

Abstract
Exercise training confers sustainable protection against ischemia/reperfusion injury. However, the mechanism by which this process occurs is not fully understood. Previously, it was shown that β3-adrenergic receptors (β3-ARs) play a critical role in regulating the activation of endothelial nitric oxide synthase (eNOS) in response to exercise and play a critical role in exercise-mediated cardioprotection. Intriguingly, a deficiency in β3-ARs led to increased myocardial injury following exercise training. The purpose of the current study was to determine mechanisms by which β3-ARs are linked to eNOS activation and to determine the mechanism responsible for the exacerbated ischemia/reperfusion injury displayed by β3-AR deficient (β3-AR KO) mice after exercise training. Wild-type (n = 37) and β3-AR KO (n = 40) mice were subjected to voluntary wheel running for 4 weeks. Western blot analysis revealed that neither protein kinase B nor protein kinase A linked β3-ARs to eNOS following exercise training. However, analysis revealed a role for AMP-activated protein kinase (AMPK). Specifically, exercise training increased the phosphorylation of AMPK in the hearts of wild-type mice, but failed to do so in the hearts of β3-AR KO mice. Additional studies revealed that exercise training rendered eNOS less coupled and increased NOS-dependent superoxide levels in β3-AR KO mice. Finally, supplementing β3-AR KO mice with the eNOS coupler, tetrahydrobiopterin, during the final week of exercise training reduced myocardial infarction. These findings provide important information that exercise training protects the heart in the setting of myocardial ischemia/reperfusion injury by activating and coupling eNOS via the stimulation of a β3-AR-AMPK signaling pathway.
Keywords: heart, exercise, β3-adrenergic receptors, endothelial nitric oxide synthase, nitric oxide, AMP-activated protein kinase, myocardial ischemia/reperfusion injury, infarction
Introduction
Exercise remains one of preventive medicine's strongest therapeutic approaches, given that it reduces many risk factors related to cardiovascular disease (CVD),1 confers protection against myocardial infarction in animal models1,2,3,4,5 and improves survival following myocardial ischemia in humans.6,7 Despite the well-documented beneficial effects of exercise,8,9 the signaling mechanisms that mediate these actions have not been fully elucidated.10 Therefore, continued investigation into the unknown signaling mechanisms of exercise is extremely important given their enormous health care implications.11 Additionally, understanding how the cardiovascular system adapts to exercise is also important, because in regards to other strategies (i.e., pharmacological preconditioning), exercise appears to be unique in that it can elicit sustainable protection over the course of a long training period and provide protection for days after the cessation of the training period.11 As such, a better comprehension of the signaling cascades induced by exercise will provide the framework for developing therapeutic strategies designed to treat pathologies such as CVD.
β-adrenergic receptors (β-ARs) belong to a superfamily of G protein coupled-receptors and regulate cardiac structure and function in response to catecholamines.12,13 Three populations of β-ARs have been cloned and characterized: β1-ARs, β2-ARs, and β3-ARs.14 The effects of β1-ARs and β2-ARs are well established both in human and other mammals, as their stimulation produces positive chronotropic and inotropic effects.15,16 Additionally, targeting β1-ARs and β2-ARs with pharmacological inhibitors are well-established therapeutic strategies to treat patients with hypertension and heart failure. In regards to stimulation of the β3-AR in the cardiovascular system, the exact physiological and pathophysiological role is not fully understood. There is evidence, however, that β3-ARs have distinct physiological and pharmacological properties from β1-ARs and β2-ARs. For instance, the primary cardiovascular role for β3-ARs appears to be to act as a “brake” on the sympathetic nervous system.13 This is based on the evidence that β3-ARs are activated at high catecholamine concentrations and produce negative inotropic effects to oppose those of β1-ARs and β2-ARs.17,18 This has led to the emergence of the β3-AR as a potential target for the treatment of CVD.19 Specifically, studies using murine models of myocardial injury have reported that mice deficient in β3-ARs experience exacerbated remodeling to pressure-induced heart failure,20 whereas mice with a cardiac-specific overexpression of β3-ARs experience attenuated neurohormone-induced hypertrophic remodeling.21 Additionally, β3-AR stimulation with agonists ameliorates acute myocardial ischemia-reperfusion injury.22
The cardioprotective effects of β3-AR stimulation have been attributed to the activation of endothelial nitric oxide synthase (eNOS) and increase in nitric oxide (NO) bioavailability.13 As such, the localization of β3-ARs in the vascular endothelium and their function coupling to eNOS is of particular importance for cardiovascular homeostasis, given the physiological properties of NO. Recently a novel role for β3-ARs in exercise-mediated cardioprotection was discovered, as it was found that β3-ARs play a critical role in regulating the phosphorylation of eNOS and the generation of NO in response to exercise.23 More importantly it was shown that a deficiency in β3-ARs led to an increase in myocardial injury following exercise training. The exact mechanism by which β3-ARs are linked to eNOS activation is not completely understood. Moreover, the mechanism responsible for the exacerbated injury β3-AR deficient mice after exercise training is not known. The purpose of this study was to address the exact mechanism by which β3-ARs are linked to eNOS activation and the mechanism responsible for the exacerbated ischemia/reperfusion injury in β3-AR deficient (β3-AR KO) mice after exercise training.
