

Abstract
Nucleotide excision repair (NER) is critical for the repair of DNA lesions induced by UV radiation, but its contribution in replicating cells is less clear. Here, we show that dual incision by NER endonucleases, including XPF and XPG, promotes the S-phase accumulation of the BRCA1 and Fanconi anemia–associated DNA helicase FANCJ to sites of UV-induced damage. FANCJ promotes replication protein A phosphorylation and the arrest of DNA synthesis following UV irradiation. Interaction defective mutants of FANCJ reveal that BRCA1 binding is not required for FANCJ localization, whereas interaction with the mismatch repair (MMR) protein MLH1 is essential. Correspondingly, we find that FANCJ, its direct interaction with MLH1, and the MMR protein MSH2 function in a common pathway in response to UV irradiation. FANCJ-deficient cells are not sensitive to killing by UV irradiation, yet we find that DNA mutations are significantly enhanced. Thus, we considered that FANCJ deficiency could be associated with skin cancer. Along these lines, in melanoma we found several somatic mutations in FANCJ, some of which were previously identified in hereditary breast cancer and Fanconi anemia. Given that, mutations in XPF can also lead to Fanconi anemia, we propose collaborations between Fanconi anemia, NER, and MMR are necessary to initiate checkpoint activation in replicating human cells to limit genomic instability.
Introduction
Repair of UV irradiation–induced DNA damage depends on the nucleotide excision repair (NER) pathway. Underscoring the essential role of NER in repair of UV-induced DNA damage, inherited defects in NER genes result in the skin cancer–prone disease xeroderma pigmentosum (1). In nonreplicating cells, NER factors sense UV-induced DNA damage and excise the lesion in a multistep process. The remaining short ssDNA region serves as a template for repair synthesis, “gap” repair (2, 3). Lesions escaping NER-dependent gap repair stall replication forks and initiate checkpoint responses. Some NER factors interact with the replisome and contribute to the early S-phase checkpoint response (4, 5). In postreplication, repair lesions are managed largely through DNA-damage tolerance mechanisms (6-8). Among the recent factors found to be involved in this process is the hereditary breast cancer–associated gene product BRCA1, which function independently of NER to suppress mutations (9).
Several lines of evidence indicate that UV-induced damage is also limited by proteins of the DNA mismatch repair (MMR) pathway. MMR factors induce checkpoints, apoptosis, preserve genomic stability, and suppress cancer induced by UV irradiation (10-14). The mechanism by which MMR functions in response to UV irradiation could stem from its general role in genome surveillance and mismatch correction. Canonical MMR begins with the recognition of replication errors (15), where MSH2–MSH6 (MutSα) or MSH2–MSH3 (MutSβ) assemble and recruit the heterodimer MLH1–PMS2 (MutLα). These complexes function in the repair of mismatched bases. As such, loss of MMR confers a mutator phenotype and a predisposition to hereditary nonpolyposis colon cancer (HNPCC; ref. 16). However, it is also well appreciated that MMR proteins respond to DNA damage from exogenous sources, such as to DNA alkylating agents, known to induce mismatches following DNA replication (17). In response to UV irradiation, MMR factors could have an alternative noncanonical role in UV lesion processing given that the MSH2–MSH6 complex directly binds UV lesions (18, 19). Clarifying how MMR contributes to genomic stability in the UV response will be central to understanding the HNPCC variant, Muir–Torre syndrome that is characterized by skin cancers (20-22).
Both the MMR protein MLH1 and BRCA1 bind directly to the DNA helicase FANCJ, which has essential functions in activating checkpoints following replication stress, although it has not hitherto been linked to the UV-induced damage response (23-27). FANCJ is mutated in hereditary breast and ovarian cancer as well as in the rare cancer-prone syndrome Fanconi anemia (24, 28). Complementation studies using FANCJ-deficient (FA-J) patient cells demonstrated that MLH1 binding is critical for FANCJ function in the repair of DNA interstrand crosslinks (24, 29). Here, we reveal that MLH1 binding to FANCJ is also essential for the response to UV-induced damage, in which FANCJ promotes an S-phase checkpoint point and limits UV-induced mutations. Because dual incision by NER also promotes FANCJ accumulation at sites of UV-induced damage, and the NER endonuclease XPF was recently shown to be a Fanconi anemia gene (30, 31), our analysis suggests that Fanconi anemia, MMR, and NER pathways collaborate to process UV lesions in S-phase cells to preserve the genome.
Materials and Methods
Cell culture
A549, MCF7, and U2OS cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco; Life Technologies) supplemented with 10% FBS and 1% penicillin/streptomycin. FA-J, 48BR, MEFs, GM04429 XP-A, XP2YO XP-F, and XPCS1RO XP-G cells, and their respective complements were cultured in DMEM supplemented with 15% FBS and 1% penicillin/streptomycin. Patient cell lines XP2YO XP-F, XPCS1RO XP-G, and their respective complements were generated by Dr. O. Schärer and MSH2−/− or MSH2+/+ MEFs were a generous gift of J. Stavnezer.
DNA constructs
FA-J cells were infected with pOZ retroviral vectors (32), expressing FANCJWT, FANCJK141/142A, FANCJS990A, or empty vector as described earlier (23, 24). Stable cell lines were generated by sorting with anti-IL-2 magnetic beads (Dyna Beads). The pCDNA-3myc-6xhis vectors were generated with a QuickChange Site-Directed Mutagenesis Kit (Stratagene) using published primers for FANCJK52R (23). GFP-polh was expressed in U2OS cells as described in refs. 28, 33, and 34.
RNA interference
The packaging cell line 293TL was used to produce lentiviral particles containing pGIPZ or pLKO.1 vectors and 293TD cells were used to produce retroviral particles containing pStuffer vector. Cells were transfected with 1:1:2 μg of DNA packaging versus insert using Effectene Transfection Reagent (Qiagen) 48 hours before harvesting retroviral or lentiviral supernatants. Supernatants were filtered and added to recipient cell lines with 1 μg/mL polybrene. Cells infected with short hairpin RNA (shRNA) vectors were selected with either puromycin (pGIPZ, pLKO.1) or hygromycin (pStuffer). For shRNA-mediated silencing, the mature antisense was used for pLKO.1 shNSC 5′-CCGCAGGTATGCACGCGT-3′, shMLH1 5′-AATACAGAGAAAGAAGAACAC-3′, shMSH2 5′-AAACTGAGAGAGATTGCCAGG-3′, shXPF 5′-AAATCACTGATACTCTTGCGC-3′, shFANCJ 5′-TATGGATGCCTGTTTCTTAGCT-3′, for pGIPZ shNSC 5′-ATCTCGCTTGGGCGAGAGTAAG-3′, shMSH2-1 5′-ATTACTTCAGCTTTTAGCT-3′, shMSH2-2 5′-GCATGTAATAGAGTGTGCTAA-3′, shMSH6-1 TTCAACTCGTATTCTTCTGGC, and shMSH6-2 TTTCAACTCGTATTCTTCTGG. The pStuffer vectors were a generous gift of Dr. J. Chen. The pStuffer shRNA-targeting luciferase was 5′-GUGCGCUGCUGGUGCCAAC-3′, shFANCJ-1 5′-GUACAGUACCCCACCUUAU-3′, and shFANCJ-2 5′-GAUUUCCAGAUCCACAAUU-3′. RNAi-mediated depletion of Luciferase, FANCJ, or RAD18 using siRNA reagents was performed as described previously (28).
