

Abstract
A simple classification scheme that uses only the presence or absence of a protein domain architecture has been used to determine the phylogeny of 174 complete genomes. The method correctly divides the 174 taxa into Archaea, Bacteria, and Eukarya and satisfactorily sorts most of the major groups within these superkingdoms. The most challenging problem involved 119 Bacteria, many of which have reduced genomes. When a weighting factor was used that takes account of difference in genome size (number of considered folds), small-genome taxa were mostly grouped with their full-sized counterparts. Although not every organism appears exactly at its classical phylogenetic position in these trees, the agreement appears comparable with the efforts of others by using sophisticated sequence analysis and/or combinations of gene content and gene order. During the course of the study, it emerged that there is a core set of ≈50 folds that is found in all 174 genomes and a single fold diagnostic of all Archaea.
Keywords: fold superfamily
The advent of the era of complete genome sequences has led to a variety of approaches for determining the evolutionary history of organisms over and beyond the comparison of the sequences themselves (1–4), including the use of such features as concatenated protein sequences (5, 6), gene content (1–3, 7), gene order (8–10), and the distribution of structural folds (11–15). Such efforts have continued even though there are those who feel the construction of a unified phylogeny is a hopeless task, horizontal gene transfers having been too pervasive to allow a singular depiction (16). In this vein, it is fair to say that the resulting phylogenies have not been entirely consistent between one method and another, and certainly none on its own has resulted in a wholly satisfactory classification. Attempts to filter out anomalies (17) or the use of combinations of various approaches (9, 10) have been more satisfactory, but incongruities remain.
The principal goal of these endeavors is to generate a phylogeny that best represents the evolutionary histories of the taxa represented, and that resolves previous incongruities. It is generally agreed that three major forces are at work in modifying the genetic information in any genome: (i) expansion (gene duplication), (ii) deletion (gene loss), and (iii) exchange (horizontal transfer) (18–22). Additionally, there must be some degree of de novo “gene genesis,” the concoction of new genes by various means (23). The challenge is to find the level of informational bundling that best accounts for this combination of events.
Here we report a simple scheme that uses a structural attribute, the protein domain content, as the principal determinant of relatedness. In particular, we have focused on the fold superfamily level (FSF) as opposed to the fold grouping itself that has been used by many other workers in the past (11–15). It is a subtle but critical distinction (14). The mere presence or absence of an FSF in a genome, as opposed to its overall abundance, was used as the raw material for classification. In an examination of 174 organisms whose complete genomes have been determined, the method readily distinguishes the three major groupings of life. Beyond that, it correctly divides the Archaea into crenarchs and euryarchs and groups the Eukarya into animals, plants, fungi, and others (protists). The most challenging part of the phylogeny reconstruction involved the 119 Bacteria, many of which are parasites and have greatly reduced genomes compared with their nearest relatives. When a weighting scheme that takes account of genome size (actually specific domain content) was used, these organisms were mostly clustered together with their proper groups.
Methods
Data Sources. The Structural Classification of Proteins (SCOP) database (24, 25) provides a hierarchical listing of all protein domains published in the Protein Data Bank (26). Version 1.65 has sorted 50,000 domains into 800 defined folds that are assigned to 1,294 superfamilies and that are further subdivided into 2,327 families. The superfamily level of folds is defined as those folds for which there is good evidence of common ancestry (24). It is distinguished on the one hand from the fold level itself, for which there is either weak or no evidence for common ancestry over and beyond a similar arrangement of secondary structure elements, and on the other from the family level, where sequence similarities on their own are strong enough to indicate common ancestry. To this end, the Superfamily database (27, 28) was the source of all domain assignments. Release 1.65 covers 212 complete genomes, but many of the entries are for strains of the same species. In the study described here, only a single strain of any given species was used, reducing the number of genomes to 174. These genomes include 19 Archaea, 36 Eukarya, and 119 Bacteria.