Materials and Methods
Animals
Two strains of mice were utilized in this study: (1) Male C57BL6/J mice (Jackson Labs, Bar Harbor, ME, USA; 8–10 weeks of age; n = 37) and (2) male β3-AR KO mice (8-10 weeks of age; n = 40). The generation of β3-AR KO has been described previously.24 All experimental procedures were approved by the Institute for Animal Care and Use Committee at Emory University School of Medicine and conformed to the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health (NIH Publication No. 86-23, Revised 1996) and with federal and state regulations.
Exercise training
Mice were placed in custom designed cages fitted with running wheels (Mini Mitter, Bend, OR, USA) for a period up to 4 weeks. The mice were unrestricted to run as much as they could. Running distances were monitored daily. After the exercise-training period, the running wheel was removed from the cage and the mice were allowed to rest for a 24-hour period before further experimentation was conducted.
Myocardial ischemia-reperfusion protocol and myocardial injury assessment
Surgical ligation of the left coronary artery (LCA) and myocardial infarct size determination were performed similar to methods described previously.25
Western blot analysis
Whole cell homogenates and western blot analysis was performed as previously described.25 Immunoblots were probed with a Super Signal West Dura kit (Thermo Fisher, Waltham, MA, USA) to visualize signal, followed by exposure to X-ray film (Denville Scientific, Holliston, MA, USA). The film was scanned to make a digital copy and densitometric analysis was performed to calculate relative intensity with ImageJ software from the National Institutes of Health (version 1.40g) using the Rodbard function.
Immunoprecipitation
Heart homogenates were immunoprecipitated with an antibody to eNOS using the Dynabeads® Protein G Co-immunoprecipitation Kit (Thermo Fisher) according to manufacturer's instructions. The samples were then subjected to standard Western blot techniques and the membranes probed with antibodies to heat shock protein 90 (HSP90), AMP-activated protein kinase (AMPK), and eNOS.
eNOS monomer-dimer blots
eNOS monomers and dimers were evaluate using nonboiled lysates and low-temperature sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as described previously.26
Superoxide and nitric oxide synthase (NOS)-dependent superoxide production
Superoxide content was determined by lucigenin-enhanced luminescence as described previously.27 Parallel samples were incubated in the presence and absence of NG-nitro-L-arginine methyl ester (L-NAME). The difference in superoxide production between the two samples represents NOS-dependent production.
Tetrahydrobiopterin (BH4) intervention
BH4 was purchased from Sigma Aldrich (St. Louis, MO, USA). BH4 was added to the drinking water to provide an approximate dose of 10 mg/kg per day.28 To minimize oxidation of BH4 in the drinking water, 0.04% vitamin C was also added. This prevents BH4 oxidation for up to 24 hours,28 so the water was changed daily. As a vehicle control, all mice not receiving BH4 received only vitamin C in their drinking water.
Statistical analysis
All the data in this study are expressed as the mean ± SEM. Differences in data between groups were compared using Prism 5 (GraphPad Software Inc., San Diego, CA, USA) with one-way analysis of variance (ANOVA), or two-way ANOVA. For the one-way ANOVA, if a significant variance was found, the Tukey test was used as the post hoc analysis. When we compared data between the wild-type and β3-AR KO mice under sedentary and exercise settings, we used a 2-way non-repeated measures ANOVA with a Bonferroni test as the posthoc analysis. In all cases, a P value less than 0.05 was considered statistically significant and P-values were two-sided. Sample size estimates were calculated using G*Power software (version 3.1.9.2; Heinrich-Heine University of Dusseldorf, Dusseldorf, Germany).
Results
β3-AR does not regulate eNOS via protein kinase B (Akt) in response to exercise.
Initial studies focused on the contribution of Akt. In response to exercise training, the phosphorylation of Akt was significantly increased in the hearts of both Wild-type and β3-AR KO mice when compared to hearts collected from their respective sedentary controls (Figure 1A, B). Similarly, the expression of phosphorylated eNOS at serine residue 617 (p-eNOSSer617) was equally increased in both strains following exercise training (Figure 1C, D). Together this data suggests that the activation of cardiac Akt in response exercise training is not dependent on the β3-AR.
Figure 1.
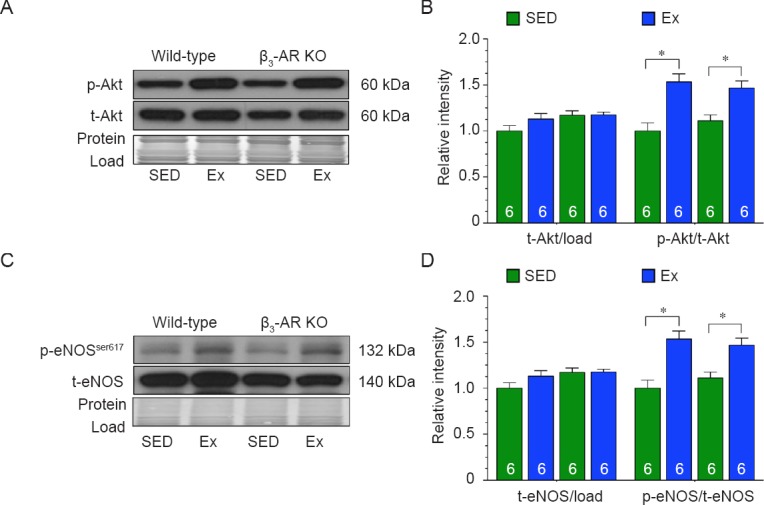
β3-adrenergic receptor (β3-AR) does not regulate endothelial nitric oxide synthase (eNOS) via protein kinase B (Akt) in response to exercise.