Local UV irradiation and immunofluorescence
Local UV irradiation was performed as described (35) using a 254 nm UV lamp (UVP Inc.) with a dose of 100 J/m2, although 3-or 5-μm Isopore polycarbonate membrane filters (Millipore). Cells were fixed for 10 minutes with either ice cold methanol or 3% paraformaldehyde/2% sucrose in PBS, permeabilized for 5 minutes with 0.5% Triton X-100, and treated with 0.08M NaOH for 2 minutes only before using 6-4 pyrimidine-pyrimidone (6-4 PP) or cyclobutane pyrimidine dimer (CPD) Abs. Coverslips were rinsed 3× in 1× PBS before each step. For primary and secondary staining, cells were incubated for 40 minutes each in a humid chamber, face down on a 100 μL meniscus of Abs diluted in 3% bovine serum albumin (BSA) in PBS. Primary Abs used were anti-FANCJ (1:500; Sigma; Lot #051M4759, #014K4843), anti-phospho-S4/S8 RPA32 (1:500; Bethyl), anti-MLH1 (1:200; BD Bioscience), anti-MSH2 (1:200; Calbiochem), anti-XPF (1:200; Neomarkers), anti-ERCC1 (1:500; Santa Cruz), anti-XPC (1:500; Abcam), anti-6-4 PP, and anti-CPD (both 1:1,000; CosmoBio). Secondary Abs used include Rhodamine Red-X–conjugated AffiniPure Goat anti-rabbit or anti-mouse immunoglobulin G (IgG) and fluorescein (FITC)-conjugated AffiniPure Goat anti-rabbit IgG (Jackson Immuno-Research Laboratories Inc.). Coverslips were mounted on slides using Vectashield mounting media with 4′,6-diamidino-2-phenylindole (DAPI; Vecta laboratories, Inc.) and analyzed on a fluorescence microscope (Leica DM 5500B) with a Qimaging Retiga 2000R Fast 1394 camera. For each experimental time point, ≥400 DAPI-positive cells (≥1,200 in triplicate) were analyzed using Q-Capture Pro line intensity profile software with the intensity gated at ≥0.1 for positive localized UV damages (LUD) for 6-4 PP or CPD staining. The accumulation of a protein at an LUD was considered positive if its intensity was 10-fold greater than the line drawn over the rest of the nucleus.
Mutation frequency assays
The hypoxanthine phosphoribosyltransferase (HPRT) assay was performed in A549 cells as described (36) with the following modifications. After culturing cells for 1 week in media containing hypoxanthine, aminopterin, and thymidine (HAT selection) to eliminate background HPRT mutations, cells were stably depleted of FANCJ with 2 unique shRNA targets versus a nonsilencing control (NSC) and selected with hygromycin. UV-induced HPRT mutants were obtained by seeding 6 plates at a confluence of 1 × 106 cells/10-cm dish 24 hours before either mock treatment, 5, or 10 J/m2 UV irradiation in a 254-nm Spectrolinker XL-1500 (Dot Scientific, Inc.). Posttreatment cells were allowed to recover to 6 × 106 cells or with mock cells 6 population doublings and 6 × 106 cells were seeded at a confluence of 1 × 106/10-cm dish in media containing 24 μM 6-thioguanine (6-TG) to select for HPRT-inactivated colonies. At the same time, 200 cells were also seeded in 6-TG–free media to determine colony-forming efficiency. The frequency of inactivating mutations at the HPRT locus was calculated as the [(no. of total 6-TG–resistant colonies)/(6 × 106 cells seeded)] × the colony-forming efficiency. HPRT inactivation frequency represents the mean of 3 independent experiments. Individual colonies were picked and grown until enough cells were obtained for RNA isolation using TRizol reagent (Life Technologies) according to the manufacturer’s protocol. The HPRT gene was subjected to reverse transcription-PCR using SuperScript (Invitrogen) followed by sequencing using overlapping primers HPRT1 5′-CTTCCTCCTCCTGAGCAGTC-3′, HPRT2 5′-AAGCAGATGGC-CACAGAACT-3′, HPRT3 5′-CCTGGCGTCGTGATTAGTG-3′, HPRT4 5′-TTTAC-zTGGCGATGTCAATAGGA-3′, HPRT5 5′-GACCAGTCAACAGGGGACAT-3′, and HPRT6 5′-ATGTCCCCTGTTGACTGGTC-3′. Patient-derived FA-J cells were complemented with empty vector or FANCJWT and treated as described with A549 cells. As FA-J cells do not make colonies, the % increase in 10 μmol/L 6-TG survival was calculated as the % of UV-irradiated cells surviving 6-TG minus the % of untreated cells surviving 6-TG. The % increase in 6-TG survival represents the mean of 3 independent experiments.
EdU labeling
EdU incorporation was performed as described previously (37), except cells were seeded on coverslips and left untreated or UV-irradiated through 5-μm filters before 3-hour incubation in 10 μmol/L EdU diluted in serum-free media. When using global UV irradiation, cells were left untreated and pulsed 45 minutes in 10 μmol/L EdU or UV irradiated and pulsed 16 hours later for 45 minutes with 10 μmol/L EdU. Cells were processed by Click-iT EdU Imaging Kit (Invitrogen) using the manufacturer’s instructions immediately followed by the above immunofluorescence protocol.
