

Significance
The metabolism of xenobiotics is a critical aspect of drug discovery; nowadays, aldehyde oxidase (AOX) has emerged as a key metabolic enzyme having a pivotal role in the failures of several clinical candidates. The lack of homogenous data on possible substrates and not substrates of this enzyme represents a serious limit for the development of an in silico model for metabolism prediction. Here, we present a database of 270 chemically diverse compounds containing aza-aromatic and/or amide moieties (susceptible to human AOX), experimentally tested in vitro. The results herein reported should be useful in the development of a reliable prediction model, which should be of wide interest in chemistry, biology, biotechnology, and medicine.
Keywords: aldehyde oxidase, amide hydrolysis, site of metabolism, variously decorated heterocycles, structure–metabolism relationship
Abstract
Aldehyde oxidase (AOX) is a metabolic enzyme catalyzing the oxidation of aldehyde and aza-aromatic compounds and the hydrolysis of amides, moieties frequently shared by the majority of drugs. Despite its key role in human metabolism, to date only fragmentary information about the chemical features responsible for AOX susceptibility are reported and only “very local” structure–metabolism relationships based on a small number of similar compounds have been developed. This study reports a more comprehensive coverage of the chemical space of structures with a high risk of AOX phase I metabolism in humans. More than 270 compounds were studied to identify the site of metabolism and the metabolite(s). Both electronic [supported by density functional theory (DFT) calculations] and exposure effects were considered when rationalizing the structure–metabolism relationship.
In recent years, the cytochromes P450 (CYP450)-mediated metabolism has been intensively studied. As a consequence, the development of new and more efficient in silico and in vitro screening systems contributed to decrease the discontinuation rates of new drugs in clinical studies by 10% in recent years (1). In particular, several models for in silico prediction of human CYP450-mediated metabolism have been reported so far (2–4) and, more recently, for human FMO3 (5). Nevertheless, a deeper understanding of non-CYP drug enzyme metabolism is advisable to further improve screening and models efficacy (6). Among all non-CYP metabolic enzymes (7), emerging importance has been attributed to human aldehyde oxidase (hAOX), a cytosolic drug-metabolizing enzyme expressed in human liver that, similarly to CYPs, contributes to new chemical entities’ oxidation, but acting in the absence of NADPH cofactor. Drugs that are substrates for AOX often exhibit high metabolic clearance, resulting in low exposure and hence in decreased drug efficacy in humans. In particular, AOX catalyzes the oxidation of a wide range of aza-aromatic scaffolds at the unsubstituted carbon in ortho to the nitrogen (SI Appendix, Fig. S1 A and B), usually the most electron deficient (8). Several marketed drugs are well-known substrates of AOX (e.g., methotrexate, famciclovir, and zaleplon) (9), and several compounds have failed due to undetected AOX oxidation (e.g., BIBX1382, RO-1, FK3453, and carbazeran) (10–14). The role of AOX in drug development has become more relevant in the last few years as a result of organic synthesis strategies designed to reduce cytochromes P450-related metabolism, based on chemical modifications and decorations that have subsequently increased the drug reactivity toward AOX (11, 15). In addition, AOX was recently found to be responsible for the rapid hydrolysis of an amide bond in GDC-0834 (SI Appendix, Fig. S1C), a potent inhibitor of Bruton’s tyrosine kinase (16). Although the development of structure–metabolism relationships (SMRs) for hAOX metabolism prediction would strongly reduce the risk of clinical failure in late stages of drug discovery, such SMRs are scarcely reported in literature, and general rules for hAOX SMR are missing (17–19). For instance, Ghafourian and Rashidi (18) developed a quantitative structure–activity relationship method for phthalazine and quinazoline scaffold, based on COSMIC force field, AM1 semiempirical method, and structural parameters generated by MOPAC and found a strong dependence of the oxidation rate with the electronic nature of substituents; Torres et al. (17) reported a qualitative method based on density functional theory (DFT) to predict the product of AOX metabolism by examining the energetics of likely tetrahedral intermediates resulting from nucleophilic attack on carbon and confirmed the primary role of the electronic effects of the substituents; later, Jones and Korzekwa (19) developed a mechanistic computational model to predict intrinsic clearance by DFT studies, taking into account electronic and steric effect of the substituents. However, in all papers, the relationships and models were based on a small number of compounds of similar structure (e.g., phthalazine and quinazoline), and an attempt made by us to apply the same strategies in new different chemical entities proved to be inadequate (for further details, see SI Appendix).
Therefore, we performed in-house AOX-mediated metabolism experiments covering a diverse chemical domain of commonly used drugs and drug candidates to obtain a sound base for further mechanistic investigation and modeling studies.
Regarding AOX oxidation, 198 aza-aromatic compounds were acquired or synthesized to assess AOX susceptibility, and examples of the scaffolds investigated are shown in Fig. 1. The full dataset with more experimental details is reported in SI Appendix, Tables S1–S5.
Fig. 1.

Examples of the studied aza-aromatic scaffolds: the number of tested compounds is reported in brackets. The complete dataset is provided in SI Appendix.
