtsQ | NWG413 | 44 | 49.4 | 0.36 | 0.5 | 103.6 | |
| 15 | GFP—FtsN | + | - | ZapA—FtsQ | NWG257 | 207 | 7.0 | 0.85 | 1.0 | 7.1 |
| 16 | + | - | FtsQ | NWG391 | 107 | 10.5 | 0.7 | 1.1 | 9.1 | |
| 17 | - | + | ZapA—FtsQ | NWG342 | 46 | 38.9 | 0.70 | 1.1 | 35.8 | |
| 18 | - | + | FtsQ | NWG419 | 27 | 46.3 | 0.85 | 1.4 | 32.9 | |
| 19 | - | - | ZapA—FtsQ | NWG269 | 41 | 38.8 | 0.8 | 1.2 | 33.1 | |
| 20 | - | - | FtsQ | NWG428 | 32 | 42.0 | 0.75 | 1.2 | 36.3 | |
| 21 | YFP—FtsK | - | + | ZapA—FtsQ | NWG352 | 28 | 40.0 | 1.00 | 4.7 | 8.5 |
| 22 | - | + | FtsQ | NWG421 | 30 | 42.6 | 0.83 | 1.6 | 26.6 | |
| 23 | - | + | ZapA | NWG446 | 24 | 31.9 | 0.89 | 2.2 | 15.3 | |
| 24 | GFP—FtsQ | + | - | ZapA—FtsQ | NWG255 | 99 | 21.4 | 0.44 | 0.5 | 38.4 |
| 25 | - | + | ZapA—FtsQ | NWG343 | 41 | 43.5 | 0.85 | 1.3 | 32.4 | |
| 26 | - | - | ZapA—FtsQ | NWG261 | 26 | 55.2 | 0.50 | 0.7 | 79.8 | |
| 27 | - | + | ZapA—FtsQ | NWG414 | 31 | 22.8 | 0.45 | 0.5 | 41.6 | |
| 28 | - | +++ | ZapA—FtsQ | NWG415 | 50 | 21.1 | 0.9 | 1.9 | 11.0 | |
| 29 | - | +++ | FtsQ | NWG423 | 23 | 41.9 | 0.87 | 1.6 | 26.8 |
+ indicates endogenous FtsK expression; +++ indicates endogenous FtsK plus expression from pBAD44.
Ring spacing = total length (microns)/number rings; bold indicates positive localization score based on comparison with control cells (FtsQ or ZapA) and with published values of 8.1-14.5 μm/ring for localization of fts proteins in FtsN-depleted cells (Chen and Beckwith 2001).
It remained possible that this ability of ZapA–FtsQ to recruit FtsL was specific in its ability to bypass FtsK but still depended on proteins farther upstream. We considered FtsA as the most likely candidate for such a contributor as two-hybrid data suggest it may contact downstream proteins FtsI and FtsN (Di Lallo et al. 2003; G. Karimova and D. Ladant, in prep.) and is widely conserved among bacteria (Margolin 2000). To test the ability of ZapA–FtsQ to recruit FtsL in the absence of FtsA, we combined the ZapA–FtsQ fusion and GFP–FtsL constructs with the ftsA12(Ts) mutation and analyzed localization of GFP–FtsL at the restrictive temperature. Under these conditions, expression of ZapA–FtsQ is able to restore localization of GFP–FtsL to regularly spaced rings (Fig. 4D; Table 1, row 4). As is the case in FtsK-depleted cells, expression of either FtsQ or ZapA alone failed to restore localization of GFP–FtsL, as indicated by reduced frequency and fluorescence intensity of observed rings (Fig. 4E,F; Table 1, rows 5–6). Thus, neither FtsA nor FtsK is required for recruitment of FtsL in cells expressing the ZapA–FtsQ fusion.
Finally, it remained formally possible that FtsL requires FtsQ and either FtsA or FtsK for its localization. To rule out such redundant roles for FtsA and FtsK, we performed the same experiment in cells depleted for both FtsK and FtsA. To do this, we introduced the ftsA12(Ts) mutation into the FtsK depletion strains expressing GFP–FtsL and either the ZapA–FtsQ fusion or FtsQ alone as described above. By growing the resulting cells at 42°C to deplete FtsA and in glucose to repress expression of FtsK from the complementing plasmid, we were able to analyze recruitment in cells missing both proteins. GFP–FtsL localized in doubly depleted cells expressing ZapA–FtsQ (Fig. 4G; Table 1, row 5), indicating that ZapA–FtsQ is in fact capable of recruiting FtsL in the absence of both FtsA and FtsK. In light of previous work indicating that FtsB and FtsL are dependent on each other for localization and can be coimmune-precipitated (Buddelmeijer et al. 2002; Buddelmeijer and Beckwith 2004), these data are consistent with a model in which FtsL, FtsB, and FtsQ exist in a stable complex that can form independently of proteins upstream of FtsQ. Hence, under normal circumstances, the simple presence of FtsQ at the septum is sufficient to allow localization of FtsL and, presumably, FtsB.