Western blot analysis
Cells were harvested and lysed in 150 mmol/L NETN (0.5% NP-40 detergent, 1 mmol/L EDTA, 20 mmol/L TRIS, 150 mmol/L NaCl) lysis buffer [20 mmol/L Tris (pH 8.0), 150 mmol/L NaCl, 1 mmol/L EDTA, 0.5% NP-40, 1 mmol/L phenylmethylsulfonyl fluoride, and 1 × protease inhibitor cocktail] for 30 minutes on ice. Cell extracts were clarified by centrifugation at 14,000 rpm, protein was quantified by Bradford assay, and lysates were boiled in SDS-loading buffer. Chromatin extracts were prepared as described (38). For CPD immunoprecipitation, cells were lysed in 150 mmol/L NETN buffer, spun down, and the insoluble pellet was resuspended in radioimmunoprecipitation assay buffer (RIPA) and sonicated. The RIPA fraction was spun down and chromatin lysate was quantified by Bradford assay. Lysates were then precleared with Protein A beads and immunoprecipitated overnight with CPD Abs. Proteins were separated by SDS-PAGE on 4% to 12% bis Tris or 3% to 8% Tris Acetate gels (Novex; Life Technologies) and electrotransferred onto nitro-cellulose membranes. Membranes were blocked in 5% milk diluted in PBS. Antibodies used for Western blot analysis included anti-FANCJ [1:1,000; Sigma, 1:1,000; E67 (previously described; ref. 39)], anti-Bactin (1:5,000; Sigma), anti-MLH1 (1:500; BD Bioscience), anti-MSH2 (1:500; Calbiochem), anti-MSH2 (mouse specific; Santa Cruz), anti-XPF (1:1,000; Neomarkers), anti-ERCC1 (1:500; Santa Cruz), anti-XPC (1:1,000; Abcam), anti-CHK1 (1:500; Bethyl), anti-p317 CHK1 (1:500; Bethyl), anti-RPA32 (1:500; Bethyl), and anti-phospho-S4/S8 RPA32 (1:500; Bethyl). Membranes were washed and incubated with horseradish peroxidase-linked secondary antibodies (Amersham; 1:5,000), and detected by chemiluminescence (Ambersham). The ratio of phospho-protein to total protein was measured and quantified using Image J software.
Fluorescence-activated cell sorting analysis
FA-J cells cultured to ~80% confluency were left untreated or globally irradiated with 5 J/m2 before collecting and fixing in 70% ethanol 4 hours after UV irradiation. For antibody labeling, cells were rinsed with 1 × PBS, permeabilized with 0.5% Triton-X 100 in PBS 20 minutes at room temperature, and then washed with 1% BSA/0.25% Tween-20 in PBS (PBS-TB) before resuspending 1 hour in PBS-TB with phospho-S4/S8 replication protein A (RPA) 32 antibody (1:250; Bethyl). Cells were then collected and washed 2× in PBS-TB before 1-hour incubation in PBS-TB containing FITC-conjugated AffiniPure Goat Anti-Rabbit IgG (1:200; Jackson Immuno-Research Laboratories, Inc.). After washing with PBS, cells were resuspended in RNAse A solution (100 μg/mL in PBS) for 20 minutes at room temperature and again washed with PBS before fluorescence-activated cell sorting (FACS) analysis. Cells were labeled with propidium iodide before analysis on a FACSCaliber flow cytometer (Becton-Dickinson) performed at the University of Massachusetts Medical School flow cytometry core facility using Cellquest software. The fluorescence intensity of phospho-S4/S8 RPA 32–positive cells was gated as FITC-positive cell populations compared with no antibody control.
Results
FANCJ accumulation at sites of UV-induced damage is dependent on NER dual incision
We examined the response of FANCJ to UV irradiation by assessing whether FANCJ accumulated at sites of UV-induced damage. Following UV irradiation through 3 or 5 μm filters to generate sites of LUDs (35, 40), we found that FANCJ colocalized with UV-induced 6-4 PPs, CPDs, and the NER endonu-clease XPF in the breast cancer cell line, MCF7. FANCJ localization to 6-4 PP- or XPF-positive LUDs peaked ~3 hours after UV irradiation and diminished by ~12 hours (Fig. 1A and B).
Figure 1.
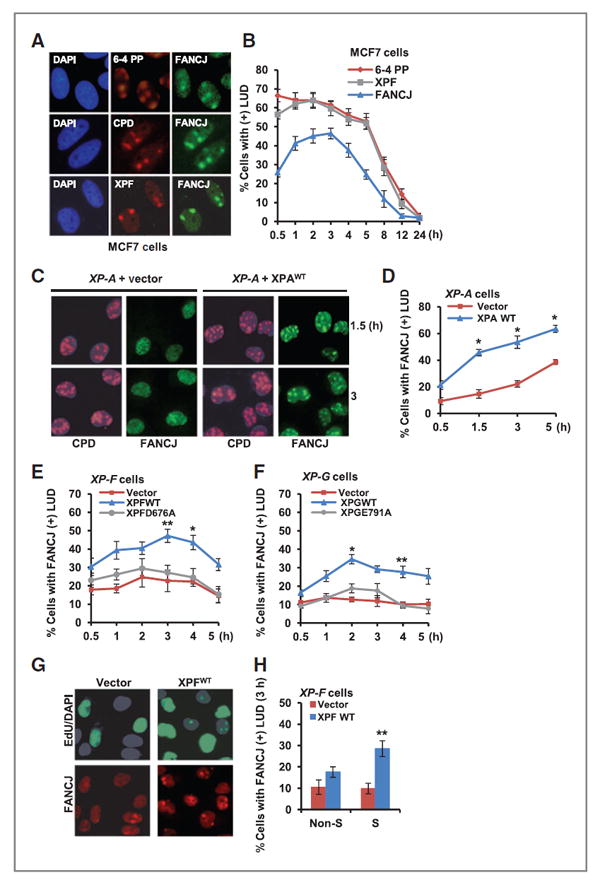
FANCJ recruitment to sites of LUDs is dependent on NER dual incision and is predominantly in S phase. A, MCF7 cells were UV irradiated through 5-μm micropore filters to generate LUDs and co-immunostained with the indicated Abs. Representative images are shown 1 hour after UV irradiation. B, quantification of MCF7 cells positive for FANCJ, XPF, or 6-4 PP LUDs. C, XP-A cells complemented with empty vector or XPAWT were UV irradiated through 3-μm micropore filters to generate LUDs and co-immunostained with the indicated Abs. D, quantification of XP-A cells with FANCJ-positive LUDs. E, XP-F cells complemented with empty vector, XPFWT, or XPFD676A were treated as in C and FANCJ-positive LUDs were quantified. F, XP-G cells complemented with empty vector, XPGWT, or XPGE791A were treated as in C and FANCJ-positive LUDs were quantified. G, XP-F cells complemented with empty vector or XPFWT were UV irradiated through 5-μm micropore filters, incubated with EdU, and co-immunostained with the indicated Abs. H, quantification of XP-F cells with FANCJ-positive LUDs. Where shown, error bars represent the standard deviation of the mean of 3 independent experiments, asterisks denote Significance from Student 2-tailed, unpaired t test: *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.005.