Chemistry
The derivatives of phthalazine were prepared starting from phthalazine and phthalazone, as summarized in SI Appendix, Fig. S2 A and B, respectively. 1-Phenylphthalazine (1) was prepared by direct arylation of phthalazine with phenylboronic acid in very mild condition (20). Nitration of phthalazine with NaNO3/H2SO4 solution gave 5-nitrophthalazine (2), which was reduced to aminophthalazine 3, in turn converted to amide 4 upon 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU)-mediated coupling with 2-picolinic acid. The phthalazine ammonium salts 5 and 6 were obtained from commercial phthalazine after alkylation with methyl and benzyl halide. Phthalazine 2-oxide (7) was obtained by phthalazine oxidation with meta-chloroperoxybenzoic acid (mCPBA) (21).
1-Chlorophthalazine hydrochloride 8 was obtained by POCl3-mediated chlorination of phthalazone (22). 1-Methoxyphthalazine 9 was produced during an attempt of chromatographic purification on SiO2 (with a methanol-containing eluent) of chloro derivative 8. 1-Chlorophthalazine produced 4-(phthalazin-1-yl)morpholine (10) and 1-phenoxyphthalazine (11) by nucleophilic substitution in alkaline condition. Purine derivatives 12–15 were obtained from 6-chloropurine (2-amino,6-chloropurine for 13d) as described in SI Appendix, Fig. S2C. Chloro compounds 12a–b were obtained upon alkylation of purine with benzyl bromide and propargyl bromide in the presence of K2CO3. The nucleophilic substitution of 6-chloroderivative with phenol, aniline, or cyclohexylamine according to literature procedures (23, 24) afforded compounds 13a–d. In turn, bromination of 13b and 13c with Br2 saturated aqueous Na2HPO4 solution gave 14 and 15 (25). Moreover, hypoxanthine and allopurinol reaction with benzyl bromide and K2CO3 in DMF (26) gave compounds 16–17 and 19–20 (SI Appendix, Fig. S2 D and E). Furthermore, chlorination of allopurinol with POCl3 gave 4-chloro-1H-pyrazolo[3,4-d]pyrimidine (18) (SI Appendix, Fig. S2E). Finally, pyrazine based compounds 21–23 were obtained according to SI Appendix, Fig. S2F. Chloropyrazine 21a was prepared according to literature procedure (27). Amino derivative 22 was obtained from chloro 21a by nucleophilic substitution with aniline (28). Finally, pyrazine 23 was obtained by Suzuki coupling from bromo derivative 21b, in turn prepared by chloro derivative 21a and trimethylsilyl bromide (29).
Evaluation of AOX-Mediated Metabolism by in Vitro Assays on Human Liver Cytosol
Metabolism of the 273 selected compounds was evaluated in vitro according to a modification of a literature procedure (30), using human liver cytosol (HLC) instead of S9 fraction, due to its higher content of AOX. After 0- and 60-min exposure, samples were analyzed by liquid chromatography–tandem mass spectrometry (LC-MS/MS), and then processed with MetaSite and WebMetabase (Molecular Discovery, Ltd.; www.moldiscovery.com/) (31). In addition, carbazeran, cinchonine, and famciclovir, well-known AOX substrates, together with a representative example of each scaffold, were also tested in the presence of 2,6-dichlorophenolindophenol (DCPIP), a selective inhibitor of AOX to ensure the reliability of our method (SI Appendix, Figs. S3 and S4, respectively). The AOX content of several cytosolic batches was quantified (32) and the average value was 0.9 µM ± 0.1 µM. Further details can be found in Materials and Methods.
DFT Calculation
As mentioned above, the electronic effect is reported to play a major role in AOX substrate susceptibility; therefore, partial atomic charges (ACs) were calculated to support the experimental trend observed and to determine the site of metabolism (SoM). DFT calculations were run with Gaussian 09 (33) and consisted of three steps: (i) creation of 3D structures of the substrates, (ii) ground-state geometry optimization by DFT at a B3LYP level of theory with 6-31G basis sets, (iii) AC calculations according to the Merz–Singh–Kollman scheme (34). Interestingly, for the investigated substrates, the AOX SoM (highlighted by the * symbol in Tables 1–4) usually corresponds to the most positive unsubstituted aromatic carbon atom (for more details, see SI Appendix, Fig. S5), thus suggesting that MK charges can be conveniently used for estimating the SoM also where multiple site of reactions are possible. This computational workflow has been chosen after evaluating the performance of alternative computational approaches that compare reaction energies to account for the multiple possible reaction sites (35) (SI Appendix, Table S6). However, this last approach is less versatile because strictly dependent on the mechanism of reaction/nature of the intermediate (not fully elucidated yet), more computational demanding, and slower compared with AC calculations. For all of these reasons and bearing in mind the goal of a comprehensive analysis on a large number of compounds, we chose to proceed with AC calculations (the most relevant results are reported in SI Appendix, Table S7).
Table 1.
AOX susceptibility for pyrimidine-based compounds
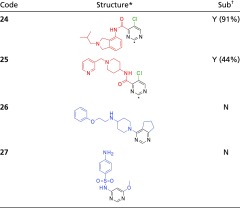 |
Compounds 24–26 were analyzed according to LC-MS method B (Materials and Methods); for 27, method A was used instead.
The predicted SoM by DFT calculations is highlighted by a star.
N, not substrate; Y, substrate (the percentage of substrate consumed after 60-min incubation is reported between brackets).
Table 4.
hAOX susceptibility of miscellaneous bicyclic compounds
 |
All compounds were analyzed according to LC-MS method B, with the exception of compound 19, for which method A was used instead (Materials and Methods).
The predicted SoM by DFT calculations is highlighted by a star.
N, not substrate; Y, substrate (the percentage of substrate consumed after 60-min incubation is reported between brackets).