The Superfamily database depends on a hidden Markov model homology searching algorithm (29) to search the National Center for Biotechnology Information Entrez Genome database for identification of superfamily fold members. At this point ≈60% of the ORFs in the 174 completed genomes have been assigned to domain superfamilies (28). The Superfamily database hidden Markov model searching protocol employs a probability cutoff of E = 2 × 10-2 for identifying likely members of a group; it also provides a confidence level (in the form of an E value) for every candidate identified. At the outset of our study, we had found that a plot of the number of alleged superfamily members versus e value for each of the major life groupings (Archaea, Bacteria, and Eukarya) showed a sharp inflection point for each group at an E value of 10-4, suggesting that a surer gauge of superfamilies needed a more stringent cutoff. Accordingly, we omitted all entries with E values greater than E = 10-4. This cutoff reduced the total number of superfamilies assigned to any organism by 8–10%. More recent postings at the Superfamily web site have data which, when plotted as described above, show a smooth increase of the domains included, meaning no inflection point is evident. Uncertain as to what cutoff to use, we studied three sets of superfamily folds where the cutoffs were set at E = 10-6, E = 10-4, and E = 2 × 10-2, respectively, and made phylogenetic trees from them. The differences in trees were negligible, and in the end we stuck to our original cutoff point of E = 10-4.
Data Management. The Superfamily listing of folds was downloaded and the information stored in a simple matrix with 174 columns corresponding to the organisms and 1,294 rows corresponding to the FSFs. The presence or absence of a particular FSF in a given organism was denoted with a value of 1 or 0, respectively. Similar approaches have been used in the past (11). Pairwise distances were calculated by considering pairs of taxa and subtracting the values in each cell from the corresponding pairmate. Nonzero values were tallied and stored in a distance matrix. A distance matrix compiled from abundances was treated similarly after normalization across all pairs.
The great differences in genome size and gene content among the Bacteria can confound the protein domain content approach, and a weighting factor was needed that could take account of massive gene loss. In those cases, distances were corrected according to the following relationship:
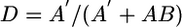 |
[1] |
where A′ is number of unique superfamily folds in the smaller of two genomes, A and B, and AB is the number of superfamily folds they share. As an example, consider the case of two closely related bacteria, one free-living and the other an intracellular parasite with a reduced genome. Clearly, the free-living species should contain most of the domains found in the parasite, and the number unique to the parasitic species (A′) ought to be very small. The two tendencies are acknowledged by setting the evolutionary distance equal to the ratio of the unique domains in the smaller genome (A′) to its total number of domains (A′ + AB). In the limit, if the parasite has no unique domains relative to the free-living species, which is to say it had only experienced massive domain losses, the evolutionary distance would remain at zero. Similar (but different) procedures for weighting have been used by others in the past (9, 14).
Phylogenetic Methods. Phylogeny construction was performed with programs available from the phylip (30) web site (http://evolution.genetics.washington.edu/phylip.html). The procedures we used were the unweighted pair group method with arithmetic mean (31) and the neighbor-joining method (NJ) (32), in each case with and without bootstrapping.
Trees were drawn with treeview (taxonomy.zoology.gla.ac.uk/rod/treeview.html). Although bootstrapping was applied to all trees, the large numbers of taxa involved made it difficult to append bootstrap values to every node in every illustration. As such, values have been appended to the figures selectively at major branch points.
In the illustrations, taxa are mostly abbreviated with an uppercase letter for genus and three lowercase letters for species (e.g., Ecol). Some exceptions to this rule were needed to avoid duplication.
Supporting Information. Figs. 6–10 and Tables 1–5 are published as supporting information on the PNAS web site.
Results
Eukarya. A simple NJ phylogenetic tree based on the presence or absence of FSFs had major clades consisting of animals, fungi, and plants, respectively, all with high bootstrap values (Fig. 1). Within the animal clade, organisms were well positioned, the single exception being that Xenopus tropicalis branched off below the two fish. However, a check of the Xenopus superfamily listing in Superfamily revealed 74 entries not found in any other vertebrate, 40 of which were not present in any other animal, and 19 not present in any other eukaryote. The majority of these entries were bacterial in nature, and it seemed possible that the anomalous position of X. tropicalis was due to contaminating sequences. Accordingly, three additional Eukarya trees were regenerated in which the 19, 40, and 74 suspect FSFs were omitted, respectively. When the 19 FSFs not found in other eukaryotes were omitted, the branching order remained the same, but when the 40 not found in other animals were put aside, X. tropicalis assumed its correct position, branching off between fish and birds (chicken, Ggal). When all 74 suspect sequences were removed, X. tropicalis branched off between the bird and mammals. The experiment strongly suggests that the preliminary X. tropicalis genome sequence is contaminated. Alternative tree contructions are found in Figs. 6 I–L.