To address the relationship of FANCJ to NER, we used XP fibroblast cell lines and their functionally complemented counterparts. We found that the accumulation of FANCJ at LUDs was reduced ~2- to 3-fold in NER-deficient XP-A, XP-F, and XP-G cells when compared with the wild-type complemented cells (Fig. 1C–F). Contributing to FANCJ localization was XPF and XPG endonuclease activity as complementation with XPF or XPG nuclease-defective mutant species, XPFD676A or XPGE791A (41) failed to restore robust FANCJ accumulation at LUDs between ~1 and 5 hours after UV-induced damage (Fig. 1E and F). Following global UV irradiation, FANCJ foci were also more prominent in XP-F cells complemented with wild-type XPF endonuclease (Supplementary Fig. S1A and S1B). By contrast, FANCJ depletion did not affect the localization dynamics of NER factors (Supplementary Fig. S1C and S1D). Collectively, these data indicate that NER incision events potentiate the accumulation of FANCJ to sites of UV-induced damage.
NER promotes the accumulation of FANCJ at UV-induced damage in S phase
Next, we investigated whether NER contributed to FANCJ accumulation at LUDs in a specific cell-cycle phase. Cells within S phase and non-S phase can be easily distinguished after local UV irradiation by staining for 5-ethynyl-2′-deoxyur-idine (EdU) incorporation into genomic DNA (37). In S-phase cells, EdU staining is bright and pan nuclear. In non-S-phase cells, EdU staining is restricted to sites of LUDs, representing sites of unscheduled DNA synthesis that occurs during gap repair in NER (42). Following localized UV irradiation, cells were incubated in media with EdU for 3 hours and immunostained with FANCJ antibodies. Consistent with a role for XPF in NER-dependent gap filling, EdU-positive LUDs in non-S-phase cells were only present in XPFWT cells (Fig. 1G and H ). FANCJ recruitment to LUDs was not significantly improved in non-S-phase XPFWT cells, however it was significantly enhanced in S-phase XPFWT cells, indicating that XPF potentiates FANCJ accumulation in cells undergoing DNA synthesis (Fig. 1G and H).
FANCJ localization to sites of UV-induced damage is MMR dependent
FANCJ directly binds BRCA1, which functions in the response to UV irradiation selectively in S–G2 phase cells (9, 23). FANCJ also directly binds MLH1 (24), which along with other MMR factors function in the response to UV irradiation and preserve genomic integrity (43). Because both BRCA1 and MLH1 contribute to FANCJ localization and function in the DNA-damage response (24, 28, 29, 44), we investigated whether BRCA1 or MLH1 interactions were required for FANCJ localization to LUDs. We analyzed FANCJ recruitment in FANCJ-deficient FA-J patient cells complemented with empty vector, FANCJWT, the BRCA1-interaction defective mutant (FANCJS990A; ref. 45), or the MLH1-interaction defective mutant (FANCJK141/142A; ref. 24). Although the FANCJ species expressed at similar levels, we found that FANCJK141/142A localization was dramatically reduced as compared with FANCJS990A, which localized to LUDs just as efficiently as FANCJWT (Fig. 2A–C). Importantly, FANCJ-positive LUDs were not detected in FANCJ-null FA-J cells unless complemented with wild-type FANCJ, confirming the specificity of our FANCJ antibody (Fig. 2A–C). Further validating that FANCJ localization to LUDs requires functional MMR, we found that as compared with a NSC, FANCJ recruitment to LUDs was severely reduced in U2OS cells depleted of MLH1, MSH2, or MSH6 (Fig. 2D–I and Supplementary Fig. S2A and S2D). In contrast, XPC and ERCC1 recruitment to LUDs was not affected by MSH2 depletion (Supplementary Fig. S2E and S2F), indicating that MMR is required for accumulation of FANCJ at LUDs, but not XPC or ERCC1. Similarly, MSH2 recruitment to LUDs was similar in vector and XPFWT complemented XP-F cells whereas as expected ERCC1 was only present in the XPFWT complemented XP-F cells, suggesting that MMR and NER accumulation at LUDs is not inter-dependent (Supplementary Fig. S2G and S2H). We also noted that the residual accumulation of FANCJ found at LUDs in XP-F cells was eliminated by depletion of MSH2 (Fig. 2J–L), suggesting that NER and MMR operate in a parallel manner to support FANCJ localization. In the XP-F cells, however MSH2 depletion did not perturb FANCJ nuclear or chromatin localization, suggesting MMR and NER contribute to FANCJ localization to LUDs as opposed to nuclear import (Supplementary Fig. S2I).
Figure 2.