Hydrolysis reaction contributes to substrate consumption.
Docking Analysis
Compounds identified as substrates by in vitro assay were analyzed by docking approach with FLAP software (36, 37) using the 4UHX hAOX 3D structure (4UHX) (38). The protein structure 4UHX lacks two important polar amino acids (Asp881, Glu882) located in the loop between the residues Leu880–Ser883. However, these amino acids are 4.5 Å from the phthalazine substrate; thus, they may play a relevant role in molecular interactions with substrates. To avoid misleading results, the two missing amino acids were added to the 4UHX X-ray structure loop in between Leu880–Ser883 residues and gently minimized before any computation. The resulting AOX catalytic cavity encompassed 30 aa that, despite their different chemophysical nature and polarity, generated large hydrophobic interaction pathways, with minor occupancy for protein H-bonding acceptor interaction pathways and almost absent protein H-bond donor interactions hot spots (Fig. 2). Compounds were docked in the catalytic active site, allowing flexibility on both ligands and cavity amino acids.
Fig. 2.

(A) Hydrophobic molecular interaction fields generated using the GRID (41) DRY probe in 4UHX AOX protein catalytic cavity; (B) H-bond donor molecular interaction fields generated using the GRID N1 probe. Phthalazine (in green) is added for reference.
Results and Discussion
Oxidation of Aza-Heterocycles by hAOX.
All data concerning the oxidation by hAOX are collected in SI Appendix, Tables S1–S5. Among the scaffolds investigated, pyridines were the only class that resulted always in not being susceptible to AOX metabolism (SI Appendix, Table S1). In this section, a number of scaffolds that provided interesting insights on the structure–metabolism relationship in hAOX enzyme are discussed.
For example, the study of the 15 pyrimidines and 4 pyrazines (SI Appendix, Table S2) suggested that compounds bearing these scaffolds are barely substrates. When metabolism occurs, the C2 position seems to be the preferred SoM for oxidation, with all 2-substituted compounds being stable. A comparison of the four pyrimidine-based compounds in Table 1 exemplifies a general trend observed for this class of compounds. Indeed, compounds having EWG group at C4 and a chlorine at C5 (24 and 25) resulted as substrates, whereas pyrimidines with EDG substituents at C4 and C6 (26 and 27) were not substrates. This finding highlights the importance of electronic effect in AOX susceptibility.
Concerning the 13 quinolines and 15 isoquinolines tested (SI Appendix, Table S3), they were easily metabolized by AOX at C2 and C1/C3, respectively, regardless of the substituents’ nature. Additional insights were extracted from compounds in Table 2. About quinolines, similar structures were compared. Quinine ester 32 was more stable than quinine 30; a rationale for this behavior is provided by the correspondent calculated AC at the SoM: the higher the charge, the more reactive the SoM. Moreover, comparing sulfonamides 31 and 33, the presence of the benzotriazole moiety seems to be detrimental for metabolic susceptibility, suggesting that this chemical group has an impact on both electronic and exposure effects. This hypothesis was supported by the calculated charges (the AC of 31-SoM is higher than that of 33-SoM; SI Appendix, Table S7) and by docking analysis. Indeed, benzotriazole was found to compete with quinoline to give a π–π stacking interaction with Phe923 and Phe885 (SI Appendix, Fig. S6), previously identified as important residues for recognition in the active site (38). Concerning isoquinolines, we observed that compounds with EDG at C4, C5, and C7 were substrates, as exemplified by compounds 34 and 35 in Table 2, whereas EWG-substituted compounds were stable (e.g., 36 and 38, Table 2).
Table 2.
hAOX susceptibility for quinoline- and isoquinoline-based compounds
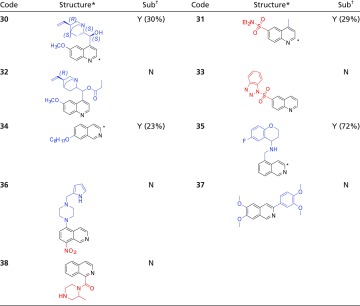 |
All compounds were analyzed according to LC-MS method B (Materials and Methods).
The predicted SoM by DFT calculations is highlighted by a star.
N, not substrate; Y, substrate (the percentage of substrate consumed after 60-min incubation is reported between brackets).
Comparing 34 to 37, both EDG-substituted compounds, only the second was stable: as suggested above, the phenyl in 37 can play the same role of benzotriazole in 33 (and again the AC of the putative 37-SoM is lower than the one for 34).
Quinoxaline and related regioisomers represent another class of compounds deeply investigated, with more than 45 compounds tested (SI Appendix, Table S4). These compounds were mostly good AOX substrates, with 17 compounds leading to more than 50% of the parent compound consumed after 60-min incubation. Both EDG- and EWG-substituted quinoxaline were good AOX substrates (as exemplified by compounds 39 and 40 in Table 3), with the exception of compounds 41 and 42 whose substituents orient the compounds in an unreactive pose in the catalytic cavity according to our docking studies (SI Appendix, Fig. S6). The steric hindrance may in this case overpower the electronic contribution, because the calculated AC values (SI Appendix, Table S7) suggest the opposite behavior. Similarly, quinazoline (e.g., 43 and 44) and phthalazine (e.g., 1, 3, and 9) were highly reactive toward AOX, with the exception of the nitro-containing compound 2. Finally, 1,6- and 1,8-naphthyridines (e.g., 45 and 46) were substrates, whereas 1,7-naphthyridines like 47 and 48 were not.