Recruitment of FtsI and FtsN
Having established the ability of ZapA–FtsQ to recruit FtsL in the absence of upstream proteins, we proceeded to examine the extent to which simply localizing FtsQ to the septum is able to restore localization of the remainder of downstream proteins in the cell division pathway, including FtsI and FtsN. To this end, we repeated the above experiments with GFP–FtsI and GFP–FtsN in ZapA–FtsQ-expressing cells that were depleted for FtsK, FtsA, or both.
FtsI was efficiently localized in cells expressing ZapA–FtsQ but depleted for either FtsK or FtsA (Fig. 5A,B). In cells depleted for both FtsA and FtsK, localization also seems to occur at regular intervals in filaments (Fig. 5C). Such localization was not seen in control cells expressing FtsQ (Table 1). This suggests that ZapA–FtsQ not only restores FtsL and FtsB to the septum in FtsA/FtsK-depleted cells, but also restores the localization pathway from FtsQ through FtsI as shown in Figure 8A (below). Since FtsI strictly requires FtsL, FtsB, and FtsW to localize to the septum under normal conditions, we assume these proteins are also required for recruitment of FtsI when FtsQ is localized via fusion to ZapA. If such is the case, FtsQ, FtsL, FtsB, FtsW, and FtsI are likely capable of assembling into a stable complex in the cell independently of upstream proteins and hence independently of the proteins' localization to midcell.
Figure 5.

ZapA–FtsQ restores localization of FtsI, but not FtsN. Cells express either GFP–FtsI (A–C) or GFP–FtsN (D–F) from chromosomal constructs and are depleted for the cell division proteins FtsA and FtsK as indicated. All cells express ZapA–FtsQ from the λatt site. Representative images from paired samples are shown. Note the short cells in D, indicating suppression of the FtsK depletion by expression of GFP–FtsN. (C) Examples of rings in a doubly depleted cell are indicated by white arrowheads. A quantitative summary of results including control cells expressing wild-type FtsQ from the λatt site is presented in Table 1, rows 9–20. Bar, 10 μm.
Figure 8.

Summary of observed recruitment by prematurely targeted FtsQ in cells depleted for FtsA and FtsK (A) or FtsA alone (B). (A) ZapA–FtsQ is able to recruit FtsL and likely FtsB, which restores the pathway through FtsI. (B) ZapA–FtsQ is also able to interact with FtsK, and this interaction with FtsK in turn supports association of multiple FtsQ molecules. FtsB and FtsW are shown in outline as their presence is not explicitly tested in this work, but implied from previous data, which indicate FtsI requires both FtsB and FtsW to localize to the septum.
FtsN presents quite a different story. ftsN was originally identified as a multicopy suppressor of the ftsA12(Ts) mutation at 37°C. Subsequently, overexpression of FtsN was shown to suppress Ts mutations in ftsA, ftsK, ftsQ, and ftsI as well as partially suppressing FtsK depletion strains (Dai et al. 1993; Draper et al. 1998). This property of FtsN complicates the analysis of FtsN recruitment by ZapA–FtsQ in FtsK depletion strains. Notably, in cells depleted for FtsK, GFP–FtsN localizes well regardless of whether FtsQ or ZapA–FtsQ is expressed (Fig. 5; J. Chen and J. Beckwith, unpubl.). In fact, average cell length is much reduced compared to FtsK depletion strains expressing GFP fusions to other cell division proteins (Fig. 5D; Table 1, rows 15–16 vs. 1–3, 9–10), and these cells readily form colonies on glucose plates, suggesting gfp–ftsN is acting as an overexpression suppressor in these strains. We have some indications that this apparent suppression may only be possible because of residual FtsK expression under non-inducing conditions (data not shown).
To obviate the problem of ftsN overexpression suppression, we analyzed recruitment of FtsN in ZapA–FtsQ-expressing strains depleted for FtsA or both FtsK and FtsA, since, at 42°C, the GFP–FtsN fusion is unable to suppress the cell division defects of these strains. In both strains, GFP–FtsN did not localize to regularly spaced rings (Fig. 5E,F; Table 1, rows 17, 19). In contrast, under identical conditions, GFP–FtsN localizes normally in wild-type cells and can complement a ΔftsN-null mutation (data not shown), indicating the GFP–FtsN fusion protein is stable and functional under our experimental conditions.
The fact that FtsN fails to localize under conditions in which FtsQ, FtsL, FtsB, and FtsI are recruited suggests that whether or not FtsN makes direct contacts with FtsI or FtsQ, it must rely on additional signals at the septum to localize. Since FtsN requires the presence of FtsI and does not localize in cells depleted for FtsI, it is likely that its localization must integrate signals from both FtsI and early proteins. While previous studies were consistent with interactions between FtsI and FtsN being sufficient for recruitment of FtsN, our results point to a more complex process (Di Lallo et al. 2003; Wissel and Weiss 2004; G. Karimova and D. Ladant, in prep.).