Note: (A, B) Immunoblots and quantitative analysis of phosphorylated and total Akt. (C, D) Immunoblots and quantitative analysis of phosphorylated eNOS at serine residue 617 (p-eNOSSer617) and total eNOS. The results of target protein were expressed as optical density ratio to sedentary Wild-type mice. Numbers inside bars indicates sample size. Values are expressed as the mean ± SEM. SED: Sedentary; Ex: exercise. *P < 0.05.
β3-AR does not regulate eNOS via protein kinase A (PKA) in response to exercise
Next, we evaluated the role of PKA signaling. In response to exercise training, the phosphorylation of cyclic adenosine monophosphate response element binding protein (CREB - a target of PKA) was significantly increased in the hearts of both Wild-type and β3-AR KO mice when compared to hearts collected from their respective sedentary controls (Figure 2A, B). Similarly, the expression of phosphorylated eNOS at serine residue 635 (p-eNOSSer635) was equally increased in both strains following exercise training (Figure 2C, D). Together this data suggests that the activation of cardiac PKA in response exercise training is not dependent on the β3-AR.
Figure 2.
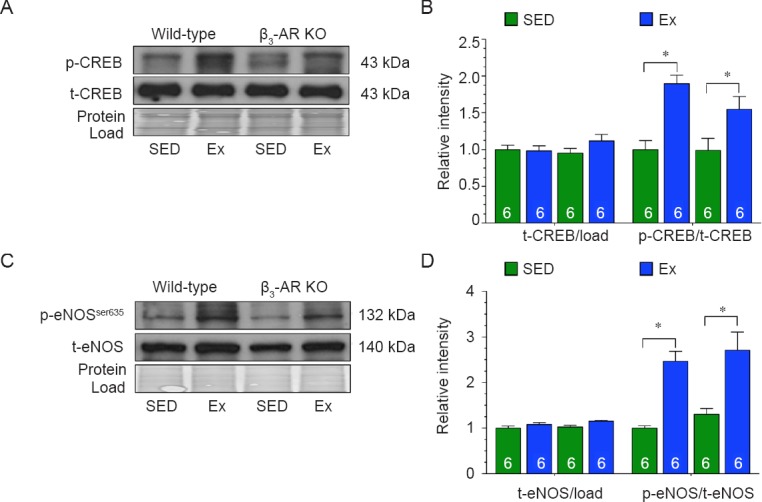
β3-adrenergic receptor(β3-AR) does not regulate endothelial nitric oxide synthase (eNOS) via protein kinase A (PKA) in response to exercise.
Note: (A, B) Immunoblots and quantitative analysis of phosphorylated and total CREB. (C, D) Immunoblots and quantitative analysis of phosphorylated eNOS at serine residue 635 (p-eNOSSer635) and total eNOS. The results of target protein were expressed as optical density ratio to sedentary Wild-type mice. Numbers inside bars indicates sample size. Values are expressed the mean ± SEM. SED: Sedentary; Ex: exercise; CREB: cyclic adenosine monophosphate response element binding protein. *P < 0.05, ***P < 0.001.
β3-AR regulates eNOS via AMPK in response to exercise
Next, we evaluated the role of AMPK signaling. In response to exercise training, the phosphorylation of AMPK was significantly increased in the hearts of Wild-type mice when compared to hearts collected from Wild-type sedentary control mice (Figure 3A, B). In contrast, exercise training failed to increase the phosphorylation of AMPK in the hearts of β3-AR KO mice. Similarly, the expression of phosphorylated eNOS at serine residue 1177 (p-eNOSSer1177) was only increased in the hearts of Wild-type mice following exercise training (Figure 3C, D).
Figure 3.
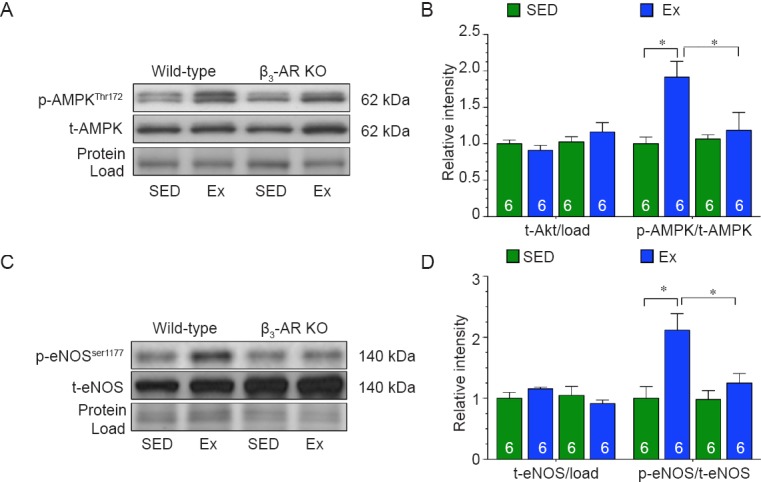
β3-adrenergic receptor (β3-AR) regulates endothelial nitric oxide synthase (eNOS) via AMP-activated protein kinase (AMPK) in response to exercise.