Table 3.
hAOX-mediated metabolism for quinoxaline and its isomers
 |
All compounds were analyzed according to LC-MS method B, with the exception of compounds 1–3, phthalazone, and 9, for which method A was used instead (Materials and Methods).
The predicted SoM by DFT calculations is highlighted by a star.
N, not substrate; Y, substrate (the percentage of substrate consumed after 60-min incubation is reported between brackets).
A number of other bicyclic scaffolds were investigated (SI Appendix, Table S5). For the eight pyridopyrazines tested, the SoM could be C2 and C3: when at least one of these positions is occupied by bulky groups the oxidation does not occur at all (50 and 51). About pyridophthalazines, only two compounds were tested, resulting as not substrates; however, 53 is already oxidized (similar to phthalazone), whereas 52 is very hindered by the presence of three phenyl groups (Table 4). The pteridine scaffold was tested in seven compounds, represented in Table 4 by 54–56. The different oxidation susceptibility of 54 and 56, only differing in the methyl substituent position, was rationalized by considering that the calculated charge on 54-C7 (the exposed site according to docking analysis; SI Appendix, Fig. S6) is higher than the 56-C6 charge (unexposed site according to docking analysis; SI Appendix, Fig. S6). Indeed, C7 is the preferred SoM for exposition reasons along with C2, with 55 being the only substrate with C7 occupied but still oxidized. Similarly, according to AC charges, C6 is the SoM in 2 out of 10 imidazopyrazines, (SI Appendix, Table S7). For example, concerning representative compounds in Table 4, 57-C6 (substrate) is more electropositive than 58-C6 (not substrate). Additionally, stability of 59 and 60, with AC charges similar to 57, can be rationalized by the presence of aromatic groups at C2 (i.e., indole and phenol, respectively). Purine scaffolds were also tested (SI Appendix, Table S5). Based on our data, 2- or 6-EDG–substituted purines were substrates of AOX (with unsubstituted N7-H or N9-H), as in compounds 12a and 12b (Table 4). Again, as shown in SI Appendix, Table S7, the calculated AC on the putative SoM for 12a and 12b (substrates) is higher than the one calculated for 15 and 16 (not substrates, Table 4), confirming that the electronic effect may dominate the exposition effects in determining the AOX oxidation susceptibility in this scaffold. On the contrary, allopurines were not substrates (e.g., 18 and 19). The three pyrazolopyridazines might be assimilated with the phthalazine scaffold: consequently, compounds 62 and 63 were substrates, whereas 64, similar to phthalazone, was not a substrate. Other miscellaneous scaffolds were investigated as reported in SI Appendix, Table S5. In summary, the presence of indole, pyrazole, and pyridine were detrimental for AOX susceptibility. Mostly, EDG substituents favor the susceptibility of compounds to oxidation. The most evident exception to this statement is represented by pyrimidines, where the opposite trend might be due to the higher pKa (>8) of EDG-substituted compounds, as the protonated species might interact differently with the catalytic site and through a different mechanism. According to our data, another important trend observed is that electrophilicity and reactivity seem strictly correlated, although in some cases highly positive-charged carbon atoms are not oxidized (e.g., 36-C3, SI Appendix, Table S7): electrophilicity alone cannot discriminate the susceptibility to AOX. This last observation can be rationalized considering that the initial nucleophilic attack by molybdenum cofactor (MoCo) requires a moderate electrophilic center, although the widely accepted mechanisms and recent experiments with kinetic isotopic effects point to hydride displacement as the critical step (39). Therefore, we suggest that there should be a trade-off between the SoM nucleophilic and electrophilic nature, requiring an in-depth modeling study, which is beyond the aim of this work.
Hydrolysis of Amides by hAOX.
Among the five pyridazoindoles tested for AOX susceptibility, all bearing an amide moiety, two of them (65 and 66), in addition to the oxidated metabolites, lead to the formation of the corresponding anilines, consistent with the amide bond hydrolysis. This finding was supported by a single recently published example of the amidic compound GDC-0834 (16). To confirm AOX contribution to both hydrolysis and oxidation, compound 66 was also tested in the presence of DCPIP inhibitor. As expected, both reactions were inhibited (Fig. 3; MS/MS spectra are reported in SI Appendix, Fig. S7).
Fig. 3.

Inhibition study on 66 with DCPIP (50 µM). The chromatograms report the substrate consumption after 0- (Top) and 60-min (Bottom) HLC incubation in the absence (Left) or presence (Right) of AOX selective inhibitor DCPIP. For each peak, the corresponding compound is also reported.
Thus, we further investigated the hydrolysis reaction, because it can cause not only substrate inactivation but also toxification (liberated anilines are potentially hemato-, nephro-, hepato-toxic, and carcinogenic) (40).
Therefore, we synthesized (SI Appendix, Fig. S2G) and acquired 75 amides to perform a SMR study (Fig. 4 and Tables 5 and 6; additional information can be found in SI Appendix, Tables S8 and S9).
Fig. 4.

Design of differently decorated amides inspired to GDC-0834 to investigate hAOX-mediated amide hydrolysis.
Table 5.
Thiophene-containing anilides tested for AOX-mediated metabolism
 |
All compounds were analyzed according to LC-MS method B, with the exception of compounds 69, 70, 81, and 82, for which method A was used instead (Materials and Methods).
N, not substrate; Y, substrate (the percentage of substrate consumed after 60-min incubation is reported between brackets).
Table 6.