Back-recruitment of FtsK
FtsK immediately precedes FtsQ in the cell division pathway and is ordinarily required for the localization of FtsQ (Chen and Beckwith 2001), suggesting that it likely plays a direct role in recruiting FtsQ. As proposed above, one can envision several models to explain how FtsQ is recruited by FtsK. In the linear association model, a simple interaction between FtsK and FtsQ would allow recruitment. In this case, upstream proteins such as FtsA would only be required in order to localize FtsK. Moreover, if a simple physical interaction links FtsK and FtsQ, localization of either protein should restore the localization of the other in cells lacking FtsA. Under alternative models, FtsK would necessarily need to localize first, either to associate with other upstream proteins or to modify the septum to produce an FtsQ recruitment signal.
To distinguish among these models, we determined whether localization of FtsQ via the premature targeting method could restore localization of FtsK. To do this, we examined localization of YFP–FtsK in ftsA12(Ts) cells expressing either ZapA–FtsQ or wild-type FtsQ constructs from the λ att site. Compared to expression of FtsQ or ZapA alone, expression of ZapA–FtsQ promotes efficient localization of YFP–FtsK, leading to both increased ring frequency (decrease in average ring spacing) and increased localization signal intensity (Fig. 6; Table 1, rows 21–23). These results indicate that, for the recruitment of FtsQ, there is no underlying fundamental temporal order or integration of upstream signals. Rather, this evidence suggests that FtsQ can physically associate with FtsK within the cell. Although further experiments will be required to address whether this association requires FtsZ, it is possible that FtsK may be part of a cell division complex along with FtsQ, FtsL, FtsB, FtsW, and FtsI that can form independently of localization to midcell.
Figure 6.

ZapA–FtsQ promotes localization of FtsK in cells depleted for FtsA. Cells express an FtsK–YFP fusion from the native ftsK locus and ZapA–FtsQ (A), FtsQ (B), or ZapA (C) as indicated from IPTG-inducible constructs integrated at the λatt site. Cells also carry the ftsA12(Ts) allele and were grown at 42°C to deplete FtsA. Representative images are shown. A quantitation of results is presented in Table 1, rows 21–23. Bar, 10 μm.
“Divisome” may contain multiple copies of FtsQ
Recent work using bacterial two-hybrid systems has raised the possibility that FtsQ interacts with itself (Di Lallo et al. 2003; G. Karimova and D. Ladant, in prep.; M. Gonzalez and J. Beckwith, unpubl.). It remains unclear from these results whether FtsQ is acting as a homodimer or higher-order oligomer, or if the two-hybrid data reflect the fact that FtsQ is simply present in multiple copies within a single divisome complex. Given these results, it seemed reasonable to ask whether ZapA–FtsQ could recruit GFP–FtsQ to the septum in cells depleted for FtsK, FtsA, or both; however, our experiments indicated that GFP–FtsQ fails to localize in such cells expressing ZapA–FtsQ, conditions under which FtsL and FtsI were efficiently localized (Table 1, rows 24–26; data not shown).
Initially, we concluded that either the two-hybrid data were not reflecting direct interactions in vivo or that for some reason our ZapA–FtsQ and GFP–FtsQ fusions were incompatible. One possible explanation for the latter hypothesis lies in the recent observation that ZapA, itself, is present as a dimer in the cell (Low et al. 2004). This could result in strongly favoring a ZapA–FtsQ homodimer rather than the ZapA–FtsQ:GFP–FtsQ heterodimer, making any potential dimerization impossible to detect.
In the course of related experiments, however, we occasionally saw localization of GFP–FtsQ in ZapA–FtsQ-expressing cells when FtsK was expressed from a multicopy plasmid, suggesting that FtsK may somehow facilitate the recruitment of multiple FtsQ molecules.
To test this idea, we introduced a multicopy plasmid expressing excess FtsK into a strain carrying the ftsA12(Ts) allele and expressing both ZapA–FtsQ and GFP–FtsQ constructs. Under conditions of FtsA depletion, GFP–FtsQ was recruited in these cells (Fig. 7B; Table 1, row 28). If we either replaced the FtsK plasmid with empty vector, or expressed wild-type FtsQ instead of ZapA–FtsQ, GFP–FtsQ localization was reduced (Fig. 7A,C; Table 1, rows 27, 29). This result suggests that FtsK may play some role in facilitating the association of multiple FtsQ molecules, perhaps through stabilizing the assembled proteins within a single large complex.
Figure 7.

