

Abstract
Carbon monoxide (CO) formed endogenously is considered to be cytoprotective, and the vast majority of CO formation is attributed to the degradation of heme by heme oxygenases-1 and -2 (HO-1, HO-2). Previously, we observed that brain microsomes containing HO-2 produced many-fold more CO in the presence of menadione and its congeners; herein we explored these observations further. We determined the effects of various drugs on CO production of rat brain microsomes and recombinant human cytochrome P450 reductase (CPR); CO was measured by gas chromatography with reductive detection. Brain microsomes of Sprague-Dawley rats or recombinant human cytochrome P450 reductase (CPR) were incubated with NADPH and various drugs in closed vials in phosphate buffer at pH 7.4 and 37°C. After 15 minutes, the reaction was stopped by cooling in dry ice, and the headspace gas was analyzed for CO production using gas chromatography with reductive (mercuric oxide) detection. We observed drug-enhanced CO production in the presence of both microsomes and recombinant CPR alone; the presence of HO was not required. A range of structurally diverse drugs were capable of amplifying this CO formation; these molecules had structures consistent with redox cycling capability. The addition of catalase to a reaction mixture, that contained activating drugs, inhibited the production of CO. Drug-enhanced CO formation can be catalyzed by CPR. The mechanism of CPR activation was not through classical drug-receptor mediation. Redox cycling may be involved in the drug-induced amplification of CO production by CPR through the production of reactive oxygen species.
Keywords: carbon monoxide, cytochrome P450 reductase, enzyme activation, menadione, heme, NADPH, brain microsomes, redox
Introduction
Heme is an essential component of several key molecules in animals, and is necessary for oxygen transport, electron transfer and sensing oxygen. Nevertheless, it is often seen as a double-edged sword–necessary for life, but toxic if left uncontrolled and free to interact with a variety of other molecules in ways that are deleterious to an organism. Heme (ferroprotoporphyrin)/hemin (ferriprotoporphyrin) may generate reactive oxygen species (ROS), induce hemolysis and promote inflammation as reviewed by Kumar and Bandyopadhyay.1 Cells mitigate heme toxicity by minimizing the availability of free heme for such detrimental activities by ensuring that the vast majority of the heme present is sequestered in hemoproteins, and by the catabolism of free heme by heme oxygenase (HO). HO catalyzes the degradation of heme into three products–ferrous iron, carbon monoxide (CO) and biliverdin; the latter is rapidly converted to bilirubin by biliverdin reductase. HO is considered to be cytoprotective because it eliminates a prooxidant (heme) while generating biliverdin/bilirubin and CO, which have anti-oxidant, anti-apoptotic and anti-inflammatory properties. Much of the progress in understanding the physiology and pathology of HO has been garnered through the use of research tools such as genetic variants and genetically engineered animal models as well as drugs that induce HO expression or inhibit HO activity.
Our laboratory has contributed to the pharmacology of HO inhibitors through the design of non-porphyrin, second-generation HO inhibitors, which differ in both mechanism of action and isozyme selectivity from the first-generation porphyrin-based HO inhibitors. We have also revealed the structural basis of the inhibition mechanism through structure determination of HO-1 in complex with various inhibitors.2 Our assay of HO activity has been based on the in vitro incubation of the heme substrate aerobically in the presence of NADPH, HO and its companion enzyme, cytochrome P450 reductase (CPR, EC 1.6.2.4, NADPH-hemoprotein reductase); the latter two were often used in their native forms as spleen microsomes for HO-1 (inducible)3 and brain microsomes for HO-2 (constitutive).4 Our endpoint was the determination of CO in the headspace above the reaction mixture. In the course of screening molecular skeletons for candidates as novel HO inhibitors, we observed that menadione (MD) and some other drugs enhanced the production of CO by incubation mixtures containing rat brain microsomes, as a source of HO-2. This was interpreted to represent the activation of HO-2 as described earlier.5,6
In seeking to understand how brain microsomes might be activated by MD and other compounds to produce copious quantities of CO, we were reminded that the often-overlooked companion enzyme, cytochrome P450 reductase (CPR), could do more than simply support other enzymes such as HO and P450. Accordingly, in their review, Riddick et al.7 described a role for CPR in donating electrons to cytochrome b5, squalene monoxygenase and a number of bioreductive prodrugs. As early as 1978, Guengerich8 showed that purified, rat liver CPR could destroy heme, when added to the reaction mixture in the form of methemalbumin (MHA), free hemin or P450. Thus, although the HOs are widely considered to be the degradation enzymes for heme, there is longstanding evidence that CPR per se shares this ability to some extent. Despite this early observation, CPR-catalyzed heme degradation has not been fully characterized using purified recombinant CPR derivatives, an important concern given that CPR is often associated with other hemoproteins that could co-purify. Herein, we address the hypothesis that CPR mediates the drug-induced activation of CO production from heme in rat brain microsomes.