Fig. 1.

NJ tree of 36 Eukarya based exclusively on presence or absence of SCOP superfamily folds. Bootstrap numbers are limited to the major nodes.
Archaea. The 19 Archaea genomes were readily divided into the four Crenarchaeota and 15 Euryarchaeota (Fig. 2). Beyond that, all of the methanogens fell into a single clade, which also included the sulfate-reducing Archaeoglobus fulgidus. The three Pyrococci bunched together (Pfur, Paby, and Phor), as did the three Thermoplasmata (Tvol, Taci, and Ptor). The small-genomed and enigmatic Nanoarchaeum equitans, reportedly the only known archaeal parasite (33), appeared near the root of the branch leading to the Pyrococci. When the weighting factor was used, N. equitans moved higher on to the same branch, and the bootstrap value improved from 0.595 to 0.968.
Fig. 2.

NJ tree of 19 Archaea based exclusively on presence or absence of SCOP superfamily folds. Bootstrap numbers are limited to the major nodes.
Halobacteria appeared on a separate basal branch, in agreement with trees based on gene content (1) and gene order (9) but not with trees based on ribosomal RNA, which generally cluster Halobacteria with Methanosarcinae (33). Alternative tree constructions are found in Fig. 6 A–D.
Bacteria. All told, 13 different classical phyla were represented among the 119 bacterial genomes studied. These phyla included the five classes of 53 different proteobacteria (14 α, 7 β, 25 γ, 3 δ, and 4 ε) and four different classes of 31 Fermicutes (9 Mollicutes, 10 Bacillales, 8 Lactobacillales, and 4 Clostridia). Several groups included parasitic representatives with severely reduced genomes; indeed, the range of genome sizes among the Bacteria is >20-fold.
Phylogenies were generated with both the unweighted pair group method with arithmetic mean and NJ procedures, with and without the weighting factor, and the trees evaluated with regard to how the phyla were distributed. In fact, there were significant differences among the four sets of results. With both methods, use of the weighting factor helped provide consistency in terms of keeping members of a given phylum clustered together. The phylogenetic tree constructed by the NJ method with the weighting factor was judged the best (Fig. 3). The other three trees are Fig. 6 E, G, and H.
Fig. 3.

NJ tree (with weighting) of 119 Bacteria based exclusively on the presence or absence of SCOP superfamily folds. Asterisks denote anomalously positioned taxa.
The NJ tree with weighting divided the 119 taxa into several major sectors corresponding to phyla, including one that contained all 53 proteobacteria, a subsector of the six cyanobacteria, and a branch to the lone representative of the Aquficiae (Fig. 3). The two Deinococci appear anomalously among the proteobacteria (major anomalies are denoted with asterisks). Other major groups include all 31 fermicutes, the 11 Actinobacteria, and the 5 Chlamydiae. In a significant anomaly, the four spirochaetes separate into three sectors. In the case of the Mollicutes, eight of the nine entries cluster together, but onion yellow phytoplasma forms a separate branch. Three of four Clostridia cluster together, but Thermoanaerobacter tengcongensis (Tten) is grouped with Thermotoga (Tmar). The remaining phyla are discretely and satisfactorily positioned; these are the two Bacteroidetes and the lone taxa of Chlorobi, Fusobacteria, Planctomycetes, and Thermotogae, respectively (Fig. 3).
Overall Phylogeny. Both unweighted pair group method with arithmetic mean and NJ were used to construct trees for all 174 taxa. The unweighted pair group method with arithmetic mean tree readily divided the taxa into the three superkingdoms, but many bacteria with small genomes fell into a clade separate from their nearest relatives within the bacterial zone (Figs. 6 M–P). The use of the weighting factor led to most of them being redistributed to their proper positions. In the case of the NJ tree, the three superkingdoms were mostly correct, but two organisms with reduced genomes, the archaeon N. equitans and the eukaryote E. cuniculi, fell among the Bacteria. When the weighting factor was used, the two anomalously placed small-genome organisms retreated to their correct realms, and the tripartite grouping was maintained with high bootstrap values (Fig. 4). Concomitantly, however, a number of rearrangements occurred within the three major groups; in the end, the best phylogenetic trees were generated when the taxa were restricted to a single superkingdom.
Fig. 4.