Overexpression of FtsK promotes association of multiple FtsQ molecules. (B) When FtsK was overexpressed from multicopy plasmid pBAD44 (pNG137) in cells expressing both GFP–FtsQ and ZapA–FtsQ under conditions of FtsA depletion, localization of GFP–FtsQ was seen. If either wild-type FtsQ was expressed instead of ZapA–FtsQ (C) or empty vector was substituted for the FtsK plasmid (A), localization was significantly reduced. Representative images from paired samples are shown. A quantitation of results is presented in Table 1, rows 24–29.
Prior to this work, it was unclear whether FtsA and FtsK played a role in cell division other than facilitating the recruitment of downstream proteins. Interestingly, although premature recruitment of FtsQ to the septum via fusion to ZapA was able to restore recruitment of most of the cell division proteins downstream of FtsA and FtsK, expression of ZapA–FtsQ failed to restore the ability of ftsA12(Ts) or ΔftsK strains to divide under restrictive conditions (Table 1, average cell length ZapA–FtsQ vs. FtsQ-expressing cells; data not shown).
Discussion
The study of an individual step within a sequentially ordered pathway is complicated by the fact that such a step requires the prior activity of upstream proteins. In a synthetic reaction, for example, activity of early enzymes is required to produce intermediate substrates for the enzyme of interest. In such a case, the addition of exogenous substrate divorces the reaction of interest from its normal place in the pathway, allowing the reaction to be analyzed in isolation. For a sequential assembly pathway, such as the septal recruitment pathway in E. coli, the ability of a protein to recruit downstream proteins to the septum requires a priori the localization of that protein by proteins farther upstream. Hence, within the normal in vivo pathway, it is difficult to analyze recruitment by a particular protein separately from the upstream events that are required for its localization.
In this work we have developed a method, which we term premature targeting, of divorcing a particular protein in the cell division pathway of E. coli from its normal localization requirements. Here, fusion of the FtsZ-binding protein ZapA to FtsQ allowed the premature targeting of FtsQ directly to FtsZ rings. FtsQ, when recruited prematurely to the septum by ZapA, can efficiently recruit two downstream proteins, FtsL and FtsI, in the absence of FtsK and/or FtsA, two upstream proteins that are normally strictly required for localization of FtsQ and all proteins downstream of it (Fig. 8A). We also show that prematurely targeted FtsQ is even capable of “back-recruitment” of FtsK in the absence of FtsA (Fig. 8B).
These results are consistent with prior studies using two-hybrid analysis and coimmune precipitations, which established that FtsQ, FtsL, and FtsB can form a complex within the cell and that this complex can form even in the absence of FtsK, a condition under which these proteins are not localized to the septum. Those findings suggest that these proteins are always associated within the cell regardless of localization (Di Lallo et al. 2003; Buddelmeijer and Beckwith 2004; G. Karimova and D. Ladant, in prep.). Our results indicate further that neither FtsK nor FtsA is required for the formation of this complex or necessarily for its localization to the septum if FtsQ is localized artificially (i.e., by fusion to ZapA). Rather, since the simple localization of FtsQ by premature targeting is sufficient to direct recruitment of this FtsQ–FtsL–FtsB complex to the septum, FtsK and FtsA presumably are only required for the localization of the members of this complex insofar as they allow localization of FtsQ. We also show that this complex is not limited to FtsQ, FtsL, and FtsB, but likely includes FtsI and by inference FtsW, which is “upstream” of FtsI and is normally required for FtsI localization. While the molecular mechanism by which FtsW and FtsI are recruited to this complex is unclear, the association does not appear to involve FtsK or FtsA.
Although the results presented on FtsN localization are not unambiguous, they suggest that FtsN, the last known essential protein in the pathway, may follow a more complex path to the septum. The finding that FtsN is not simply recruited by contact with FtsI raises the intriguing possibility that it plays a role in integrating signals from both early and late proteins. Prior work has speculated on such a role for FtsN in the pathway, as overexpression of FtsN is capable of suppressing temperature-sensitive mutations in various other genes within the cell division pathway including ftsA, ftsK, ftsQ, and ftsI (Dai et al. 1993; Draper et al. 1998). The apparent FtsA–FtsN interaction (Fig. 1B) raises the question as to whether FtsA directly contributes to FtsN localization, providing this “early signal.” However, the cytoplasmic and membrane-spanning segments of FtsN are not required for FtsN localization and function (Dai et al. 1996), making it difficult to imagine how a direct interaction between cytoplasmic FtsA and FtsN would be required for cell division. Instead, it seems reasonable to speculate that FtsN somehow recognizes either protein conformations or a modification of the septum that is generated only by properly assembled ring. By recognizing such a signal, FtsN could prevent activation of the division machinery until assembly of the “divisome” is complete.