The present observations indicate that there exists a group of compounds including, but not restricted to, MD, that can dramatically increase production of CO from MHA by CPR alone; furthermore, the magnitude of this elevated CO production in brain microsomes can be many times higher than that of HO-mediated CO production.
Materials and Methods
Preparation of rat brain microsomes
This was conducted as described previously.6 Briefly, rat brains of Sprague-Dawley rats (male from Charles River, Montreal, Canada, n = 20, 8–10 weeks of age) were homogenized in buffer and the microsomal pellet was obtained by differential centrifugation. After re-suspension the microsome fraction was stored at –80°C, and thawed as needed for assays.
Enzymatic assay
NADPH-dependent CO production activity was determined by quantifying headspace CO as described by Vukomanovic et al.6 In brief, a reaction mixture (150 μL) containing 100 mM phosphate buffer (pH 7.4), 50 μM MHA and enzymes (as microsomes or pure recombinant forms) was incubated with drugs at the concentrations reported in Results. The reaction was initiated by adding NADPH (Sigma-Aldrich, Toronto, Canada) to a final concentration of 1.0 mM, and allowed to proceed for 15 minutes at 37°C. The reaction was stopped by instantly freezing the reaction mixture on dry ice, and the CO generated was determined using the method of Vreman et al.,9 which is sensitive and selective for CO by virtue of headspace sampling, size exclusion gas chromatography and mercuric oxide reduction detection.
Preparation of recombinant enzymes
For our earlier experiments, purified recombinant CPR was obtained from Becton Dickinson Canada Inc., Toronto, ON, Canada; for later experiments recombinant CPR was prepared as follows. The hCPR-pET22b vector encoding full-length human CPR was a generous gift from Dr. Paul Ortiz de Montellano (University of San Francisco). Protocols for expression and purification were based on those previously described.10 The plasmid was transformed into BL21 (DE3) pLysS cells for expression in terrific broth (TB) supplemented with trace elements and riboflavin in the presence of ampicillin and chloramphenicol. Expression was induced with 0.4 M isopropyl-β-D-thiogalactopyranoside (IPTG) when the absorbance (A) at 600 nm had reached ~0.4, and the cells allowed to grow further overnight at 30°C. Cells were harvested and spheroplasts generated prior to lysis by sonication. Lysates were centrifuged at 10,000 r/min (Ti45 rotor) and the resultant supernatant subjected to centrifugation at 35,000 r/min (Ti45 rotor) to pellet the membrane fraction which was subsequently stored at –80°C for future purification.
Thawed membrane pellets were solubilized in 20 mM potassium phosphate (pH 7.4), 20% (v/v) glycerol, 0.2% Triton X-100, 0.1 mM EDTA, 1 mM PMSF, 0.2% sodium cholate with one Roche EDTA-free protease cocktail tablet. After clarification of the solubilized membranes, the supernatant was purified over a 2′5′-ADP-Sepharose column, which had been equilibrated with Buffer A (20 mM potassium phosphate (pH 7.4), 20% glycerol, 0.1 mM EDTA, 0.2% sodium cholate). The column was washed with Buffer A, followed by washes in the presence of 250 mM NaCl, and in the presence of 2 mM adenosine. The hCPR protein was eluted in the presence of 5 mM 2’AMP. Purified protein was dialyzed against 40 mM potassium phosphate, 20% glycerol overnight at 4°C, concentrated, and quantified by Bradford assay using bovine serum albumin (Sigma-Aldrich) as a standard. Protein was aliquoted, flash-frozen in liquid nitrogen and stored at –80°C.
Full-length human HO-2 (FL-hHO-2) was prepared as described previously.5 Briefly the recombinant protein was produced in BL21 (DE3) cells, transformed with the FL-hHO-2/pET28a plasmid. Cell pellets were harvested and stored at –80°C prior to purification. The protein was solubilized with n-dodecyl β-D-maltopyranoside then centrifuged and the supernatant was subjected to nickel affinity chromatography. Following dialysis, heme was conjugated to a final molar ratio of 2:1 and the enzyme was subjected to size-exclusion chromatography over an S200 column. Protein concentration was determined by A (ε405 = 171.4 ± 1.2 mM–1 cm–1).11 Purity was assessed by measurement of the ratio (A405/A280 > 2.1) and by SDS-PAGE analysis.