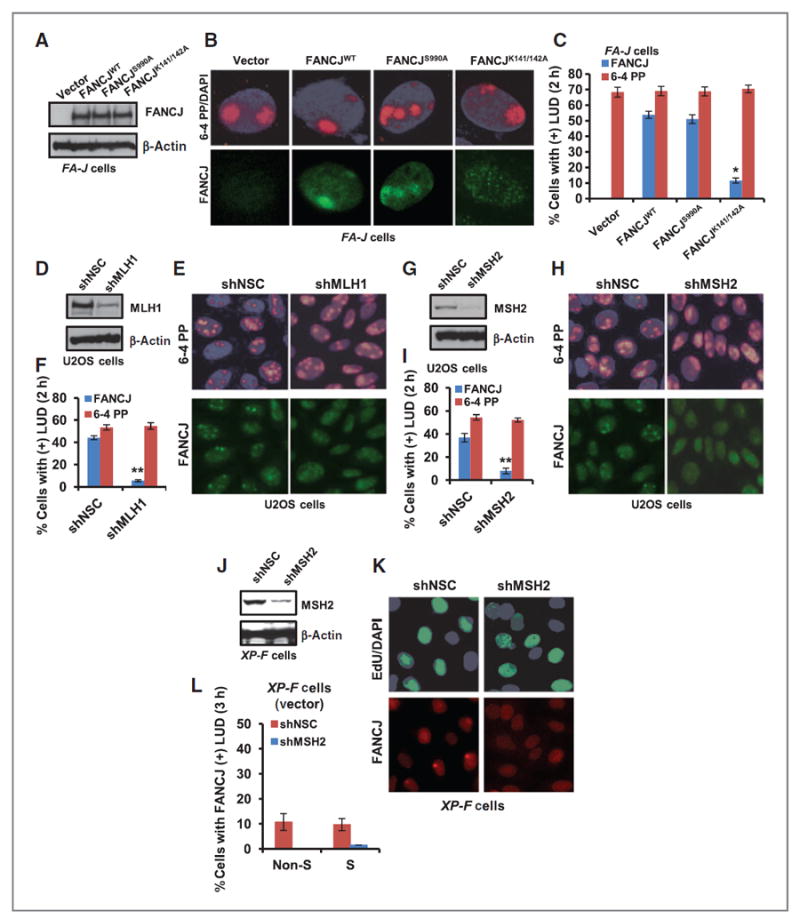
FANCJ recruitment to sites of LUDs is MMR dependent. A, FA-J cells were complemented with empty vector, FANCJWT, FANCJS990A, or FANCJK141/142A and analyzed by immunoblot. B and C, FA-J cells were UV irradiated through 5-μm micropore membrane filters, coimmunostained with the indicated Abs (B), and quantified for FA-J cells with FANCJ- and 6-4 PP–positive LUDs (C). D–F, U2OS cells containing shRNA vectors targeting MLH1 or NSC were analyzed by immunoblot (D) and UV irradiated through micropore filters (E) and quantified for cells with FANCJ- or 6-4 PP–positive LUDs (F). G–I, U2OS cells containing shRNA vectors targeting MSH2 or NSC were analyzed by immunoblot (G) and UV irradiated through micropore filters (H) and quantified for cells with FANCJ- or 6-4 PP–positive LUDs (I). J, XP-F cells complemented with empty vector were stably depleted of MSH2 versus NSC and analyzed by immunoblot. K, cells were treated as in E and processed for EdU incorporation and coimmunostained with the indicated Abs and quantified for cells with FANCJ-positive LUDs (L). Error bars represent the standard deviation of the mean of three independent experiments.
We expected that both NER and MMR would also be present in S-phase cells given that they contribute to the S-phase localization of FANCJ. However, NER proteins are best known for their UV repair function in non-S-phase cells (3) and from the literature it was not clear if MMR proteins had a cell-cycle–dependent localization to LUDs. We used primary immortalized 48BR fibroblasts that have been used to characterize NER proteins in gap repair by means of EdU incorporation (42). Although XPF was clearly present in non-S-phase cells at sites of gap filling, as expected, we also detected XPF in nearly all LUDs in S-phase cells, ~95% (Supplementary Fig. S3A–S3C). MLH1 and MSH2 were also present at LUDs with a similar percent in both non-S- and S-phase cells. Instead, FANCJ was primarily at LUDs in S-phase cells, ~86% and only in ~19% of non-S-phase cells (Supplementary Fig. S3A–S3C). Collectively, these studies show that MMR and NER proteins localize to LUDs in both non-S- and S-phase cells, whereas FANCJ localizes primarily in S-phase cells.
FANCJ promotes the UV-induced arrest of DNA synthesis and the induction of RPA phosphorylation
UV irradiation activates checkpoint responses and inhibits DNA replication in S-phase cells (46). Given the role of FANCJ in checkpoint responses (25-27) and the accumulation of FANCJ at LUDs during S phase, we tested if FANCJ contributed to the UV-induced checkpoint response. By pulsing cells with EdU, we found that FA-J cells expressing FANCJWT underwent a 10.5-fold reduction in S-phase cells when examined 16 hours after UV irradiation. By comparison, FA-J cells expressing vector underwent a 2.0-fold reduction (Fig. 3A and B), indicating that FANCJ contributes to the arrest of DNA synthesis in response to global UV irradiation.
Figure 3.
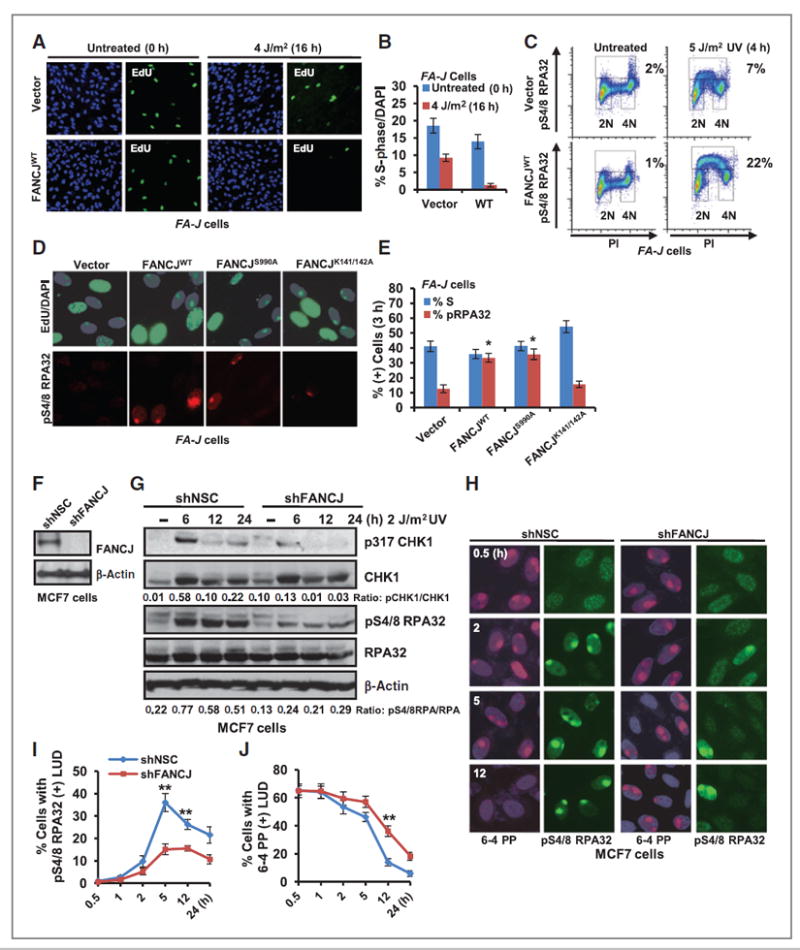
FANCJ contributes to the UV-induced checkpoint response. A, FA-J cells complemented with empty vector or FANCJWT were left untreated and pulsed for 45 minutes with 10 μmol/L EdU or globally UV irradiated and pulsed for 45 minutes with 10 μmol/L EdU 16 hours later. Cells were processed for EdU incorporation and costained with DAPI. B, quantification of EdU incorporation/total number of DAPI (+) cells. ≥1,000 DAPI cells were quantified for each experiment in triplicate. C, FA-J cells complemented with empty vector or FANCJWT were left untreated or UV irradiated and analyzed by FACS sorting for phospho-S4/8 RPA32–positive cells; representative plots are shown. D, FA-J cells were UV irradiated through 5-μm micropore filters, incubated with 10 μmol/L EdU for 3 hours, and coimmunostained with phospho-S4/8 RPA32 Ab. E, quantification of phospho-S4/8 RPA32–positive LUDs in S-phase cells. F and G, MCF7 cells containing shRNA vectors targeting FANCJ or NSC were analyzed by immunoblot with the indicated Abs (F) or at the indicated time points after UV irradiation (G). The ratio of phospho-protein/total protein by densitometry using Image J software is quantified. H, the MCF7 cells shown were UV irradiated through 5-μm filters to generate LUDs and coimmunostained with the indicated Abs at several time points. I, quantification of cells with phospho-S4/8 RPA32–positive LUDs. J, quantification of 6-4 PP–positive LUDs. Where shown, error bars represent the standard deviation of the mean of three independent experiments.