The “back-recruitment” of FtsK by ZapA–FtsQ suggests a possible interaction between these two proteins, which under normal circumstances allows forward recruitment of FtsQ by a localized FtsK. The fact that localization of either protein results in the localization of the other, argues against a model in which activity of a localized FtsK must precede FtsQ localization. Additionally, since this interaction is independent of FtsA and since FtsQ localization under normal circumstances is independent of FtsL, FtsB, FtsW, FtsI, and FtsN (Chen et al. 1999; Chen and Beckwith 2001; Buddelmeijer et al. 2002; Di Lallo et al. 2003), this FtsK–FtsQ interaction is likely not dependent on any other identified proteins in the cell division pathway. Given these results in combination with those discussed above, it is reasonable to speculate that FtsQ, FtsL, FtsB, FtsW, and FtsI could assemble into a complex, which is then recruited to the septum via interaction of FtsQ with FtsK or that all of these proteins could possibly exist as a preassembled complex within the cell, even when the cell is not dividing.
Finally, we have presented preliminary evidence that multiple copies of FtsQ protein are localized together within the cell division complex and that this association of multiple FtsQ proteins is dependent on the amount of FtsK in the cell. One must interpret these results with some degree of caution as we have been unable to determine whether our data reflect the stabilization of FtsQ dimers or oligomers by FtsK. Models in which FtsK is capable of multimerization or in which a single FtsK molecule can recruit multiple FtsQ molecules are equally plausible interpretations of these data. Rather, we include these data only to raise the idea that the “divisome” complex could include multiple copies of the FtsQ protein, suggesting the divisome may, in fact, represent an assembly of subcomplexes.
Recent bacterial two-hybrid results suggested an unexpectedly high number of interactions among cell division proteins within the E. coli cell (Di Lallo et al. 2003). The interpretation of these results suggested that despite the linear assembly pathway in E. coli, the cell division proteins may actually be assembled in a cooperative manner into a large divisome complex and hence assembly may in fact be more similar to that in B. subtilis than originally thought. Our results suggest these two-hybrid interactions should be interpreted with caution as they do not necessarily reflect direct interactions that are required for recruitment or assembly of the ring complex. For example, although FtsI appears to interact with both FtsK and FtsA, neither protein appears to be required for FtsI localization provided FtsQ is localized efficiently. Hence, although our data support a view based on two-hybrid data that these proteins ultimately are assembled within a large divisome complex, assembly of the divisome in E. coli would seem to be driven ultimately by a specific subset of these observed interactions.
It is interesting that ZapA–FtsQ, although restoring localization of downstream cell division proteins (with the exception of FtsN) in cells depleted for FtsA and/or FtsK, does not restore cell division in these cells. This result implies that FtsK and FtsA perform critical functions at the division site, including but not necessarily limited to facilitating the localization of FtsN. If FtsA and FtsK were only required to facilitate localization of FtsK and FtsQ, respectively, restoring the localization to the septum of FtsQ and the proteins it interacts with would seem to be sufficient to rescue cells deficient for either protein. Whatever this function is, it must be lacking in cells in which cell division proteins are recruited by ZapA–FtsQ rather than by the endogenous pathway.
This method of recruiting proteins prematurely to the septum using ZapA should be applicable to the study of other cell division proteins. Presumably, any given cytoplasmic or transmembrane cell division protein could be targeted to FtsZ independently of normally required proteins. This would allow for analysis of recruitment by prematurely targeted cell division proteins as performed here for FtsQ. We think this approach will be particularly useful in analyzing the recruitment steps among the late proteins in the cell division pathway.
In trying to dissect the role of a particular protein in the cell division pathway, our lab and others have turned to genetics to isolate mutations in cell division proteins that affect localization, recruitment, and potential enzymatic activity. The analysis of such mutants in vivo is often complicated by the fact that these proteins are essential and it is often difficult to determine which of a protein's functions are affected by a given mutation. The premature targeting method provides a potential tool for characterizing mutants affected in localization and recruitment. For example, one could analyze the behavior of mutant alleles of cell division genes when incorporated into a ZapA fusion protein (which would be predicted to restore function of mutant proteins specifically affected in localization) or when introduced into strains expressing ZapA fusions to downstream cell division proteins (which would be predicted to restore function of mutant proteins specifically defective in recruitment).
Materials and methods
Bacterial strains, plasmids, and media
The bacterial strains and plasmids used in this work are listed in Supplementary Table S1. All experiments were performed in NZY medium (Guzman et al. 1992). Antibiotics were added when appropriate at the indicated concentrations: ampicillin, 200 μg/mL (plasmid) or 25 μg/mL (chromosome); chloramphenicol, 10 μg/mL; kanamycin, 40 μg/mL; spectinomycin, 100 μg/mL (plasmid) or 50 μg/mL (chromosome); and tetracycline, 15 μg/mL. D-Glucose and L-arabinose were added at 0.2% to repress or induce the expression of genes under control of the PBAD promoter. IPTG (isopropyl-β-D-thiogalactoside) was added at the appropriate concentrations as indicated.