Note: (A, B) Immunoblots and quantitative analysis of phosphorylated and total AMPK. (C, D) Immunoblots and quantitative analysis of phosphorylated eNOS at serine residue 1177(p-eNOSser1177) and total eNOS. The results of target protein were expressed as optical density ratio to sedentary Wild-type mice. Numbers inside bars indicates sample size. Values are expressed as the mean ± SED. SED: Sedentary; Ex: exercise. *P < 0.05, ***P < 0.01.
β3-AR couples eNOS in response to exercise
eNOS can exist in two states: (1) coupled as a dimer, where it produces NO, and (2) uncoupled as a monomer, where it produces superoxide.29 In addition to its ability to alter the phosphorylation status of eNOS, AMPK can also influence the coupling of eNOS by promoting its interaction with HSP90.30,31 We found that exercise training did not alter the expression of HSP90 in the hearts of either strain (Figure 4A, B). Exercise training did however induce the interaction between HSP90, AMPK and eNOS in the hearts of Wild-type mice (Figure 4C, D). In contrast, exercise training did not induce the interaction between HSP90, AMPK, and eNOS in the hearts of β3-AR KO mice. Further analysis revealed that eNOS existed more in the monomeric form (uncoupled) in the hearts of β3-AR KO mice following exercise training (Figure 5A, B). This was associated with an increase in the production of total and NOS-dependent superoxide (Figure 5C, D). Together, this data suggest that exercise induces the uncoupling of eNOS in the absence of β3-ARs.
Figure 4.
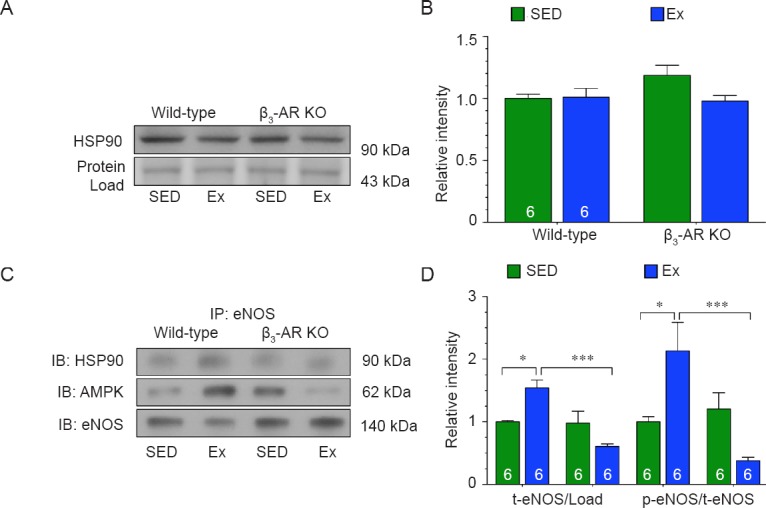
β3-adrenergic receptor (β3-AR) couples endothelial nitric oxide synthase (eNOS) via AMP-activated protein kinase (AMPK) in response to exercise.
Note: (A, B) Immunoblots and densitometric analysis of heat shock protein 90 (HSP90). (C, D) Immunoblots and densitometric analysis of the interaction between HSP90, AMPK, and eNOS. Numbers inside bars indicates sample size. The results of target protein were expressed as optical density ratio to sedentary Wild-type mice. Values are expressed as the mean ± SEM. SED: Sedentary; Ex: exercise. *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 5.
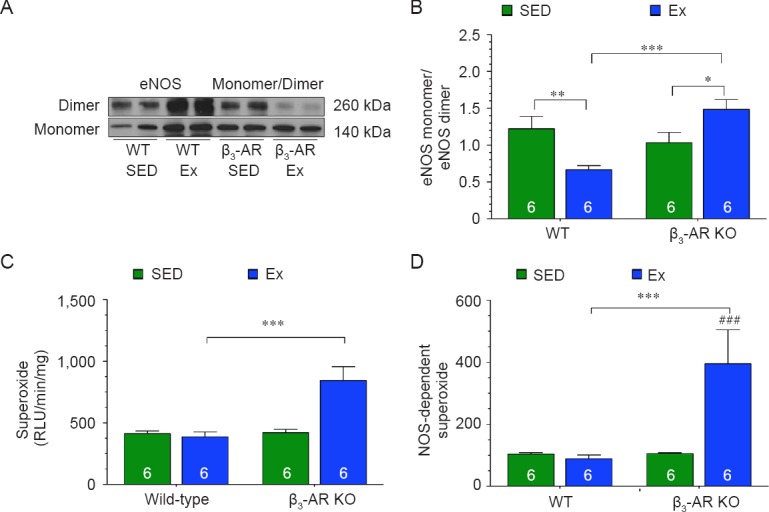
Endothelial nitric oxide synthase (eNOS) exists more in the monomeric form in the hearts of mice following exercise training.