Thiophene-free anilides tested for hAOX-mediated metabolism
 |
All compounds were analyzed according to LC-MS method B, with the exception of compounds 91–93, for which method A was used instead (Materials and Methods).
N, not substrate; Y, substrate (the percentage of substrate consumed after 60-min incubation is reported between brackets).
Substrate consumption is also due to oxidation reaction.
First, amides 67–90 sharing the same acyl thiophene moiety of GDC-0834 were tested (Table 5). Amides 67–69 carrying only an EDG in the aniline ortho position resulted as substrates of AOX, with the parent compound being consumed by 81–90% after 60 min of incubation. Conversely, 70–71 with EWG (i.e., carbonyl) were stable to AOX exposition. On the contrary, meta or para substitution with both EDG and EWG led to more stable compounds (72–83).
Thus, provided the same acidic group, the best metabolism rate is obtained when the amine counterpart is substituted in the ortho position with an EDG. Moreover, tertiary amides seemed more stable (84 and 85). Finally, the introduction of more sterically hindered and less flexible anilines (86 and 87), or aliphatic amines (88–90), gave stable compounds. Thus, we concluded that the nature of acyl moiety is less important to ensure AOX susceptibility. To confirm our hypothesis, furane, indole, 2-methylenindole, (3-fluorophenyl)propane, cyclohexane, pyrazine, and pyridine compounds 91–100 were tested, resulting as substrates (Table 6). Regardless of the presence of only an EDG ortho substituent, isoquinoline 104 was stable (Table 6), possibly as a consequence of an intramolecular H bond (8). Acquired aliphatic benzamides were also tested (SI Appendix, Table S9), but they were not AOX substrates. This finding correlates well with the more favorable hydrolysis of aromatic amides with respect to the aliphatic ones, due to resonance effects. Para-substituted or heterocyclic anilides (e.g., 102–103, Table 6) and amide-like moieties (hydrazides, ureas, and carbamates) were not susceptible to AOX metabolism (see SI Appendix, Table S9, amide-like bond). Furthermore, the docking analysis of compounds herein reported confirmed the experimental trends. Indeed, when substrate and nonsubstrate molecules are docked into the AOX catalytic site, molecules tend to interact with the AOX cavity in three different ways (far from MoCo or close to MoCo in correct orientation or not correct orientation). Generally, the most populated poses are nonproductive poses because the molecules interact with the AOX cavity far from the reactive MoCo. This behavior is common for both substrate and nonsubstrate molecules. However, substrates tend to show a higher number of productive poses, that is, the poses that expose the reactive carbon moiety to the activated oxygen atoms of the MoCo, with respect to the nonproductive poses. Conversely, nonsubstrates show a higher number of nonproductive poses, that is, the poses that expose unreactive atom moieties to the MoCo, with respect to the productive ones. Moreover, often the nonsubstrate molecules do not adopt any of the potential reactive poses. This behavior is exemplified in Fig. 5 where the substrate 65 and nonsubstrate 105 are reported.
Fig. 5.

Comparison of compounds 65 and 105 by docking analysis. (A) Productive poses for substrate 65 (not all poses are reported for clarity). The most reactive carbon atom of the pyridazino ring is well exposed and oriented toward the MoCo. (B) Nonproductive poses for 65 where the unreactive cyanophenyl moiety is exposed to MoCo. (C) Productive poses for hydrolytic reactions of substrate 65 (not all poses are reported for clarity). The amide moiety is well oriented toward the MoCo. (D) Nonproductive poses for nonsubstrate 105 exposing unreactive dimethyl-1H-pyrazol toward MoCo. (E) Nonproductive poses for nonsubstrate 105 showing a suboptimal orientation of the pyrimidine moiety. The phthalazine substrate was added to the picture for reference. (F) 65 (Top) and 105 (Bottom) structures.
About 10% of the overall poses of 65 are productive for carbon ring oxidation because, as reported in Fig. 5, the reactive carbon atom of the pyridazino ring is well exposed and oriented toward the MoCo. Interestingly, a similar number of poses show a potential interaction of the amide moiety with the MoCo. Indeed, 65 is also hydrolyzed by hAOX. These poses are mostly stabilized by proficuous hydrophobic interactions and to a lesser extent by some substrate H-bond donor–protein H-bond acceptor interactions. Poses where the cyanophenyl moiety is exposed to MoCo are nonproductive because the cyanophenyl ring is not reactive to MoCo. Conversely, for the nonsubstrate 105 the great majority of poses expose unreactive moieties toward MoCo (the figure reports the exposure of the unreactive dimethyl-1H-pyrazol and of poorly oriented pyrimidine moieties). As an example, we decided to test three additional compounds (106a–c, Fig. 6) with two possible SoM according to DFT analysis, but exposing only one site toward the MoCo in the majority of the poses.
Fig. 6.

Metabolism of additional designed and synthesized quinazolines 106a–c. * and ** are the sites with the highest AC (SI Appendix, Table S7). According to docking analysis, * is most favorable exposed toward the MoCo.
Quinazolines 106a–c and their predicted 4-(3H)-quinazolone metabolites (107a–c) were synthesized (SI Appendix, Fig. S9). The EDG-substituted quinazoline resulted as AOX substrates (percentage substrate consumed, 85, 85, and 68%, respectively), and their metabolism was confirmed by comparison of the RT and LC-MS/MS spectra of the obtained metabolite and synthesized quinazolone. Thus, protein–ligand recognition elements able to correctly expose the reactive ligand moiety to the MoCo cofactor appear to be the key factors in predicting the hAOX isoform selectivity and site of metabolism.