Statistical analysis
Data were expressed as the mean ± SD. Means were compared by Student's t-test. A P value less than 0.05 was considered to be significant.
Results
MD-enhanced CO production by rat brain microsomes
The production of CO from heme by a reaction mixture containing brain microsomes (a constitutively rich source of HO-2) was determined in the presence of increasing amounts of MD (Sigma-Aldrich) from 0.01 to 100 μM (Figure 1). The basal activity of microsomes in the absence of MD is considered to represent CO formation by the membrane bound HO-2. The addition of MD resulted in a significant increase in the production of CO relative to the basal rate (P < 0.05), with the maximal concentration of 100 μM resulting in an eight-fold increase to ~800 pmol produced. A MD concentration of ~0.1 μM appeared to be the threshold for enhancing CO production. In the presence of the HO inhibitor, QC-321, baseline production of CO was blocked (Figure 1) resulting in decreased CO production at the lowest concentrations of MD (P < 0.05); nevertheless, its presence did not prevent the enhancement of CO production at MD concentrations over 1 μM.
Figure 1.
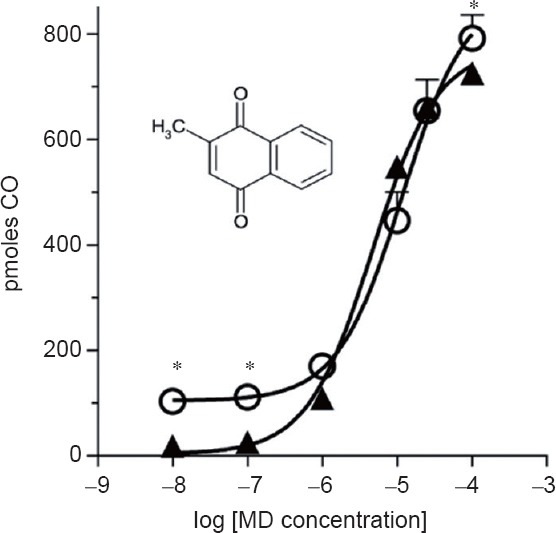
Amplification of carbon monoxide (CO) production by rat brain microsomes by menadione (MD).
Note: The open circles show the CO production when reaction mixture contained 50 μM methemalbumin (MHA), 1 mg microsomal protein and MD as indicated on the abscissa in 100 mM phosphate buffer (pH 7.4). The solid triangles show the effect of 25 μM QC-321 (heme oxygenase inhibitor) in addition to MHA, microsomes and MD. Bars represent SD, where bars are not shown the SD is smaller than the symbol; n = 4. Significantly differences, *P < 0.05. The inset shows the structure of MD.
Rat brain microsomal CO production – effect of representative drugs/compounds with diverse structures
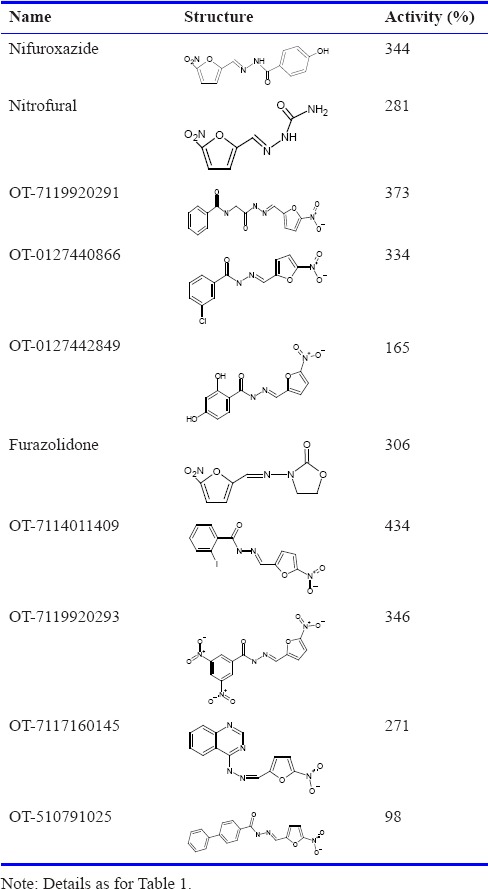
Tables 1 and 2 show that the ability of MD to increase CO production in rat brain microsomes was shared by a broad range of compounds. In these experiments, the baseline activity represents HO activity. Within the naphthoquinone family, we found that many members acted similarly to MD by increasing CO formation while some others did not; our results of testing ~50 other naphthoquinones were reported earlier.6 The presence of a variety of chemical entities increased CO production many-fold; for example, OT-7116980337 (Otava, Toronto, Canada), SI-1606 (Sniekus Innovations, Kingston, Canada) and VV-101 (St. Sava Bioanalytical & Medical Research, Kingston, Canada) (Table 1). The nitro-furanyl-methylene-hydrazides, nifuroxazide (MP Biomedicals, Solon, OH, USA) and furazolidone (Sigma-Aldrich), as well as a number of OT compounds increased CO generation by approximately 3- to 4-fold (Table 2).