The UV-induced arrest of DNA synthesis is also associated with changes in phosphorylation of the ssDNA binding protein RPA (47). Following UV irradiation, the 32 kDa subunit of RPA is phosphorylated on several serine residues in the N-terminal of the protein in a cell-cycle–dependent manner by DNA-PK and cyclin-dependent kinases (48, 49). By examining phosphorylation of serines4/8 on RPA32 with a specific antibody, we found that in response to global UV irradiation, FANCJ complementation was sufficient to enhance RPA serines4/8 phosphorylation in FA-J cells by FACS (Fig. 3C) and immunoblot (Supplementary Fig. S3D) analyses. By FACS analysis, basal phospho-S4/8 RPA32 was ~1% to 2% in both untreated vector and wild-type FANCJ complemented FA-J cells. Following UV irradiation, phospho-S4/8 RPA32 was induced to ~22% in FANCJWT FA-J cells as compared with only ~7% in vector FA-J cells (Fig. 3C). Similarly, using phospho-S4/8 RPA32 immunostaining in conjunction with EdU pulse, we uncovered that phospho-S4/8 RPA32 staining was detected only in S-phase cells (Fig. 3D and E). Furthermore, we found that FA-J patient cells complemented with FANCJWT or the BRCA1-interaction defective mutant (FANCJS990A) had significantly greater EdU-positive S-phase cells with phospho-S4/8 RPA32–positive LUDs as compared with the FA-J cells complemented with empty vector or the MLH1-interaction defective mutant (FANCJK141/142A; Fig. 3D and E). This finding further suggested that FANCJ and the FANCJ–MLH1 interaction, but not the BRCA1 interaction, contributes to checkpoint responses in S-phase cells.
Immunoblotting in FANCJ-depleted MCF7 cells also revealed that phospho-S4/8 RPA32 as well as the soluble checkpoint factor phospho-317 CHK1 was reduced compared with NSC whereas total CHK1 and RPA levels were unchanged (Fig. 3F and G). Moreover, co-immunostaining with phospho-S4/8 RPA32 and 6-4 PP antibody was used to visually mark UV-induced LUDs and revealed that phospho-S4/8 RPA32 was significantly reduced in FANCJ-depleted cells as compared with NSC (Fig. 3H–J). Interestingly, by 12 hours post-UV damage, 6-4 PP LUDs persisted in FANCJ-depleted cells (Fig.3H–J). FANCJ or MSH2 depletion also consistently enhanced the persistence of 6-4 PP–positive LUDs in the male lung cancer cell line, A549 in which the formation of phospho-S4/8 RPA32-positive LUDs was also significantly reduced (Supplementary Fig. S4A–S4C). Furthermore, the combination of FANCJ and MSH2 depletion was not additive (Supplementary Fig. S4A–S4C), suggesting that FANCJ and MSH2 function in a common pathway that is not cell-type specific.
Recently, NER factors were shown to promote the S-phase checkpoint response, including RPA phosphorylation in response to UV irradiation (4, 5, 50). Given that the mechanism by which NER promotes the S-phase checkpoint is unclear, we considered whether the NER-dependent accumulation of FANCJ at LUDs in S phase was required. As before, XP-F patient cells were segregated into non-S- and S-phase cells by labeling with EdU and phospho-S4/8 RPA32 staining was detected only in S-phase cells (Fig. 4A and B). We found that phospho-S4/8 RPA32 induction was greatest in XP-F cells complemented with XPFWT (Fig. 4A–C). Strikingly, depletion of FANCJ or MSH2 profoundly reduced the phospho-S4/8 RPA32 induction of XPFWT complemented XP-F cells (Fig. 4A–E). Notably, the residual phospho-S4/8 RPA32–positive LUDs found in vector complemented XP-F cells were also reduced by depletion of FANCJ or MSH2 (Fig. 4A–E). Thus, FANCJ promotes S-phase checkpoint responses in not only cancer cell lines, but also in nontransformed fibroblasts. Together, these data suggest that MSH2 and FANCJ contribute to NER-dependent and -independent UV-induced phospho-S4/8 RPA32 induction at LUDs in S-phase cells. In contrast, when either FANCJ or MSH2 were depleted, we found gap filling was proficient in the XP-F cells complemented with XPFWT (Fig. 4A and B and E–H). Gap filling was also proficient in 48BR cells depleted of FANCJ, MLH1, or MSH2, but reduced in cells depleted of XPF (Supplementary Fig. S4D). Msh2−/− and Msh2+/+ mouse embryonic fibroblasts also had similar levels of gap filling (Supplementary Fig. S4E), suggesting that FANCJ and MMR factors are not required for NER-dependent gap filling.
Figure 4.