In summary, preliminary SMR guidelines are given in Fig. 7. Although in the majority of aza-heterocycles the ortho C-H is often oxidized by hAOX, the metabolism can be tuned not only by addition of substituents at the predicted most positive C–H, but also in other position (mostly EWG, excepting pyrimidine) able to reduce the AC. Furthermore, hindered and highly hydrophobic substituents are often able to drastically change the SoM exposition, so that metabolic stability can be achieved. Regarding amides, the amine counterpart is more relevant to establish hAOX susceptibility: substituents in the ortho < meta < para position of the anilide counterpart enhance the stability, EWG stabilizing the most the amidic bond.
Fig. 7.

Stability of compounds toward AOX metabolism oxidation (A) and hydrolysis (B) may be enhanced following the criteria reported in the arrows.
Conclusion
In summary, a drug-like database of variously decorated aza-aromatic scaffolds has been constructed, and all compounds subsequently tested using the same experimental conditions. Moreover, the in-depth study on several amides highlighted the ability of this enzyme to hydrolyze the amidic bond, especially in EDG-substituted anilides. As with the cytochromes, the biotransformation ability of AOX seems to depend on a combination of electronic and exposure factors. DFT calculations and docking procedure have been performed to support this finding. In some cases, comparison of calculated atomic charges on the putative site of reaction also allowed rationalizing the AOX susceptibility outcome (Y/N), but this is true only for very similar structures for which we can assume that the exposure factor play a similar role. From all of these experiments, it emerges that it is not at all simple to predict whether a compound is a substrate of AOX or not. We noted that, although the role of physicochemical molecular descriptors may be relevant in case of congeneric series of compounds, when different molecules are compared, their contribution to isoform selectivity resulted in being relatively modest.
In general, when pharmaceutical compounds bearing different scaffolds are compared, more sophisticated modeling techniques that combine electrophilicity calculation with productive spatial interaction with the enzyme catalytic site are required to rationalize SMRs.
To conclude, we believe the study herein reported represents a solid base to construct an in silico model to predict the AOX susceptibility of lead compounds and drug candidates.
Materials and Methods
Materials.
Solvents and pooled mixed-gender cryopreserved HLC (1 mg/mL) were obtained from Sigma Aldrich. Tested compounds were obtained through chemical synthesis (see above) or acquired from SPECS and ENAMINE. All tested compounds were >98% pure as determined by ultrahigh-performance liquid chromatography on Agilent Technologies 6540 UHD accurate-mass quadrupole time-of-flight (QTOF) LC/MS system.
Metabolism Assays on HLC.
Metabolism of selected compounds was evaluated upon incubation with HLC according to a modified Dalvie et al. (30) procedure. Test compound (10 μM) was incubated at pH 7.4 and 37 °C in the presence of magnesium chloride hexahydrate (MgCl2·6H2O, 1 mM) and HLC. After 0 and 60 min of incubation, the reaction was stopped by addition of 250 μL of ice-cold acetonitrile (containing 0.6 μM labetalol as internal standard). Blank was prepared similarly, but in absence of the investigated compounds. Similarly, in the inhibition study, test compound (10 μM) was incubated in the presence (50 μM) or absence of selective inhibitor DCPIP. Proteins were precipitated by centrifugation at 5,000 rpm for 10 min (Eppendorf, Italy; centrifuge 5810 R; rotor F-45-30-11) at room temperature (RT), and an aliquot of supernatant (1 μL) was analyzed by LC-MS/MS on an Agilent 1200 series HPLC coupled to an Agilent 6540 UHD accurate-mass QTOF with a dual Jet Stream electron spray ionization source. Analytical separation was carried out according to the methods listed below. The mobile phase was a mixture of water (solvent A) and acetonitrile (solvent B), both containing formic acid at 0.1%. Method A was as follows: Aeris Widepore 3.6-µm (C4, 100 × 4.6-mm) column at 30 °C using a flow rate of 0.850 mL/min in a 10-min gradient elution. Gradient elution was as follows: 100:0 (A/B) to 70:30 (A/B) over 9 min, 5:95 (A/B) for 1 min, and then reversion back to 100:0 (A/B) over 0.1 min. Method B was as follows: Acquity UPLC BEH C18 1.7-µm (C18, 150 × 2.1-mm) column at 40 °C using a flow rate of 0.650 mL/min in a 10-min gradient elution. Gradient elution was as follows: 99.5:0.5 (A/B) to 5:95 (A/B) over 8 min, 5:95 (A/B) for 2 min, and then reversion back to 99.5:0.5 (A/B) over 0.1 min. The MS/MS data were processed using “MetaSite 4.2.2 Mass 3.0.22” and “WebMetabase release-3.1.9” (Molecular Discovery, Ltd.). LC-MS chromatograms in the reported figures were extracted by Mass Hunter (MassHunter Workstation Software B.06.00 Qualitative Analysis; Agilent).
Supplementary Material
Acknowledgments
We gratefully acknowledge Molecular Discovery, Ltd., for Mass-MetaSite and WebMetabase software and financial support; Nahong Qiu (F. Hoffmann-La Roche) for technical assistance; Istituto di Scienze e Tecnologie Molecolari–Consiglio Nazionale delle Ricerche (Perugia) for DFT calculation support; and Istituto di Biostrutture e Bioimmagini–Consiglio Nazionale delle Ricerche (Naples) for providing the YIQDIVASTLK peptide.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1618881114/-/DCSupplemental.