Note: (A) Immunoblots of eNOS dimers and monomers. (B) Ratio of eNOS monomer to dimer. (C, D) Total superoxide (C) and nitric oxide synthase-dependent superoxide (D) production rates. Numbers inside bars indicates sample size. Values are expressed as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. ###P < 0.001, vs. β3-AR KO mice (β3-AR). SED: Sedentary; Ex: exercise; RLU: relative light unit; WT: Wild-type mice.
Finally, we sought to determine if the uncoupling of eNOS contributed to the exacerbated myocardial ischemia-reperfusion injury observed in β3-AR KO mice subjected to exercise training. For these experiments, Wild-type and β3-AR KO mice were allowed to exercise voluntarily for 4 weeks. A subset of β3-AR KO mice was given the eNOS coupler, BH4, in their drinking water after 3 weeks of exercise training. This supplementation continued for a week (Figure 6A). After the training period, the exercised mice along with sedentary controls were subjected to 45 minutes of myocardial ischemia followed by 24 hours of reperfusion. Exercise training decreased infarct size in Wild-type mice and increased infarct size in β3-AR KO mice when compared to their respective sedentary controls (Figure 6B). BH4 supplementation reduced infarct size in β3-AR KO subjected to exercise training to a size similar to that observed in sedentary mice.
Figure 6.

Uncoupling of endothelial nitric oxide synthase (eNOS) contributes to the exacerbated myocardial ischemia/reperfusion injury (MI/R).
Note: (A) Experimental protocol. (B, C) Myocardial area-at-risk (AAR) as a percentage (%) of total left ventricle (LV) (B) and infarct size (INF) as a percentage of AAR(C). Numbers inside bars indicates sample size. Values are expressed as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. SED: Sedentary; VE: voluntary exercise; BH4: tetrahydrobiopterin intervention; WT; Wild-type mice.
Discussion
When β3-ARs were first characterized, they were thought to only mediate metabolic effects in adipocytes. However, recent evidence noting the expression and functional coupling of β3-ARs in cardiovascular tissue, such as the heart and endothelium, has led to the re-examination of their physiological role. For instance, the primary cardiovascular role for β3-ARs appears to be to oppose those of β1- and β2-ARs. This suggests that β3-ARs may serve as endogenous “beta blockers”. Therefore, it is important to understand the consequences of β3-AR signaling in both health and disease. As noted, studies using murine models of myocardial injury have reported that mice deficient in β3-ARs experience exacerbated remodeling to pressure-induced heart failure,20 whereas mice with a cardiac-specific overexpression of β3-ARs experience attenuated neurohormone-induced hypertrophic remodeling.21 Additionally, β3-AR stimulation with agonists ameliorates acute myocardial ischemia-reperfusion injury.22 Together this data indicates that stimulation of the β3-AR is cytoprotective in the setting of cardiac injury. This data also promotes the use and development of selective β-blockers such as Nebivolol22 that activate the β3-AR and antagonize the β1-AR and β2-AR. Previous findings23 and the findings for the current study expand on this earlier data and provide a physiological context for the stimulation of the β3-AR.
In a previous study, it was discovered that β3-ARs play a critical role in regulating the phosphorylation of eNOS and the generation of NO in response to exercise.23 However, a mechanism was not explored. Moreover, the exact mechanism by which β3-ARs in general are linked to eNOS activation is not completely understood. There is some evidence from in vitro experiments with endothelial cells to suggest that stimulation of the β3-AR activates eNOS via Akt.32 Additionally, it is widely accepted that increases in blood flow across the endothelial cell surface are potent stimuli for the production of NO from eNOS.33 Studies using cultured endothelial cells and isolated blood vessels suggest that shear stress induces a signaling cascade involving Akt, PKA and/or AMPK.34,35,36 Importantly, these kinases appear to act independently of each other, suggesting that if one pathway is compromised the other two can still increase NO.34 There is also evidence that epinephrine leads to the activation of Akt, PKA, and AMPK under certain conditions.32,37 Given that epinephrine stimulates β3-ARs and is increased in response to exercise,23 a main purpose of this study was to determine the contribution of each kinase in linking β3-AR signaling to eNOS phosphorylation in response to exercise training. Our studies revealed that the exercise-induced phosphorylation of AMPK was attenuated in the hearts of β3-AR KO mice, whereas the exercise-induced phosphorylation of Akt and CREB remained intact. Further, using phosphorylation site-specific antibodies to eNOS, we found that exercise increased the p-eNOSSer617 and p-eNOSSer635 in the absence of the β3-AR, but failed to alter the p-eNOSSer1177. Given our findings related to Akt, PKA, and AMPK and the evidence that the p-eNOSSer1177 is influenced by all three kinases, we suggest that AMPK is the main regulator of cardiac p-eNOSSer1177 in this mouse model of exercise.