Table 1.
Amplification of carbon monoxide (CO) production by representative naphthoquinones and phenazines in rat brain microsomes
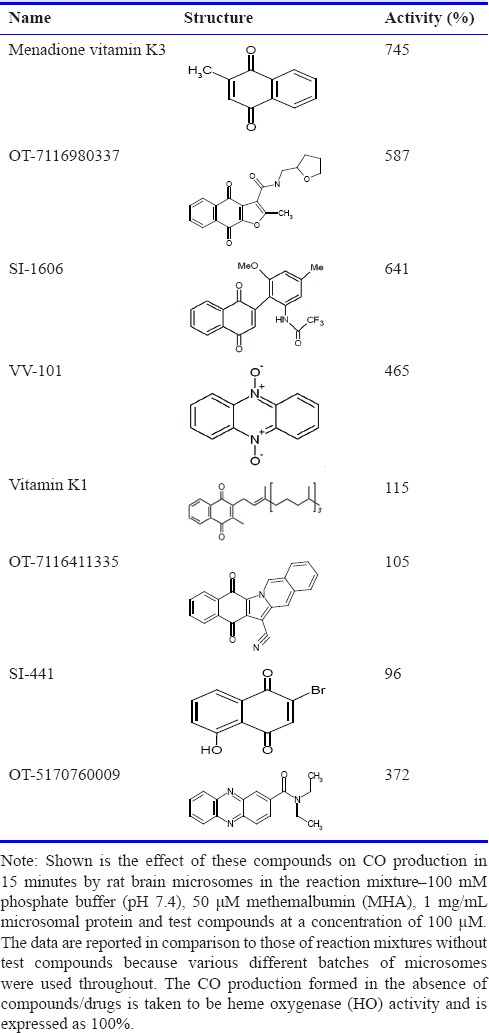
Table 2.
Amplification of carbon monoxide (CO) production by nitro-furanyl-methylene-hydrazides in rat brain microsomes
MD-enhanced CO Production by CPR
Experiments using microsomal preparations are useful to provide physiologically relevant observations for HO-mediated reactions; however, given the crude nature of the preparations, it was difficult to confirm definitively that the observed activation of CO production was due to HO activity. As such, to investigate the source of the enhanced CO production and the protein(s) involved, we moved to a system comprising purified components.
The effect of MD on CO production by reaction mixtures containing either recombinant, human CPR (rhCPR) alone or rhCPR plus full-length human HO-2 (FL-hHO-2) is shown in Figure 2. The rhCPR responded to the presence of increasing concentrations of MD with large increases in CO production (similar to that shown in Figure 1 for the membrane-associated native enzymes). Similar observations were made when the reaction mixture contained both rhCPR and FL-hHO-2. With rhCPR alone, the activity curve for increasing MD concentrations was almost identical to that with CPR plus FL-hHO-2; the small difference may represent the contribution of HO activity. The graph shows that the omission of FL-hHO-2 had virtually no effect on enhancement of CO production by MD, while the omission of rhCPR or NADPH resulted in no CO production. We have also observed (data not shown) in parallel experiments that furazolidine produced concentration-effect curves similar to that shown for MD on rhCPR.
Figure 2.
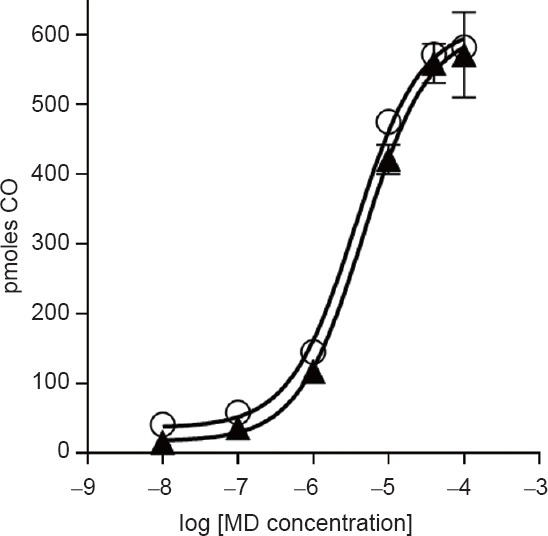
Menadione amplification of carbon monoxide (CO) production by cytochrome P450 reductase (CPR) in the presence or absence of heme oxygenase (HO).