References
- 1.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 2.Cruciani G, et al. MetaSite: Understanding metabolism in human cytochromes from the perspective of the chemist. J Med Chem. 2005;48:6970–6979. doi: 10.1021/jm050529c. [DOI] [PubMed] [Google Scholar]
- 3.Sheridan RP, Korzekwa KR, Torres RA, Walker MJ. Empirical regioselectivity models for human cytochromes P450 3A4, 2D6, and 2C9. J Med Chem. 2007;50:3173–3184. doi: 10.1021/jm0613471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cruciani G, Baroni M, Benedetti P, Goracci L, Fortuna CG. Exposition and reactivity optimization to predict sites of metabolism in chemicals. Drug Discov Today Technol. 2013;10:e155–e165. doi: 10.1016/j.ddtec.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 5.Cruciani G, et al. Flavin monooxygenase metabolism: Why medicinal chemists should matter. J Med Chem. 2014;57:6183–6196. doi: 10.1021/jm5007098. [DOI] [PubMed] [Google Scholar]
- 6.Cerny MA. Prevalence of non-cytochrome p450-mediated metabolism in Food and Drug Administration-approved oral and intravenous drugs: 2006–2015. Drug Metab Dispos. 2016;44:1246–1252. doi: 10.1124/dmd.116.070763. [DOI] [PubMed] [Google Scholar]
- 7.Di L. The role of drug metabolizing enzymes in clearance. Expert Opin Drug Metab Toxicol. 2014;10:379–393. doi: 10.1517/17425255.2014.876006. [DOI] [PubMed] [Google Scholar]
- 8.Pryde DC, et al. Aldehyde oxidase: An enzyme of emerging importance in drug discovery. J Med Chem. 2010;53:8441–8460. doi: 10.1021/jm100888d. [DOI] [PubMed] [Google Scholar]
- 9.Sanoh S, Tayama Y, Sugihara K, Kitamura S, Ohta S. Significance of aldehyde oxidase during drug development: Effects on drug metabolism, pharmacokinetics, toxicity, and efficacy. Drug Metab Pharmacokinet. 2015;30:52–63. doi: 10.1016/j.dmpk.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 10.Zhang X, et al. In silico and in vitro pharmacogenetics: Aldehyde oxidase rapidly metabolizes a p38 kinase inhibitor. Pharmacogenomics J. 2011;11:15–24. doi: 10.1038/tpj.2010.8. [DOI] [PubMed] [Google Scholar]
- 11.Hutzler JM, Obach RS, Dalvie D, Zientek MA. Strategies for a comprehensive understanding of metabolism by aldehyde oxidase. Expert Opin Drug Metab Toxicol. 2013;9:153–168. doi: 10.1517/17425255.2013.738668. [DOI] [PubMed] [Google Scholar]
- 12.Akabane T, Tanaka K, Irie M, Terashita S, Teramura T. Case report of extensive metabolism by aldehyde oxidase in humans: Pharmacokinetics and metabolite profile of FK3453 in rats, dogs, and humans. Xenobiotica. 2011;41:372–384. doi: 10.3109/00498254.2010.549970. [DOI] [PubMed] [Google Scholar]
- 13.Kaye B, Offerman JL, Reid JL, Elliott HL, Hillis WS. A species difference in the presystemic metabolism of carbazeran in dog and man. Xenobiotica. 1984;14:935–945. doi: 10.3109/00498258409151492. [DOI] [PubMed] [Google Scholar]
- 14.Dittrich Ch, et al. Phase I and pharmacokinetic study of BIBX 1382 BS, an epidermal growth factor receptor (EGFR) inhibitor, given in a continuous daily oral administration. Eur J Cancer. 2002;38:1072–1080. doi: 10.1016/s0959-8049(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 15.Barr JT, Choughule K, Jones JP. Enzyme kinetics, inhibition, and regioselectivity of aldehyde oxidase. Methods Mol Biol. 2014;1113:167–186. doi: 10.1007/978-1-62703-758-7_9. [DOI] [PubMed] [Google Scholar]
- 16.Sodhi JK, et al. A novel reaction mediated by human aldehyde oxidase: Amide hydrolysis of GDC-0834. Drug Metab Dispos. 2015;43:908–915. doi: 10.1124/dmd.114.061804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torres RA, Korzekwa KR, McMasters DR, Fandozzi CM, Jones JP. Use of density functional calculations to predict the regioselectivity of drugs and molecules metabolized by aldehyde oxidase. J Med Chem. 2007;50:4642–4647. doi: 10.1021/jm0703690. [DOI] [PubMed] [Google Scholar]
- 18.Ghafourian T, Rashidi MR. Quantitative study of the structural requirements of phthalazine/quinazoline derivatives for interaction with human liver aldehyde oxidase. Chem Pharm Bull (Tokyo) 2001;49:1066–1071. doi: 10.1248/cpb.49.1066. [DOI] [PubMed] [Google Scholar]
- 19.Jones JP, Korzekwa KR. Predicting intrinsic clearance for drugs and drug candidates metabolized by aldehyde oxidase. Mol Pharm. 2013;10:1262–1268. doi: 10.1021/mp300568r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seiple IB, et al. Direct C–H arylation of electron-deficient heterocycles with arylboronic acids. J Am Chem Soc. 2010;132:13194–13196. doi: 10.1021/ja1066459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leclerc JP, Fagnou K. Palladium-catalyzed cross-coupling reactions of diazine N-oxides with aryl chlorides, bromides, and iodides. Angew Chem Int Ed Engl. 2006;45:7781–7786. doi: 10.1002/anie.200602773. [DOI] [PubMed] [Google Scholar]