An intriguing finding of the previous study was that β3-AR KO mice subjected to exercise training displayed exacerbated myocardial injury following an ischemic insult.23 It was predicated that exercise would fail to provide cardioprotection in the absence of the β3-AR, not that exercise would induce injury in its absence. A review of the literature suggested that β3-AR signaling not only linked to the phosphorylation of eNOS, but also to the coupling of eNOS.20 Specifically, the study by Moens and colleagues20 suggested that the lack of β3-AR signaling exacerbates cardiac pressure-overload-induced remodeling by enhancing eNOS uncoupling. eNOS uncoupling is a term used to refer to the condition where eNOS exists predominately in a monomeric form.29 In this state, eNOS transfers electrons to oxygen instead of L-arginine resulting in the production of superoxide instead of NO.38 This not only results in a decline in NO bioavailability, but also contributes to the development of oxidative and nitrosative stress. Therefore, the coupling of eNOS is essential to its proper function. Several years ago, HSP90 was shown to be a physiological binding partner and regulator of eNOS activity and NO production.39 Subsequent studies demonstrated that HSP90 bound to eNOS in a manner that promoted eNOS coupling.30,40 Conversely, the prevention of this interaction was shown to promote eNOS uncoupling and superoxide production from eNOS. More recently, it was shown that AMPK influenced the coupling of eNOS by promoting its interaction with HSP90.31 Here, we provide the first evidence that in the absence of β3-AR, the interaction of eNOS, HSP90, and AMPK was lost in response to exercise training. This was associated with an increase in the monomeric form of eNOS and the production of NOS-dependent superoxide. Finally, we found that this uncoupling and subsequent superoxide production was responsible for the enhanced injury observed in the hearts of β3-AR KO mice following exercise training. Together this suggests that β3-AR-AMPK signaling is critical for the regulation of eNOS activation and NO production in the setting of exercise training. As such, this evidence provides important insights into the signaling mechanism that links the physiological stimuli of exercise to the coupling and activation of eNOS.
There are a few caveats that need to be noted. First, there is some evidence that stimulation of the β3-AR activates eNOS via Akt.32 Our findings do not support this signaling cascade, at least in the context of cardiac β3-AR stimulation and exercise training. The previous study used an in vitro model to evaluate β3-AR signaling as it pertains to eNOS in cultured endothelial cells.32 Our study examined β3-AR-eNOS signaling in hearts taken from Wild-type and β3-AR KO mice subjected to voluntary exercise training. Therefore, the different results could be attributed to the tissue examined or the experimental approach. As such, further work is needed to examine the mechanisms by which β3-AR stimulation regulates eNOS in different tissue (i.e., heart vs. vasculature). Second, the exact mechanism by which AMPK facilitates the interaction of HSP90 with eNOS is not fully understood. Schulz et al.31 suggested that the chaperone function of HSP90 could be regulated by AMPK. However, evidence for this idea has not been tested. Therefore, further studies are needed to determine if this postulate is correct or if other mechanisms are in play.
In summary, the current study provides important information that exercise training protects the heart in the setting of myocardial ischemia/reperfusion injury by activating and coupling eNOS via the stimulation of a β3-AR-AMPK signaling pathway. Future research will be aimed at further elucidation of the signaling mechanisms, which couple the β3-AR to AMPK and eNOS.
Footnotes
Funding: This work was supported by funding from the Carlyle Fraser Heart Center of Emory University Hospital Midtown, USA.
Conflicts of interest
None.
Plagiarism check
This paper was screened twice using CrossCheck to verify originality before publication.
Peer review
This paper was double-blinded and stringently reviewed by international expert reviewers.