Overall phylogeny (NJ) of 174 organisms for which complete genomes have been determined. Bootstrap number was limited to the major branch point.
Genomic Occurrence of FSFs. Of the 1,294 FSFs examined, 50, mostly found in viruses, did not occur in any of the 174 genomes under study. The Venn diagram in Fig. 5A shows the census of the remaining 1,244 FSFs among the three superkingdoms. In line with previous reports (13, 15), a fold is counted if it occurs even once in a given realm. The numbers follow a similar pattern to those earlier reports (13, 15), about half of all folds occurring at some frequency in all three superkingdoms. Eukaryotes have the largest number of folds not found in the other two superkingdoms and Archaea the fewest. Bacteria share more folds with eukaryotes than they do with Archaea. Showing that these trends are more or less independent of the different numbers of taxa representing the three groups was conducted by counting the smaller sets of genomes, beginning with one that included all 19 Archaea and 19 selected genomes from each of the other two realms (Fig. 5B). In subsequent counts, the number of taxa included for the Bacteria and Eukarya was progressively increased until the full set was reached (Tables 1–3).
Fig. 5.

Venn diagrams showing occurrence of 1,244 FSFs in the three superkingdoms. The first value in any sector reflects the occurrence of a particular FSF in any genome in that sector; values in parentheses indicate numbers found in all genomes in a sector. (A) Census based on 174 complete genomes. (B) Census based on complete genomes of 19 Archaea, 19 selected Eukarya, and 19 selected Bacteria.
As an added, and arguably a more interesting feature, we noted the counts for those folds that are both unique and ubiquitous for a given sector (Fig. 5A). Thus, 49 folds occur in all 174 genomes, and 14 folds are found in all 36 eukaryotic genomes but not elsewhere. Among the bacteria, there are no unique folds that occur in all bacterial genomes but nowhere else or even that they share uniquely with only Archaea or only Eukarya. This finding was true even when only 19 bacterial genomes were used in the count with 19 each of the other superkingdoms (Fig. 5B). Interestingly, if the count is made at the fold level, the number of folds shared by all 174 genomes is 52, hardly changed from the 49 found for the FSF level (Tables 4 and 5).
A single FSF was identified that occurs in all 19 Archaea but nowhere else. The unique FSF (SCOP superfamily d.17.6) is found in an enzyme involved in the synthesis of archaeosine, a modified base (7-formamidino-7-deazaguanosine) found exclusively in the Archaea (34). The SCOP entry has been assigned to a fold level that includes six superfamilies, including cystatin-like folds. Direct visual comparison of the six types of structure shows that the archaeal domain in question is clearly distinctive from the other superfamilies in this fold group (Fig. 9).
Discussion
Fold Level vs. Superfamily Level. A number of previous reports have used the fold level of classification for taxonomic purposes (11–15), and it may be useful to consider the subtle differences between the “fold level” (800 listed in SCOP release 1.65) and the “superfamily level” (1,294 listed). As the numbers imply, many FSFs have only one member, in which case the FSF is the fold. In contrast, other fold-level categories embrace several superfamilies. By definition, the fold level of the SCOP hierarchy lists entries that have the same secondary structure and chain topology but for which there is little or no evidence of common ancestry (28). The superfamily level is defined as comprising those folds for which there is structural and functional evidence of common ancestry, and the next level of clustering, the family, is reserved for members that have obvious sequence resemblance.
The obvious advantage of using the superfamily level is that it offers a higher level of certainty that the members of each group do in fact share common ancestry; it also provides a finer grid for classification purposes. Although many superfamily entries share common ancestry with other superfamilies assigned to the same fold, the consequences for phylogenetic reconstruction in those cases where common ancestry is not the case can be dire. In this regard, Lin and Gerstein (14) found that the superfamily level performed more ably than the fold level when the number of genomes under study was increased from eight to 20.
Presence/Absence vs. Abundance. Remarkably, the mere presence or absence of protein folds (at the superfamily level) in genomes more accurately reconstructs most of the phylogenies examined here than when the overall abundance of each domain superfamily fold in a genome was used. This result was true even with regard to the threefold distribution of Archaea, Bacteria, and Eukarya because some taxa went astray when abundance was used (Fig. 7).