Standard laboratory techniques were used for cloning and analysis of DNA, PCR, electroporation, transformation, and P1 transduction (Miller 1992). Chromosomal constructs were integrated using λ InCh (Boyd et al. 2000). Individual bacterial strains and plasmids were constructed as described or as indicated in Supplementary Table S1. To transduce GFP and CFP fusions integrated at the attφ80 site into ftsK depletion strains, JOE563 was made trp– by transduction of a linked Tn5 (tetR) and screening for tryptophan auxotrophy. The resulting strain was transduced with P1 lysates from trp+ strains carrying the appropriate attφ80 integrated fusion constructs and transductants selected on minimal media. Resulting transductants were screened for expression of GFP/CFP fusions by growth in liquid NZY containing 10 μM IPTG. In all other strains, attφ80 integrated constructs were transduced by P1 using resistance to spectinomycin. λatt integrated constructs were transduced by P1 using resistance to ampicillin (25 μg/mL). The ftsA12(Ts) allele was transduced from the strain MM61 by linkage to leu::Tn10 (tetR), and resulting transductants were screened for temperature sensitivity.
To construct pNG52 and pNG53, zapA (pNG52) or yfp–zapA (pNG53) was PCR-amplified from pFG53 using primers NWG43 (YFP_ygfE_fwd, 5′-CGGATCGAATTCACTACTATG AGTAAAGGAGAAG-3′) or NWG44 (YgfE_Eco_fwd, 5′-CGGA TCGAATTCATGTCTGCACAACCCGTCGATA-3′) and NWG45 (YgfE_Xba_rev, 5′-CGGGCCTCTAGATTCAAAGTTTTGGTT AGTT-3′). The products were digested with EcoRI and XbaI and ligated into the same sites of pTrc99a-ftsN-3xMyc. To construct pNG93, yfp–zapA was PCR-amplified from pNG53 using primers pTrcFwd (5′-CAAGGCGCACTCCCGTTCTGG-3′) and NWG58 (YgfE_Eco_Rev, 5′-CGGGCCGAATTCTTCAAAGTT TTGGTTAGTT-3′). The product was digested with NcoI, EcoRI, and ligated into the same sites of pNG40.
To facilitate cloning steps, ZapA–FtsQ fusions use a previously described allele of FtsQ (QQQ). Mutations were introduced into the wild-type sequence on either side of the membrane-spanning segment to generate restriction sites, resulting in several conservative amino acid changes (Guzman et al. 1997). This allele was shown to have no effect on growth, expression level, or localization (Chen et al. 1999).
Growth conditions
To test the dependence of ZapA localization on FtsZ and FtsA, strains were grown overnight at 30°C in NZY containing appropriate antibiotics. Strains were diluted 1:100 in NZY and grown for 3 h, at which time cells were diluted 1:5 into preheated NZY media (30°C or 42°C) containing IPTG at either 1 μM (NWG128, NWG129) or 10 μM (JOE95, JOE97) and grown an additional 60–70 min.
For localization of GFP fusions under conditions of FtsK depletion, strains were inoculated into NZY + 0.2% arabinose from an overnight culture and grown to midlog phase at 37°C. Cells were subsequently diluted 1:250 into NZY + 0.2% glucose and depleted for 3–3.5 h at 37°C.
For localization of GFP fusions in cells depleted for both FtsK and FtsA, strains were inoculated from an overnight culture into NZY + 0.2% arabinose and grown to midlog phase at 30°C. Cells were diluted 1:250 into NZY + 0.2% glucose, grown 2 h at 30°C to begin depletion of FtsK, and transferred to 42°C for the final hour of growth to deplete FtsA. For cells depleted only of FtsA, cells were treated as above, but were grown in NZY without sugar for the entire growth period. In some cases as described in the results, FtsK expression was induced from a low-copy plasmid with 0.2% arabinose.
In all cases, to minimize potential toxic effects of ZapA–FtsQ or FtsQ overexpression, expression of GFP, CFP, or YFP fusion constructs and zapA–ftsQ, ftsQ, or zapA λ InCh constructs was induced by adding IPTG to a final concentration of 10–20 μM for the final 30 min of growth. Also, in order to maintain consistency, ZapA–FtsQ isolates were always paired with the FtsQ control and, if possible, all strains for a given depletion/induction condition were grown together.