References
- 1.Brown DA, Jew KN, Sparagna GC, Musch TI, Moore RL. Exercise training preserves coronary flow and reduces infarct size after ischemia-reperfusion in rat heart. J Appl Physiol. 2003;95:2510–2518. doi: 10.1152/japplphysiol.00487.2003. [DOI] [PubMed] [Google Scholar]
- 2.Chicco AJ, Johnson MS, Armstrong CJ, et al. Sex-specific and exercise-acquired cardioprotection is abolished by sarcolemmal KATP channel blockade in the rat heart. Am J Physiol Heart Circ Physiol. 2007;292:H2432–2437. doi: 10.1152/ajpheart.01301.2006. [DOI] [PubMed] [Google Scholar]
- 3.Akita Y, Otani H, Matsuhisa S, et al. Exercise-induced activation of cardiac sympathetic nerve triggers cardioprotection via redox-sensitive activation of enos and upregulation of inos. Am J Physiol Heart Circ Physiol. 2007;292:H2051–2059. doi: 10.1152/ajpheart.01102.2006. [DOI] [PubMed] [Google Scholar]
- 4.Brown DA, Chicco AJ, Jew KN, et al. Cardioprotection afforded by chronic exercise is mediated by the sarcolemmal, and not the mitochondrial, isoform of the katp channel in the rat. J Physiol. 2005;569:913–924. doi: 10.1113/jphysiol.2005.095729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freimann S, Scheinowitz M, Yekutieli D, Feinberg MS, Eldar M, Kessler-Icekson G. Prior exercise training improves the outcome of acute myocardial infarction in the rat. heart structure, function, and gene expression. J Am Coll Cardiol. 2005;45:931–938. doi: 10.1016/j.jacc.2004.11.052. [DOI] [PubMed] [Google Scholar]
- 6.Hull SS, Jr, Vanoli E, Adamson PB, Verrier RL, Foreman RD, Schwartz PJ. Exercise training confers anticipatory protection from sudden death during acute myocardial ischemia. Circulation. 1994;89:548–552. doi: 10.1161/01.cir.89.2.548. [DOI] [PubMed] [Google Scholar]
- 7.Morris JN, Everitt MG, Pollard R, Chave SP, Semmence AM. Vigorous exercise in leisure-time: protection against coronary heart disease. Lancet. 1980;2:1207–1210. doi: 10.1016/s0140-6736(80)92476-9. [DOI] [PubMed] [Google Scholar]
- 8.Powers SK, Lennon SL, Quindry J, Mehta JL. Exercise and cardioprotection. Curr Opin Cardiol. 2002;17:495–502. doi: 10.1097/00001573-200209000-00009. [DOI] [PubMed] [Google Scholar]
- 9.Shephard RJ, Balady GJ. Exercise as cardiovascular therapy. Circulation. 1999;99:963–972. doi: 10.1161/01.cir.99.7.963. [DOI] [PubMed] [Google Scholar]
- 10.Bostrom P, Mann N, Wu J, et al. C/Ebpbeta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell. 2010;143:1072–1083. doi: 10.1016/j.cell.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calvert JW, Lefer DJ. Role of beta-adrenergic receptors and nitric oxide signaling in exercise-mediated cardioprotection. Physiology. 2013;28:216–224. doi: 10.1152/physiol.00011.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Germack R, Dickenson JM. Induction of beta3-adrenergic receptor functional expression following chronic stimulation with noradrenaline in neonatal rat cardiomyocytes. J Pharmacol Exp Ther. 2006;316:392–402. doi: 10.1124/jpet.105.090597. [DOI] [PubMed] [Google Scholar]
- 13.Moens AL, Yang R, Watts VL, Barouch LA. Beta 3-adrenoreceptor regulation of nitric oxide in the cardiovascular system. J Mol Cell Cardiol. 2010;48:1088–1095. doi: 10.1016/j.yjmcc.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tavernier G, Toumaniantz G, Erfanian M, et al. Beta3-adrenergic stimulation produces a decrease of cardiac contractility ex vivo in mice overexpressing the human beta3-adrenergic receptor. Cardiovasc Res. 2003;59:288–296. doi: 10.1016/s0008-6363(03)00359-6. [DOI] [PubMed] [Google Scholar]
- 15.Steinberg SF. The molecular basis for distinct beta-adrenergic receptor subtype actions in cardiomyocytes. Circ Res. 1999;85:1101–1111. doi: 10.1161/01.res.85.11.1101. [DOI] [PubMed] [Google Scholar]
- 16.Dessy C, Balligand JL. Beta3-adrenergic receptors in cardiac and vascular tissues emerging concepts and therapeutic perspectives. Adv Pharmacol. 2010;59:135–163. doi: 10.1016/S1054-3589(10)59005-7. [DOI] [PubMed] [Google Scholar]
- 17.Gauthier C, Leblais V, Kobzik L, et al. The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J Clin Invest. 1998;102:1377–1384. doi: 10.1172/JCI2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitamura T, Onishi K, Dohi K, Okinaka T, Isaka N, Nakano T. The negative inotropic effect of beta3-adrenoceptor stimulation in the beating guinea pig heart. J Cardiovasc Pharmacol. 2000;35:786–790. doi: 10.1097/00005344-200005000-00016. [DOI] [PubMed] [Google Scholar]
- 19.Rozec B, Gauthier C. Beta3-adrenoceptors in the cardiovascular system: putative roles in human pathologies. Pharmacol Ther. 2006;111:652–673. doi: 10.1016/j.pharmthera.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Moens AL, Leyton-Mange JS, Niu X, et al. Adverse ventricular remodeling and exacerbated nos uncoupling from pressure-overload in mice lacking the beta3-adrenoreceptor. J Mol Cell Cardiol. 2009;47:576–585. doi: 10.1016/j.yjmcc.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belge C, Hammond J, Dubois-Deruy E, et al. Enhanced expression of beta3-adrenoceptors in cardiac myocytes attenuates neurohormone-induced hypertrophic remodeling through nitric oxide synthase. Circulation. 2014;129:451–462. doi: 10.1161/CIRCULATIONAHA.113.004940. [DOI] [PubMed] [Google Scholar]