Note: The open circles show the formation of CO when the reaction mixture contained 50 μM methemalbumin, 0.01 μM purified human NADPH-P450 reductase (recombinant; Becton Dickinson Canada Inc.), 0.7 μM full-length human HO-2 (FL-hHO-2) and various concentrations of menadione (MD) in 100 mM phosphate buffer (pH 7.4). The closed triangles show the formation of CO in the same reaction without FL-hHO-2 present. No CO production was detected in the absence of CPR. Bars represent SD, where bars are not shown the SD is smaller than the symbol; n = 4.
We also conducted experiments in which the UV/Vis spectrum of heme was monitored in the presence of the reaction mixture and rhCPR. Herein the Soret band for heme decreased under circumstances that resulted in the formation of CO; the presence of MD augmented this diminution of the Soret band. When rhCPR was omitted, no changes to the Soret band were observed; similar negative results were obtained in the absence of NADPH (results not shown).
Mechanism of activation of CPR
The ability of naphthoquinones such as MD to form ROS (O2‾ and H2O2) through redox cycling has been known for some time.12,13 Criddle et al.14 reported that metabolism of MD by one-electron (1e‾/H+) reducing enzymes generates a semi-quinone radical which then forms superoxide (O2‾) by re-oxidation. Also, Guengerich8 reported the destruction of heme/Fe++ PPIX and P450 in the presence of NADPH and O2 , conditions that were favourable to the formation of H2O2. These observations prompted us to conduct experiments in the presence of superoxide dismutase (to remove superoxide) or catalase (to remove H2O2) to determine if they might decrease the production of CO from rat brain microsomes or CPR.
When superoxide dismutase (Sigma-Aldrich) was added to the reaction mixture containing brain microsomes, there was a small decrease in the CO produced at higher concentrations of MD (Figure 3A). In contrast, the effect of catalase (Sigma-Aldrich) was substantial (Figure 3B); the presence of 300 U of catalase changed the entire response to MD such that approximately 80% of the activation was eliminated at the highest concentrations of MD. Similar observations were made in the case of rhCPR; Figure 3C shows that catalase blocked almost all of the MD-induced increase in CO formation observed with rhCPR. In contrast, the effect of superoxide dismutase in the presence of rhCPR differed from that with brain microsomes; herein superoxide dismutase resulted in decreased CO production that almost matched that of catalase (Figure 3D). This is in concert with a related recent report that recombinant CPR in the presence of MD generated both superoxide and H2O2 that could be eliminated by superoxide dismutase and catalase, respectively.15
Figure 3.

The effects of superoxide dismutase and catalase on production of carbon monoxide (CO) by rat brain microsomes and recombinant human cytochrome P450 reductase (rhCPR).
Note: (A) Brain microsome production of CO in the absence (open circles) and presence (closed triangles) of superoxide dismutase (300 U). (B) Brain microsome production of CO in the absence (open circles) and presence (closed triangles) of catalase (300 U). (C) rhCPR production of CO in the absence (open circles) and presence (closed triangles) of superoxide dismutase (300 U). (D) rhCPR production of CO in the absence (open circles) and presence (closed triangles) of catalase (300 U). Values are the mean of two experiments. MD: Menadione.
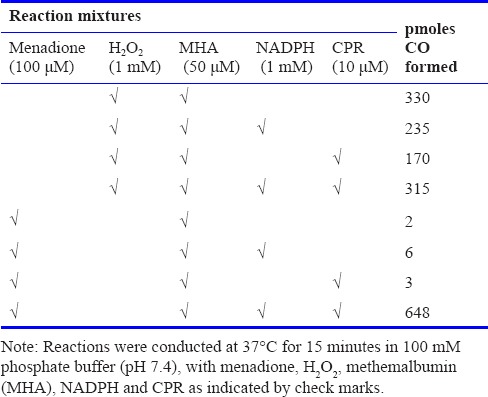
To dissect this MD-mediated mechanism further, we examined the effects of externally added H2O2 on CO formation, compared with that which might be formed in reaction mixtures comprising MHA-NADPH-rhCPR-MD. Table 3 shows that 1 mM H2O2 incubated with 50 μM MHA generated 330 pmol of CO in 15 minutes. The addition of NADPH and CPR into the same reaction mixture did not augment heme degradation. Replacement of H2O2 with MD did not generate any measurable CO, nor did addition of NADPH or CPR (to that reaction mixture). However, the combination of MD, MHA, NADPH and CPR generated almost 700 pmol of CO.