Microscopy
Cells were harvested, fixed (Chen et al. 1999), and mounted on agarose cushions for microscopy as described (van Helvoort and Woldringh 1994). In some cases, cells were stained with 0.2 μg/mL DAPI (4′,6-diamidino-2-phenylindole) in PBS and washed twice in PBS before mounting. Cells were examined for fluorescence using an Axioskop 2 microscope (Zeiss) equipped with 63× or 100× plan-Apochromat oil immersion objective and a 100-W mercury lamp. Filter sets to visualize GFP (HQ:FITC/Bodipy/Fluo3), EYFP, ECFP, and DAPI (UV) were from Chroma Technology Corp. Images were captured using an Orca-100 CCD camera (Hamamatsu Photonics) and Openlab (Improvision) and subsequently processed and analyzed in Openlab. All measurements were limited to intact cells exhibiting normal segregation of nucleoids as evidenced by DAPI staining. Final processing for presentation was performed in Adobe Photoshop.
Acknowledgments
We thank members of the Beckwith laboratory for critical comments and general assistance. We are grateful to F.X. Barre for his generous gift of the ftsK–yfp construct. We thank R. Losick and W. Margolin for helpful discussions and M. Gonzalez, G. Karimova, and D. Ladant for sharing unpublished results. N.W.G. was supported by a Howard Hughes Predoctoral Fellowship. F.G.-F. was a Helen Hay Whitney Postdoctoral Fellow. This work was supported by grants from the National Institute of General Medical Sciences (GM18568, GM38922).
Supplemental material is available at http://www.genesdev.org.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.1253805.
References
- Anderson D.E., Gueiros-Filho, F.J., and Erickson, H.P. 2004. Assembly dynamics of FtsZ rings in Bacillus subtilis and Escherichia coli and effects of FtsZ-regulating proteins. J. Bacteriol. 186: 5775–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi E.F. and Lutkenhaus, J. 1991. FtsZ ring structure associated with division in Escherichia coli. Nature 354: 161–164. [DOI] [PubMed] [Google Scholar]
- Bork P., Sander, C., and Valencia, A. 1992. An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and hsp70 heat shock proteins. Proc. Natl. Acad. Sci. 89: 7290–7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd D., Weiss, D.S., Chen, J.C., and Beckwith, J. 2000. Towards single-copy gene expression systems making gene cloning physiologically relevant: λ InCh, a simple Escherichia coli plasmid–chromosome shuttle system. J. Bacteriol. 182: 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buddelmeijer N. and Beckwith, J. 2002. Assembly of cell division proteins at the E. coli cell center. Curr. Opin. Microbiol. 5: 553–557. [DOI] [PubMed] [Google Scholar]
- ____. 2004. A complex of the Escherichia coli cell division proteins FtsL, FtsB and FtsQ forms independently of its localization to the septal region. Mol. Microbiol. 52: 1315–1327. [DOI] [PubMed] [Google Scholar]
- Buddelmeijer N., Judson, N., Boyd, D., Mekalanos, J.J., and Beckwith, J. 2002. YgbQ, a cell division protein in Escherichia coli and Vibrio cholerae, localizes in codependent fashion with FtsL to the division site. Proc. Natl. Acad. Sci. 99: 6316–6321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson M.J., Barondess, J., and Beckwith, J. 1991. The FtsQ protein of Escherichia coli: Membrane topology, abundance, and cell division phenotypes due to overproduction and insertion mutations. J. Bacteriol. 173: 2187–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.C. and Beckwith, J. 2001. FtsQ, FtsL and FtsI require FtsK, but not FtsN, for co-localization with FtsZ during Escherichia coli cell division. Mol. Microbiol. 42: 395–413. [DOI] [PubMed] [Google Scholar]
- Chen J.C., Weiss, D.S., Ghigo, J.M., and Beckwith, J. 1999. Septal localization of FtsQ, an essential cell division protein in Escherichia coli. J. Bacteriol. 181: 521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai K., Xu, Y., and Lutkenhaus, J. 1993. Cloning and characterization of ftsN, an essential cell division gene in Escherichia coli isolated as a multicopy suppressor of ftsA12(Ts). J. Bacteriol. 175: 3790–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ____. 1996. Topological characterization of the essential Escherichia coli cell division protein FtsN. J. Bacteriol. 178: 1328–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lallo G., Fagioli, M., Barionovi, D., Ghelardini, P., and Paolozzi, L. 2003. Use of a two-hybrid assay to study the assembly of a complex multicomponent protein machinery: Bacterial septosome differentiation. Microbiology 149: 3353–3359. [DOI] [PubMed] [Google Scholar]
- Draper G.C., McLennan, N., Begg, K., Masters, M., and Donachie, W.D. 1998. Only the N-terminal domain of FtsK functions in cell division. J. Bacteriol. 180: 4621–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington J., Daniel, R.A., and Scheffers, D.J. 2003. Cytokinesis in bacteria. Microbiol. Mol. Biol. Rev. 67: 52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghigo J.M., Weiss, D.S., Chen, J.C., Yarrow, J.C., and Beckwith, J. 1999. Localization of FtsL to the Escherichia coli septal ring. Mol. Microbiol. 31: 725–737. [DOI] [PubMed] [Google Scholar]