Domain abundance is greatly affected by gene and chromosome duplication, which although contributing to the evolutionary distance between genomes is not a uniform process. It has long been recognized that genetic duplication begets more duplication, the natural consequence of more opportunities for homologous recombination. As a result, excessive duplication can lead to inflated distances that mask the more crucial differences in the form of gain or loss of individual domains. The protein domain content of a given genome is changed whenever (i) a new fold evolves during a long-term divergence, (ii) a fold is lost as a result of deletion of all or part of a gene, or (iii) a new fold is acquired by horizontal transfer. Ordinarily, genetic duplication on its own does not give rise abruptly to new folds.
Fold Content vs. Gene Content. It is fair to ask why the protein domain content should be any better than gene content in classifying genomes. One answer is that proteins (gene products) are modular, and many of them are mosaics of different domains (35). Indeed, the duplicated and/or shuffled domain is the foundational unit from which new protein equipment is fashioned. Genes may be retained even when the domain content changes and vice versa. Certainly protein domain content measures evolutionary change differently from gene content.
There are some other intrinsic advantages to using the simple presence or absence of a structural attribute for phylogenetic purposes. For one, there is less concern about mistaken paralogy, as so often occurs when comparing protein sequences. Moreover, the rate of sequence change and its attendant problems of site-specific variation do not play a role, and arbitrary decisions about gene designation and function are not issues. As a general rule, also, three-dimensional structures are more highly conserved than primary sequences, allowing one to see further into the evolutionary past. It is noteworthy that in the present study, all of the decisions, arbitrary or not, about what constitutes a particular structural element and its presence or absence in a genome have been made by others (i.e., SCOP and Superfamily). As such, the results are completely objective and should be easily reproduced by anyone.
Superkingdom Fold Census. Past reports have represented the distribution of folds, shared and unique, in Venn diagrams similar to that depicted in Fig. 5, although in those reports, the count was based on folds (as opposed to fold superfamilies) (13, 15) and were necessarily limited to fewer completed genomes. Nonetheless, the number of domains in the different sectors have continued to follow a similar pattern as more genomes have been reported and more folds identified. Most reports in the past have limited the census to any occurrence among a superkingdom's genomes. A more interesting number may be a count of those folds that are both unique and ubiquitous to a superkingdom or set of superkingdoms.
In this regard, the existence of ≈50 folds that are common to all 174 genomes is a more important consideration than the overall abundance when it comes to the matter of common ancestors. It represents a core set of structures from which the most essential gene products are constructed.
Although these counts are not informative with regard to the detailed phylogenies, they do provide an interesting insight into related matters, especially when compared with those FSFs that occur uniquely and ubiquitously in any one of three superkingdoms (Fig. 5A). Although the 49 ubiquitous FSFs are mostly found in proteins involved in translation, including eight found in ribosomal proteins and six in aminoacyl-tRNA synthetases, several are clearly associated with major metabolic pathways. A full list of the 49 FSFs is included in Table 4.
That no unique and ubiquitous FSFs were found for Bacteria must be a reflection of their great diversity. The followup study with smaller numbers of bacterial and eukaryotic genomes showed that this finding was not merely attributable to the greater number of bacterial genomes available (Fig. 5B).
The single fold found in all 19 Archaea but nowhere else is noteworthy because its discovery underscores the advantage of using FSFs for phylogenetic studies. With only 19 Archaeal genomes completed, it would appear that a single (superfamily) fold, found in an enzyme responsible for modifying tRNAs, could be definitively diagnostic for membership in the Archaea (we are not suggesting such a narrow criterion). It will be of great interest to find if this fold is ever found to be absent in an archaeon or present in a bacterium or eukaryote.
The question arises as to whether enough data are in hand to make final judgments about fold occurrence and the overall phylogeny. More folds are likely to be discovered, and certainly more genomes will be sequenced. Interestingly, in a prior study made when there were only eight completed genomes, Gerstein (11) found that only 30 folds were found in all of them. Now that we have identified ≈50 such ubiquitous folds is not solely because new folds have been discovered. Inspection of Gerstein's data (11) reveal that many of the folds that have been added to the inventory were listed in that earlier report among those that occurred in 7 of the 8 genomes. What has improved in the interval is the sensitivity and certainty for correlating folds with ORFs in a genome. In this regard, currently >60% of the ORFs in the wholly sequenced genomes are being correlated with FSFs (28), whereas in early efforts such identifications amounted to <25% (11).