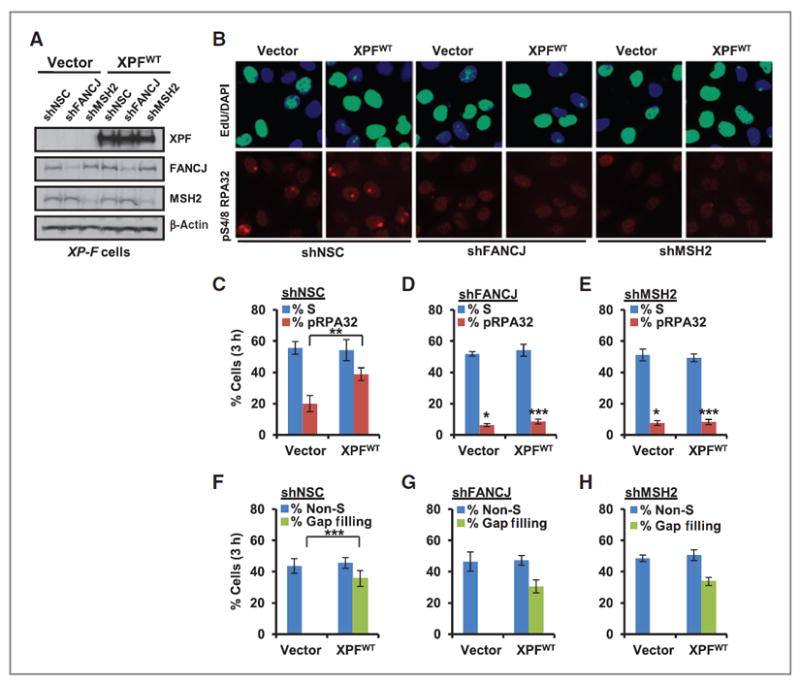
FANCJ and MSH2 are required for NER-dependent and -independent induction of RPA phosphorylation in S phase, but not for gap repair. A, XP-F cells complemented with empty vector or XPFWT were stably depleted of FANCJ, MSH2, or NSC using shRNA vectors and analyzed by immunoblot. B, cells were UV irradiated through 5-μm micropore filters, incubated with EdU, and coimmunostained with the indicated Abs 3 hours after treatment. C–E, quantification of phospho-S4/8 RPA32–positive LUDs in S-phase cells expressing shNSC (C), shFANCJ (D), and shMSH2 (E). F-H, quantification of NER-dependent gap filling in non-S-phase cells expressing shNSC (F), shFANCJ (G), and shMSH2 (H). Where shown, error bars represent the standard deviation of the mean of three independent experiments.
Collectively, our data indicate that FANCJ contributes to the UV-induced arrest of DNA synthesis by potentiating checkpoint induction pathways. Although FANCJ does not contribute to NER-dependent gap repair, it influences the clearance of UV-induced lesions in a common pathway with MSH2.
FANCJ suppresses UV-induced mutations
Given that FANCJ is dispensable for survival after UV exposure (28), we sought to examine if FANCJ preserves the integrity of the genome, as has been found for MMR (14). A549 cells are useful for analyzing mutations at the endogenous HPRT locus (36). Similar to other cell lines examined, in A549 cells FANCJ localized to sites of UV-induced damage as demonstrated by co-precipitation of FANCJ with CPD and modified proliferating cell nuclear antigen (PCNA) following UV-induced damage (Supplementary Fig. S5A). Using RNAi-mediated FANCJ silencing, we confirmed that FANCJ was not essential for survival following UV irradiation, but was essential for survival following exposure to the DNA cross-linking agent cisplatin (Fig. 5A–C and Supplementary Fig. S6A and S6B). As compared with NSC, we found that FANCJ depletion enhanced UV-induced HPRT, inactivating mutations determined by clonal selection in 6-TG (36). FANCJ depletion did not affect spontaneous HPRT mutations, but the frequency of inactivating HPRT mutations after 10 J/m2 UV irradiation was enhanced ~10-fold in A549 cells (Fig. 5D). Sequencing of clones arising from HPRT inactivation indicated no gross deletions or rearrangements as a consequence of FANCJ deficiency in response to global UV irradiation. Instead, HPRT inactivation was predominated by ~8-fold more C to T transitions in both the transcribed strand and nontranscribed strand (NTS) of HPRT in FANCJ-depleted cells (Fig. 5E and F and Supplementary Table S1). These findings suggest that FANCJ is involved in a specific process that suppresses the formation of point mutations in response to UV irradiation. Further supporting that FANCJ suppresses inactivating mutations at the HPRT locus, FANCJWT complementation in FANCJ-deficient FA-J patient cells was sufficient to reduce survival in 6-TG after UV irradiation, indicating that FANCJ averted the occurrence of mutations (Supplementary Fig. S6C). Furthermore, complementation with FANCJWT enhanced resistance to DNA cross-linking agent, mitomycin C, although in accordance with the results from Fig. 4C, the resistance to UV irradiation was unchanged (Supplementary Fig. S6D and S6E). Together, these results suggest that FANCJ, similar to MMR (10-14), contributes to the prevention of mutations in response to UV irradiation, without affecting long-term survival following this treatment. Thus, we propose that in collaboration with NER, the MMR-FANCJ pathway is important for the response to UV irradiation in S phase to ensure checkpoint responses, genome stability, and limit tumorigenesis (Fig. 5G).
Figure 5.
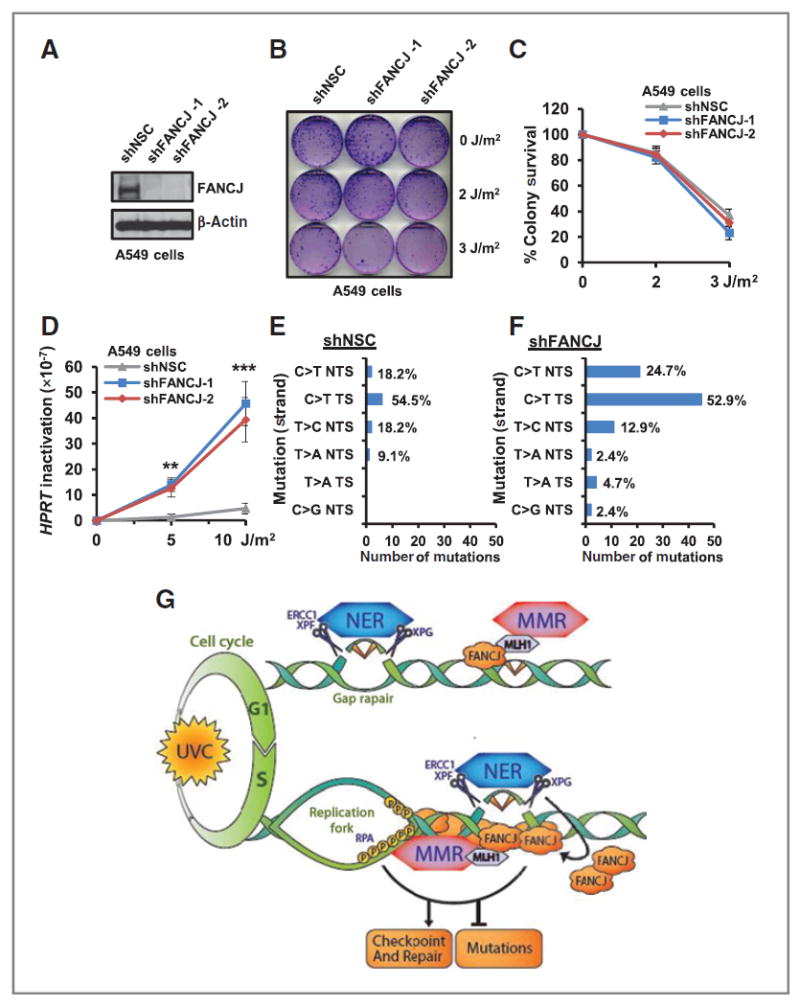
FANCJ suppresses UV-induced mutations. A and B, A549 cells expressing individual shRNA vectors targeting FANCJ or NSC were analyzed by immunoblot (A) and left untreated or globally UV irradiated and analyzed for colony survival (B). C, quantification of surviving colonies. D, quantification of 6-TG–resistant HPRT mutant colonies from mutagenesis assay. E and F, quantification of the distribution of HPRT-inactivating mutations in A549 cells expressing shRNA to NSC (E) and/or shRNAs to FANCJ (combined; F). G, model of FANCJ function in response to UV irradiation. NER and MMR factors are recruited to sites of local UV-induced damage in non-S-phase cells where NER, but not MMR, is required for gap filling. In S-phase cells, both NER and MMR factors contribute to the accumulation of FANCJ. MMR through MLH1 binding localizes FANCJ to sites of UV-induced damage. NER incision enhances the accumulation of FANCJ at the lesion site. Collectively, these events ensure a robust checkpoint response to limit the replication of damaged DNA, induction of mutations, and cancer. Where shown, error bars represent the standard deviation of the mean of three independent experiments.