Table 3.
Comparison of carbon monoxide (CO) production by externally added H2O2 and components of the menadione (MD)-cytochrome P450 reductase (CPR) reaction mixture
Discussion
The goal of this work was to explore the enhancement of CO production by MD and other compounds during in vitro incubation with rat brain microsomes with heme. The main observations made in the present study were that: (a) many compounds with a variety of structures enhanced the production of CO by reaction mixtures containing brain microsomes, NADPH and MHA, (b) MD enhanced CO production of reaction mixtures containing pure, recombinant CPR in place of brain microsomes; HO-2 was not required, (c) addition of catalase to reaction mixtures containing either brain microsomes or recombinant CPR decreased CO generation, and (d) addition of H2O2 to reaction mixtures containing MHA was sufficient to degrade heme and liberate CO.
The most notable observation in this study was that the presence of HO-2 was not necessary to observe the many-fold enhancement in CO release from heme in the presence of MD, and that CPR alone was responsible for this effect. This is interpreted to mean that CPR mediates heme degradation, and that MD is capable of magnifying the flow of electrons from NADPH. While the latter is novel, the former has been reported previously by Noguchi et al.16 In their experiments they incubated hemin and NADPH with partially purified pig liver CPR, and observed loss of hemin, NADPH oxidation and H2O2 formation. The present observations were made by incubation of NADPH and MHA with either rat brain microsomes or recombinant, human CPR; the latter revealed a modest quantity of CO generation from heme in the absence of MD, while a large quantity of CO was generated in the presence of MD. These observations were obtained by measuring CO production using a specially designed gas chromatograph with high sensitivity detection of CO by mercuric oxide reduction.9 Our results made by spectrophotometry are consistent with those made by CO determination. This CO releasing ability of CPR, especially as amplified by certain drugs, facilitates the interpretation of several other observations.
When we first observed that CO production by microsomes was increased by certain drugs, these experiments were conducted and interpreted in the context of HO. At that time, we suggested that certain drugs could increase the efficiency of HO.6 The inability of QC-325 (HO inhibitor) to alter the increased production of CO in the presence of MD, shown in Figure 1, was inconsistent with such an interpretation. In contrast, the observations are fully consistent with CPR being responsible for the observed MD-enhanced CO production. Moreover, interpretation of these results through the lens of CPR-mediated CO production facilitates the interpretation of data generated in the presence of the range of compounds presented in Tables 1 and 2, as well as discussion of the mechanism of action.
The range of drugs/compounds yielding augmented CO release through heme degradation by the microsomal reactions was notable and this broad range differs from most pharmacological observations in which drug receptors mediate activity. Typically drug receptors display considerable selectivity for both agonists and antagonists; this can demand structural similarities that are obvious even upon perusal of two-dimensional figures. Small changes to the chemical structure of an agonist can alter activity significantly. For example, in the case of one of the earliest described receptor systems, cholinergic receptors, selectivity is greatly affected by the addition of a methyl group to an agonist. Thus, the natural agonist, acetylcholine, is an effective agonist at both muscarinic and nicotinic receptors whereas methacholine is primarily active at muscarinic receptors; methacholine differs from acetylcholine by the presence of an alpha methyl group. Our present results revealed enhancement of CO production by a variety of compounds with quite different skeletons; this lack of an obvious structure-activity relationship is somewhat analogous to that seen amongst the organic nitrate vasodilators. Within this drug group the skeletal core varies from glycerol (in glyceryl trinitrate) to nicotinamide to a variety of sugars; the common feature among them is a nitrate ester. It is now thought that these compounds are prodrugs that give rise to the release of nitric oxide. With respect to our present observations, this prompts us to suggest that the structure of the skeleton is not responsible for augmentation of CO production, but rather a common function may be responsible; we suggest that redox cycling is the essential property common to all these compounds. This suggestion arises from the extensive literature on naphthoquinones and other compounds capable of redox cycling. Thus, MD and many similar compounds have been shown to be reduced by CPR/NADPH.17 This has been reported to result in the production of superoxide via redox cycling;18 in biological systems superoxide can then be converted to H2O2 and hydroxyl radical. CPR has been shown to destroy free ferriprotoporphyrin IX through a mechanism mediated by H2O2,19 and CPR was shown to produce H2O2 when incubated in the presence of hemin and NADPH.15,16