More genomes and better identifications not withstanding, it should be possible to estimate the ultimate number of FSFs on the basis of current data. To this end, the following exercise was conducted. The 174 genomes were sampled without replacement 1,000 times and the FSFs among them tallied to see how many were found in all chosen taxa. A smooth decay curve resulted (Fig. 10 Upper), the curve-fitting for which was best matched by the general exponential expression
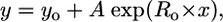 |
[2] |
where x is the number of taxa used and y the number of ubiquitous FSFs. In the limit, when x approaches infinity, y = 45.4. By this reasoning, we feel that the number of ubiquitous FSFs is not likely to change radically in the future.
The Tree of Life. In recent years, the question has been raised as to whether there is a genuine tree of life with a common ancestral organism at its root (36–39). Instead, challengers propose that the three superkingdoms arose independently from a community of primitive and undifferentiated cells that were freely exchanging their genetic components. What the three lineages shared, and what gives rise to the trichotomy provided by ribosomal RNA comparisons, was a common translation machinery but not much else. Woese (39) refers to the transition point at which vertically transmitted genes began to weigh more heavily than horizontally transferred ones as the Darwinian Threshold. The transition was marked, he feels, by a level of “componentry” that for organizational reasons could no longer survive by reckless and promiscuous exchange. He posits that the threshold came well after the three realms of life had emerged.
We feel that the census of folds sheds light on the matter. The discovery that there are 49 superfamily folds common to all 174 genomes seems to us to argue that there was a last common ancestor for all three superkingdoms, and that it had a very sophisticated genetic inventory of structural equipment, as represented by the 49 different domains being found in a wide range of gene products over and beyond those having to do with translation.
In summary, a simple tallying of FSFs for 174 fully sequenced genomes has been used for constructing phylogenies that are in good accord with those based on ribosomal RNA sequences and other genetic information. The detailed distribution of folds among the Archaea, Bacteria, and Eukarya is further revealing about the evolutionary history of life on Earth and may ultimately prove useful for detailing the routes of their diversification. Recent studies showing how structural differences in folds are only compatible with a scenario of divergence (40) underscore the promise of the approach.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health Grant P01 GMB3208.
Abbreviations: FSF, fold superfamily; NJ, neighbor joining; SCOP, Structural Classification of Proteins.
References
- 1.House, C. H. & Fitz-Gibbon, S. T. (2002) J. Mol. Evol. 54, 539-547. [DOI] [PubMed] [Google Scholar]
- 2.Wolf, Y. I., Rogozin, I. B., Grishin, N. V. & Koonin, E. V. (2002) Trends Genet. 18, 472-479. [DOI] [PubMed] [Google Scholar]
- 3.Tekaia, F., Lazcano, A. & Dujon, B. (1999) Genome Res. 9, 550-557. [PMC free article] [PubMed] [Google Scholar]
- 4.Bansal, A. K. & Meyer, T. E. (2002) J. Bacteriol. 184, 2260-2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown, J. R., Douady, C. J., Italia, M. J., Marshall, W. E. & Stanhope, M. J. (2001) Nat. Genet. 28, 281-285. [DOI] [PubMed] [Google Scholar]
- 6.Baldauf, S. L., Roger, A. J, Wenk-Siefert, I. & Doolittle, W. F. (2000) Science 290, 972-977. [DOI] [PubMed] [Google Scholar]
- 7.Snel, B., Bork, P. & Huynen, M. A. (1999) Nature Gen. 21, 108-110. [DOI] [PubMed] [Google Scholar]
- 8.Dandekar, T., Snel, B., Huynen, M. & Bork, P. (1998) Trends Biochem. Sci. 23, 324-328. [DOI] [PubMed] [Google Scholar]