- Gueiros-Filho F.J. and Losick, R. 2002. A widely conserved bacterial cell division protein that promotes assembly of the tubulin-like protein FtsZ. Genes & Dev. 16: 2544–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman L.M., Barondess, J.J., and Beckwith, J. 1992. FtsL, an essential cytoplasmic membrane protein involved in cell division in Escherichia coli. J. Bacteriol. 174: 7716–7728. [PMC free article] [PubMed] [Google Scholar]
- Guzman L.M., Weiss, D.S., and Beckwith, J. 1997. Domainswapping analysis of FtsI, FtsL, and FtsQ, bitopic membrane proteins essential for cell division in Escherichia coli. J. Bacteriol. 179: 5094–5103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale C.A. and de Boer, P.A. 1997. Direct binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E. coli. Cell 88: 175–185. [DOI] [PubMed] [Google Scholar]
- ____. 2002. ZipA is required for recruitment of FtsK, FtsQ, FtsL, and FtsN to the septal ring in Escherichia coli. J. Bacteriol. 184: 2552–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J.E., Lackner, L.L., Hale, C.A., and de Boer, P.A. 2004. ZipA is required for targeting of DMinC/DicB, but not DMinC/MinD, complexes to septal ring assemblies in Escherichia coli. J. Bacteriol. 186: 2418–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low H.H., Moncrieffe, M.C., and Lowe, J. 2004. The crystal structure of ZapA and its modulation of FtsZ polymerisation. J. Mol. Biol. 341: 839–852. [DOI] [PubMed] [Google Scholar]
- Lowe J. and Amos, L.A. 1998. Crystal structure of the bacterial cell-division protein FtsZ. Nature 391: 203–206. [DOI] [PubMed] [Google Scholar]
- Ma X. and Margolin, W. 1999. Genetic and functional analyses of the conserved C-terminal core domain of Escherichia coli FtsZ. J. Bacteriol. 181: 7531–7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin W. 2000. Themes and variations in prokaryotic cell division. FEMS Microbiol. Rev. 24: 531–548. [DOI] [PubMed] [Google Scholar]
- Miller J.H. 1992. A short course in bacterial genetics: A laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Morlot C., Noirclerc-Savoye, M., Zapun, A., Dideberg, O., and Vernet, T. 2004. The D,D-carboxypeptidase PBP3 organizes the division process of Streptococcus pneumoniae. Mol. Microbiol. 51: 1641–1648. [DOI] [PubMed] [Google Scholar]
- Mosyak L., Zhang, Y., Glasfeld, E., Haney, S., Stahl, M., Seehra, J., and Somers, W.S. 2000. The bacterial cell-division protein ZipA and its interaction with an FtsZ fragment revealed by X-ray crystallography. EMBO J. 19: 3179–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy F.J., Glasfeld, E., Mosyak, L., and Powers, R. 2000. Solution structure of ZipA, a crucial component of Escherichia coli cell division. Biochemistry 39: 9146–9156. [DOI] [PubMed] [Google Scholar]
- Mukherjee A., Dai, K., and Lutkenhaus, J. 1993. Escherichia coli cell division protein FtsZ is a guanine nucleotide binding protein. Proc. Natl. Acad. Sci. 90: 1053–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichoff S. and Lutkenhaus, J. 2002. Unique and overlapping roles for ZipA and FtsA in septal ring assembly in Escherichia coli. EMBO J. 21: 685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romberg L. and Levin, P.A. 2003. Assembly dynamics of the bacterial cell division protein FTSZ: Poised at the edge of stability. Annu. Rev. Microbiol. 57: 125–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Ent F. and Lowe, J. 2000. Crystal structure of the cell division protein FtsA from Thermotoga maritima. EMBO J. 19: 5300–5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helvoort J.M. and Woldringh, C.L. 1994. Nucleoid partitioning in Escherichia coli during steady-state growth and upon recovery from chloramphenicol treatment. Mol. Microbiol. 13: 577–583. [DOI] [PubMed] [Google Scholar]
- Wang X., Huang, J., Mukherjee, A., Cao, C., and Lutkenhaus, J. 1997. Analysis of the interaction of FtsZ with itself, GTP, and FtsA. J. Bacteriol. 179: 5551–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss D.S., Chen, J.C., Ghigo, J.M., Boyd, D., and Beckwith, J. 1999. Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J. Bacteriol. 181: 508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissel M.C. and Weiss, D.S. 2004. Genetic analysis of the cell division protein FtsI (PBP3): Amino acid substitutions that impair septal localization of FtsI and recruitment of FtsN. J. Bacteriol. 186: 490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan K., Pearce, K.H., and Payne, D.J. 2000. A conserved residue at the extreme C terminus of FtsZ is critical for the FtsA–FtsZ interaction in Staphylococcus aureus. Biochem. Biophys. Res. Commun. 270: 387–392. [DOI] [PubMed] [Google Scholar]