This leads to the suggestion that MD-induced increased production of CO by CPR-NADPH-MHA reaction mixtures is a result of redox cycling-mediated H2O2 formation, and a subsequent degradation of heme (MHA) that results in the release of CO. If this notion is valid then the introduction of conditions that interrupt any of these steps are predicted to compromise the rate of CO formation. One such test would be to replace MD with analogs in which redox cycling ability is compromised. This was the case of MD analogs such as the 2,4-dimethoxy-2-methylnaphthoquinone and 2,5,6,7,8-pentafluoro-3-methyl-1,4-naphthalenedione; in the former, quinone oxygens that are essential for redox function have been replaced by methoxy groups, and the fluorines of pentafluora analog would interfere with electron flow by virtue of their high electron affinity. Neither of these compounds resulted in an increase in CO production.6 Similarly our observation that SI-441 did not increase CO production may be attributed to preventing redox cycling; this could be due the inductive effect of the electronegative Br atom and electron delocalization of the tautomeric forms via intramolecular hydrogen bonding involving the H of the –OH and the carbonyl group as discussed in the review by Brunmark and Cadenas.20 In contrast, this simple explanation seems incompatible with the data obtained with vitamin K1, which can redox cycle21 and yet was found not to increase CO generation by brain microsomes (Table 1). As vitamin K1 has a bulky side chain, one explanation could be that steric hindrance prevents it from binding to the site on CPR at which MD acts. It may be that compounds that increase the ability of CPR to create ROS require both redox cycling and access to a binding site within CPR; the latter might be the limiting feature of vitamin K1 and OT-7116411335 (Table 1).
According to the hypothesis articulated above, H2O2, once formed, should have no difficulty diffusing to MHA which could be degraded to release CO. If the key interacting species were H2O2, the hypothesis predicts that catalase would interfere with CO formation. This is consistent with the results reported herein where treatment with this enzyme interfered with the generation of CO by both microsome and recombinant CPR-containing reaction mixtures. This interpretation is further supported by the ability of externally added H2O2 to liberate substantial quantities of CO from MHA alone. On the other hand, not all of the present observations support this hypothesis. As the function of superoxide dismutase is to decrease the concentration of superoxide by conversion to H2O2, the result shown in Figure 3D may argue against a key role for H2O2. Accordingly, the ability of superoxide dismutase to substantially interfere with CO formation by recombinant CPR argues for participation of superoxide. Another consideration is the heterogeneous makeup of microsomal preparations; they contain a plethora of enzymes, which can complicate the interpretation of observations obtained therewith.
In summary, we have reported on a novel pathway for the production of CO through heme degradation that is catalyzed by CPR in the presence of NADPH and redox cycling compounds. This action may be mediated by a ROS such as H2O2. To fully understand the mechanism of this drug-enhanced CO production by CPR and its functional implication, further experiments will be required.
Footnotes
Funding: This study was funded by the Canadian Institutes of Health Research grant MOP 119467.
Conflicts of interest
Dragic Vukomanovic and Kanji Nakatsu may file a patent.
Plagiarism check
This paper was screened twice using CrossCheck to verify originality before publication.
Peer review
This paper was double-blinded and stringently reviewed by international expert reviewers.
References
- 1.Kumar S, Bandyopadhyay U. Free heme toxicity and its detoxification systems in human. Toxicol Lett. 2005;157:175–188. doi: 10.1016/j.toxlet.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Rahman MN, Vukomanovic D, Vlahakis JZ, Szarek WA, Nakatsu K, Jia Z. Structural insights into human heme oxygenase-1 inhibition by potent and selective azole-based compounds. J R Soc Interface. 2013;10:20120697. doi: 10.1098/rsif.2012.0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braggins PE, Trakshel GM, Kutty RK, Maines MD. Characterization of two heme oxygenase isoforms in rat spleen: comparison with the hematin-induced and constitutive isoforms of the liver. Biochem Biophys Res Commun. 1986;141:528–533. doi: 10.1016/s0006-291x(86)80205-4. [DOI] [PubMed] [Google Scholar]