- 9.Korbel, J. O., Snel, B., Huynen, M. A. & Bork, P. (2002) Trends Genet. 18, 158-162. [DOI] [PubMed] [Google Scholar]
- 10.Wolf, Y. I., Rogozin, I. B., Grishin, N. V., Tatusov, R. L. & Koonin, E. V. (2001) BMC Evol. Biol. 1, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerstein, M. (1998) Proteins Struct. Funct. Genet. 33, 518-534. [DOI] [PubMed] [Google Scholar]
- 12.Gerstein, M. & Hegyi, H. (1998) Microbiol. Rev. 22, 277-304. [DOI] [PubMed] [Google Scholar]
- 13.Wolf, Y. I., Brenner, S. E., Bash, P. A. & Koonin, E. V. (1999) Genome Res. 9, 17-26. [PubMed] [Google Scholar]
- 14.Lin, J. & Gerstein, M. (2000) Genome Res. 10, 8088-8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caetano-Anolles, G. & Caetano-Anolles, D. (2003) Genome Res. 13, 1563-1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bapteste, E., Boucher, Y., Leigh, J. & Doolittle, W. F. (2004) Trends Microbiol. 12, 406-411. [DOI] [PubMed] [Google Scholar]
- 17.Clarke, G. D., Beiko, R. G., Ragan, M. A. & Charlebois, R. L. (2002) J. Bacteriol. 184, 2072-2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roelofs, J. & Van Hastert, P. J. M. (2001) Nature 411, 1013-1014. [DOI] [PubMed] [Google Scholar]
- 19.Rokas, A., Williams, B. L., King, N. & Carroll, S. B. (2003) Nature 425, 798-804. [DOI] [PubMed] [Google Scholar]
- 20.Kunin, V. & Ouzounis, C. A. (2003) Genome Res. 13, 1589-1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krylov, D. M., Wolf, Y. I., Rogozin, I. B. & Koonin, E. V. (2003) Genome Res. 13, 2229-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gogarten, J. P., Doolittle, W. F. & Lawrence, J. G. (2002) Mol. Biol. Evol. 19, 2226-2238. [DOI] [PubMed] [Google Scholar]
- 23.Snel, B., Bork, P. & Huynen, M. A. (2002) Genome Res. 12, 17-25. [DOI] [PubMed] [Google Scholar]
- 24.Murzin, A. G., Brenner, S. E., Hubbard, T. & Chothia, C. (1995) J. Mol. Biol. 247, 536-540. [DOI] [PubMed] [Google Scholar]
- 25.Lo Conte, L., Brenner, S. E., Hubbard, T. J., Chothia, C. & Murzin, A. G. (2002) Nucleic Acids Res. 30, 264-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000) Nucleic Acids Res. 28, 235-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gough, J., Karplus, K., Hughey, R. & Chothia, C. (2001) J. Mol. Biol. 313, 903-919. [DOI] [PubMed] [Google Scholar]
- 28.Gough, J. & Chothia, C. (2002) Nucleic Acids Res. 30, 268-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karplus, K., Barrett, C. & Hughey, R. (1998) Bioinformatics 14, 846-856. [DOI] [PubMed] [Google Scholar]
- 30.Felsenstein, J. (1989) Cladistics 5, 164-166. [Google Scholar]
- 31.Sokal, R. R. & Michener, C. D. (1958) Univ. Kans. Sci. Bull. 28, 1409-1438. [Google Scholar]
- 32.Saitou, N. & Nei, M. (1987) Mol. Biol. Evol. 4, 406-425. [DOI] [PubMed] [Google Scholar]
- 33.Waters, E., Hohn, M. J., Ahel, I., Graham. D. E., Adams, M. D., Barnstead, M., Beeson, K. Y., Bibbs, L., Bolanos, R., Keller, M., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 12984-12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishitani, R., Nureki, O., Fukai, S., Kijimoto, T., Nameki, N., Watanabe, M., Kondo, H., Sekine, M., Okada, N., Nishimura, S. & Yokoyama, S. (2002) J. Mol. Biol. 318, 665-677. [DOI] [PubMed] [Google Scholar]
- 35.Doolittle, R. F. (1995) Annu. Rev. Biochem. 64, 287-314. [DOI] [PubMed] [Google Scholar]
- 36.Kandler, O. (1994) Syst. Appl. Microbiol. 16, 501-509. [Google Scholar]
- 37.Koga, Y., Kyuragi, T., Nishihara, M. & Sone, N. (1998) J. Mol. Evol. 46, 54-63. [DOI] [PubMed] [Google Scholar]
- 38.Woese, C. (1998) Proc. Natl. Acad. Sci. USA 95, 6854-6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woese, C. (2002) Proc. Natl. Acad. Sci. USA 99, 8742-8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deeds, E. J., Shakhnovich, B. & Shakhnovich, E. I. (2004) J. Mol. Biol. 336, 695-706. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.