- 22.Nelson D. 2005. Synthesis of hydrazine derivatives of pyridazine. US patent publication US20050137397 A1 (June 23, 2005)
- 23.Zhang C, Shokat KM. Enhanced selectivity for inhibition of analog-sensitive protein kinases through scaffold optimization. Tetrahedron. 2007;63:5832–5838. [Google Scholar]
- 24.Keck JH, Simpson RA, Wong JL. Reactivities and electronic aspects of nucleic acid heterocycles. 7. Regiospecific substituent effects in 6-substituted purines as measured by proton magnetic resonance. J Org Chem. 1978;43:2587–2590. [Google Scholar]
- 25.Laufer SA, Domeyer DM, Scior TR, Albrecht W, Hauser DR. Synthesis and biological testing of purine derivatives as potential ATP-competitive kinase inhibitors. J Med Chem. 2005;48:710–722. doi: 10.1021/jm0408767. [DOI] [PubMed] [Google Scholar]
- 26.McCallum JEB, Huston KG, McSweeney JM, Rucker BG. Synthesis of lipophilic n-9-benzylguanine derivatives. Synlett. 2010;2010:2871–2874. [Google Scholar]
- 27.Dietz J, et al. 2009. Substituted pyrazinylmethyl sulfonamides for use as fungicides. Patent Cooperation Treaty Appl WO2009112523 A1 (September 17, 2009) and US patent publication US 12/921,504 (January 6, 2011)
- 28.Palek L, et al. Synthesis, antimycobacterial and antifungal evaluation of 3-arylaminopyrazine-2,5-dicarbonitriles. Arch Pharm (Weinheim) 2008;341:61–65. doi: 10.1002/ardp.200700119. [DOI] [PubMed] [Google Scholar]
- 29.Hu L, et al. Synthesis and activity of azaterphenyl diamidines against Trypanosoma brucei rhodesiense and Plasmodium falciparum. Bioorg Med Chem. 2009;17:6651–6658. doi: 10.1016/j.bmc.2009.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dalvie D, et al. Effect of structural variation on aldehyde oxidase-catalyzed oxidation of zoniporide. Drug Metab Dispos. 2012;40:1575–1587. doi: 10.1124/dmd.112.045823. [DOI] [PubMed] [Google Scholar]
- 31.Zamora I, Fontaine F, Serra B, Plasencia G. High-throughput, computer assisted, specific MetID. A revolution for drug discovery. Drug Discov Today Technol. 2013;10:e199–e205. doi: 10.1016/j.ddtec.2012.10.015. [DOI] [PubMed] [Google Scholar]
- 32.Barr JT, Jones JP, Joswig-Jones CA, Rock DA. Absolute quantification of aldehyde oxidase protein in human liver using liquid chromatography-tandem mass spectrometry. Mol Pharm. 2013;10:3842–3849. doi: 10.1021/mp4003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frisch MJ, et al. GAUSSIAN 09, Revision E.01. Gaussian, Inc.; Wallingford, CT: 2009. [Google Scholar]
- 34.Besler BH, Merz KM, Kollman PA. Atomic charges derived from semiempirical methods. J Comput Chem. 1990;11:431–439. [Google Scholar]
- 35.Pryde DC, et al. Medicinal chemistry approaches to avoid aldehyde oxidase metabolism. Bioorg Med Chem Lett. 2012;22:2856–2860. doi: 10.1016/j.bmcl.2012.02.069. [DOI] [PubMed] [Google Scholar]
- 36.Baroni M, Cruciani G, Sciabola S, Perruccio F, Mason JS. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for ligands and proteins (FLAP): Theory and application. J Chem Inf Model. 2007;47:279–294. doi: 10.1021/ci600253e. [DOI] [PubMed] [Google Scholar]
- 37.Cross S, Baroni M, Carosati E, Benedetti P, Clementi S. FLAP: GRID molecular interaction fields in virtual screening. validation using the DUD data set. J Chem Inf Model. 2010;50:1442–1450. doi: 10.1021/ci100221g. [DOI] [PubMed] [Google Scholar]
- 38.Coelho C, et al. Structural insights into xenobiotic and inhibitor binding to human aldehyde oxidase. Nat Chem Biol. 2015;11:779–783. doi: 10.1038/nchembio.1895. [DOI] [PubMed] [Google Scholar]
- 39.Sharma R, et al. Deuterium isotope effects on drug pharmacokinetics. I. System-dependent effects of specific deuteration with aldehyde oxidase cleared drugs. Drug Metab Dispos. 2012;40:625–634. doi: 10.1124/dmd.111.042770. [DOI] [PubMed] [Google Scholar]
- 40.Testa B, Mayer JM. Hydrolysis in Drug and Prodrug Metabolism: Chemistry, Biochemistry, and Enzymology. Chap 4 Verlag Helvetica Chimica Acta; Zurich: 2003. The hydrolysis of amides. [Google Scholar]
- 41.Carosati E, Sciabola S, Cruciani G. Hydrogen bonding interactions of covalently bonded fluorine atoms: from crystallographic data to a new angular function in the GRID force field. J Med Chem. 2004;47:5114–5125. doi: 10.1021/jm0498349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.