- 4.Trakshel GM, Kutty RK, Maines MD. Resolution of the rat brain heme oxygenase activity: absence of a detectable amount of the inducible form (HO-1) Arch Biochem Biophys. 1988;260:732–739. doi: 10.1016/0003-9861(88)90503-6. [DOI] [PubMed] [Google Scholar]
- 5.Vukomanovic D, McLaughlin BE, Rahman MN, et al. Selective activation of heme oxygenase-2 by menadione. Can J Physiol Pharmacol. 2011;89:861–864. doi: 10.1139/y11-091. [DOI] [PubMed] [Google Scholar]
- 6.Vukomanovic D, Rahman MN, Bilokin Y, et al. In vitro Activation of heme oxygenase-2 by menadione and its analogs. Med Gas Res. 2014;4:4. doi: 10.1186/2045-9912-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riddick DS, Ding X, Wolf CR, et al. NADPH-cytochrome P450 oxidoreductase: roles in physiology, pharmacology, and toxicology. Drug Metab Dispos. 2013;41:12–23. doi: 10.1124/dmd.112.048991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guengerich FP. Destruction of heme and hemoproteins mediated by liver microsomal reduced nicotinamide adenine dinucleotide phosphate-cytochrome P-450 reductase. Biochemistry. 1978;17:3633–3639. doi: 10.1021/bi00610a033. [DOI] [PubMed] [Google Scholar]
- 9.Vreman HJ, Stevenson DK. Heme oxygenase activity as measured by carbon monoxide production. Anal Biochem. 1988;168:31–38. doi: 10.1016/0003-2697(88)90006-1. [DOI] [PubMed] [Google Scholar]
- 10.Dierks EA, Davis SC, Ortiz de Montellano PR. Glu-320 and Asp-323 are determinants of the CYP4A1 hydroxylation regiospecificity and resistance to inactivation by 1-aminobenzotriazole. Biochemistry. 1998;37:1839–1847. doi: 10.1021/bi972458s. [DOI] [PubMed] [Google Scholar]
- 11.Yi L, Ragsdale SW. Evidence that the heme regulatory motifs in heme oxygenase-2 serve as a thiol/disulfide redox switch regulating heme binding. J Biol Chem. 2007;282:21056–21067. doi: 10.1074/jbc.M700664200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thor H, Smith MT, Hartzell P, Bellomo G, Jewell SA, Orrenius S. The metabolism of menadione (2-methyl-1,4-naphthoquinone) by isolated hepatocytes. A study of the implications of oxidative stress in intact cells. J Biol Chem. 1982;257:12419–12425. [PubMed] [Google Scholar]
- 13.Cadenas E, Sies H. Oxidative stress: excited oxygen species and enzyme activity. Adv Enzyme Regul. 1985;23:217–237. doi: 10.1016/0065-2571(85)90049-4. [DOI] [PubMed] [Google Scholar]
- 14.Criddle DN, Gillies S, Baumgartner-Wilson HK, et al. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J Biol Chem. 2006;281:40485–40492. doi: 10.1074/jbc.M607704200. [DOI] [PubMed] [Google Scholar]
- 15.Jan YH, Richardson JR, Baker AA, et al. Vitamin K3 (menadione) redox cycling inhibits cytochrome P450-mediated metabolism and inhibits parathion intoxication. Toxicol Appl Pharmacol. 2015;288:114–120. doi: 10.1016/j.taap.2015.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noguchi M, Yoshida T, Kikuchi G. A stoichiometric study of heme degradation catalyzed by the reconstituted heme oxygenase system with special consideration of the production of hydrogen peroxide during the reaction. J Biochem. 1983;93:1027–1036. doi: 10.1093/oxfordjournals.jbchem.a134226. [DOI] [PubMed] [Google Scholar]
- 17.Butler J, Hoey BM. The one-electron reduction potential of several substrates can be related to their reduction rates by cytochrome P-450 reductase. Biochim Biophys Acta. 1993;1161:73–78. doi: 10.1016/0167-4838(93)90198-z. [DOI] [PubMed] [Google Scholar]
- 18.Livertoux MH, Lagrange P, Minn A. The superoxide production mediated by the redox cycling of xenobiotics in rat brain microsomes is dependent on their reduction potential. Brain Res. 1996;725:207–216. doi: 10.1016/0006-8993(96)00251-x. [DOI] [PubMed] [Google Scholar]
- 19.Schaefer WH, Harris TM, Guengerich FP. Characterization of the enzymatic and nonenzymatic peroxidative degradation of iron porphyrins and cytochrome P-450 heme. Biochemistry. 1985;24:3254–3263. doi: 10.1021/bi00334a027. [DOI] [PubMed] [Google Scholar]
- 20.Brunmark A, Cadenas E. Redox and addition chemistry of quinoid compounds and its biological implications. Free Radic Biol Med. 1989;7:435–477. doi: 10.1016/0891-5849(89)90126-3. [DOI] [PubMed] [Google Scholar]
- 21.Bao D, Ramu S, Contreras A, et al. Electrochemical reduction of quinones: interfacing experiment and theory for defining effective radii of redox moieties. J Phys Chem B. 2010;114:14467–14479. doi: 10.1021/jp101730e. [DOI] [PubMed] [Google Scholar]