- 22.Aragon JP, Condit ME, Bhushan S, et al. Beta3-adrenoreceptor stimulation ameliorates myocardial ischemia-reperfusion injury via endothelial nitric oxide synthase and neuronal nitric oxide synthase activation. J Am Coll Cardiol. 2011;58:2683–2691. doi: 10.1016/j.jacc.2011.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calvert JW, Condit ME, Aragon JP, et al. Exercise protects against myocardial ischemia-reperfusion injury via stimulation of beta(3)-adrenergic receptors and increased nitric oxide signaling: role of nitrite and nitrosothiols. Circ Res. 2011;108:1448–1458. doi: 10.1161/CIRCRESAHA.111.241117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Susulic VS, Frederich RC, Lawitts J, et al. Targeted disruption of the beta 3-adrenergic receptor gene. J Biol Chem. 1995;270:29483–29492. doi: 10.1074/jbc.270.49.29483. [DOI] [PubMed] [Google Scholar]
- 25.Shimizu Y, Lambert JP, Nicholson CK, et al. DJ-1 protects the heart against ischemia-reperfusion injury by regulating mitochondrial fission. J Mol Cell Cardiol. 2016;97:56–66. doi: 10.1016/j.yjmcc.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Widder JD, Chen W, Li L, et al. Regulation of tetrahydrobiopterin biosynthesis by shear stress. Circ Res. 2007;101:830–838. doi: 10.1161/CIRCRESAHA.107.153809. [DOI] [PubMed] [Google Scholar]
- 27.Tao L, Gao E, Jiao X, et al. Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation. 2007;115:1408–1416. doi: 10.1161/CIRCULATIONAHA.106.666941. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Chen W, Rezvan A, Jo H, Harrison DG. Tetrahydrobiopterin deficiency and nitric oxide synthase uncoupling contribute to atherosclerosis induced by disturbed flow. Arterioscler Thromb Vasc Biol. 2011;31:1547–1554. doi: 10.1161/ATVBAHA.111.226456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elrod JW, Duranski MR, Langston W, et al. Enos gene therapy exacerbates hepatic ischemia-reperfusion injury in diabetes: A Role For Enos Uncoupling. Circ Res. 2006;99:78–85. doi: 10.1161/01.RES.0000231306.03510.77. [DOI] [PubMed] [Google Scholar]
- 30.Sud N, Sharma S, Wiseman DA, et al. Nitric oxide and superoxide generation from endothelial NOS: modulation by HSP90. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1444–1453. doi: 10.1152/ajplung.00175.2007. [DOI] [PubMed] [Google Scholar]
- 31.Schulz E, Anter E, Zou MH, Keaney JF., Jr Estradiol-mediated endothelial nitric oxide synthase association with heat shock protein 90 requires adenosine monophosphate-dependent protein kinase. Circulation. 2005;111:3473–3480. doi: 10.1161/CIRCULATIONAHA.105.546812. [DOI] [PubMed] [Google Scholar]
- 32.Kou R, Michel T. Epinephrine regulation of the endothelial nitric-oxide synthase: roles of RAC1 and beta3-adrenergic receptors in endothelial NO signaling. J Biol Chem. 2007;282:32719–32729. doi: 10.1074/jbc.M706815200. [DOI] [PubMed] [Google Scholar]
- 33.Jo H, Sipos K, Go YM, Law R, Rong J, Mcdonald JM. Differential effect of shear stress on extracellular signal-regulated kinase and N-terminal jun kinase in endothelial cells. Gi2- and Gbeta/gamma-dependent signaling pathways. J Biol Chem. 1997;272:1395–1401. doi: 10.1074/jbc.272.2.1395. [DOI] [PubMed] [Google Scholar]
- 34.Zhang QJ, Mcmillin SL, Tanner JM, Palionyte M, Abel ED, Symons JD. endothelial nitric oxide synthase phosphorylation in treadmill-running mice: role of vascular signalling kinases. J Physiol. 2009;587:3911–3920. doi: 10.1113/jphysiol.2009.172916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dimmeler S, Assmus B, Hermann C, Haendeler J, Zeiher AM. Fluid shear stress stimulates phosphorylation of Akt in human endothelial cells: involvement in suppression of apoptosis. Circ Res. 1998;83:334–341. doi: 10.1161/01.res.83.3.334. [DOI] [PubMed] [Google Scholar]
- 36.Iwasawa E, Ichijo M, Ishibashi S, Yokota T. Acute development of collateral circulation and therapeutic prospects in ischemic stroke. Neural Regen Res. 2016;11:368–371. doi: 10.4103/1673-5374.179033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koh HJ, Hirshman MF, He H, et al. Adrenaline is a critical mediator of acute exercise-induced AMP-activated protein kinase activation in adipocytes. Biochem J. 2007;403:473–481. doi: 10.1042/BJ20061479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sessa WC. Enos at a glance. J Cell Sci. 2004;117:2427–2429. doi: 10.1242/jcs.01165. [DOI] [PubMed] [Google Scholar]
- 39.Garcia-Cardena G, Fan R, Shah V, et al. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821–824. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- 40.Pritchard KA, Jr, Ackerman AW, Gross ER, et al. Heat shock protein 90 mediates the balance of nitric oxide and superoxide anion from endothelial nitric-oxide synthase. J Biol Chem. 2001;276:17621–17624. doi: 10.1074/jbc.C100084200. [DOI] [PubMed] [Google Scholar]