Discussion
Here, we show that both NER and MMR proteins promote the localization of the FANCJ DNA helicase to sites of UV-induced lesions to ensure a robust S-phase checkpoint response. MMR proteins initially recruit FANCJ and its further accumulation requires dual incision by the NER endonucleases XPF and XPG (Figs. 1C–H and 2D–L). Although FANCJ deficiency does not cause UV-induced sensitivity, our analysis revealed an important role for FANCJ in promoting an S-phase checkpoint response, lesion repair, and suppressing UV-induced mutations (Figs. 3A–J, 4A–E and 5A–F and Supplementary Figs. S3D, S4A–S4C, and S6C). Consistent with FANCJ and MMR functioning in a common pathway, we found that FANCJ or MMR deficiency alone or in combination generated similar defects (Supplementary Fig. S4A–S4C). Correspondingly, the direct interaction between MLH1 and FANCJ is essential for both FANCJ localization and function at sites of UV-induced damage, whereas the BRCA1 interaction is not required (Figs. 2A–C and 3E and F). Similar to NER, MMR proteins localize to LUDs in both non-S- and S-phase cells, whereas FANCJ is predominantly found at sites of UV-damage in S-phase cells (Supplementary Fig. S3A–S3C). Together, our work demonstrates that distinct pathways merge in S-phase cells to ensure a robust UV-induced DNA damage response.
These findings are important in light of the fact that defects in MMR have been associated with skin cancers found in the HNPCC variant Muir–Torre syndrome. Furthermore, we searched for both FANCJ and MMR mutations within sequenced melanoma genomes using cBioPortal (51, 52) and the Catalogue of Somatic Mutations in Cancer database (53). We identified mutations in FANCJ, MSH2, MSH6, MLH1, and PMS2 (Supplementary Fig. S7A–S7F). The majority of FANCJ mutations target the helicase domain, including domains important for enzyme function, such as the Fe–S domain and helicase boxes III to V (Supplementary Fig. S7A;. ref. 54). In addition, some of the mutations have been detected previously. The FANCJP47 residue was targeted in breast cancer and was shown to be ATPase and helicase inactive in vitro (39). The splice mutant FANCJR831 is an allele in Fanconi anemia and eliminates conserved helicase boxes required for enzyme function (39, 54, 55). To determine if loss of FANCJ ATPase/helicase/translocase activity disrupts the UV response, we attempted to express the catalytic inactive FANCJK52R mutant in FA-J cells. Because FANCJ-deficient FA-J cells are defective in the UV response and the FANCJK52R mutant has weak expression compared with FANCJWT, it was unclear if the mutant was defective in complementing FA-J cells or mediating the UV response. Thus, we overexpressed the FANCJK52R mutant in U2OS cells. Here, we found significant defects in lesion clearance at 16 hours following UV damage, but no significant affect on RPA phosphorylation at this time point (Supplementary Fig. S8A–S8C). Thus, loss of FANCJ expression could disrupt checkpoint activation, whereas expression of an enzyme inactive mutant could dominantly disrupt repair.
Further supporting that multiple pathways contribute to high-fidelity repair after UV irradiation, similar to skin tumors from XP patients (56), MMR- and FANCJ-deficient cells display an elevated frequency of UV-induced C > T point mutations (Supplementary Table S1;. ref. 14). Similar to FANCJ deficiency, MMR deficiency also has modest affects on UV sensitivity despite reduced checkpoint and apoptotic responses (12, 57). Thus, we propose that FANCJ intersects MMR- and NER-dependent repair pathways to promote efficient checkpoint activation, lesion clearance, and suppress UV-induced mutations (Fig. 5G). Conceivably, in the absence of FANCJ and its checkpoint function error-prone polymerases induce mutations at sites of UV-induced lesions. Indeed, the high-fidelity TLS polymerase, polη, has reduced foci formation in response to global UV irradiation in FANCJ-deficient cells (Supplementary Fig. S9A–S9C). Correspondingly, MSH2-deficient cells have defective UV-induced PCNA mono-ubiquitination and TLS foci formation (58).
The NER factor XPA contributes to the S-phase checkpoint following UV-induced irradiation. However, not all NER factors are required, suggesting that this checkpoint function is distinct from NER repair in G1 phase (4). Our findings further suggest that XPF promotes RPA phosphorylation in S-phase cells (Fig. 4A–C). Interestingly, XPF is the Fanconi anemia gene, FANCQ (30). Given that XPF promotes FANCJ accumulation in S-phase cells, our data also suggest that FANCJ functions downstream of this Fanconi anemia factor to promote RPA phosphorylation throughout S-phase. NER-dependent incision may provide a better substrate or change the DNA structure, enabling distribution of FANCJ at the lesion site (Fig. 5G). Here, FANCJ could facilitate repair of lesions ahead of the replication fork through checkpoint induction and the arrest of DNA synthesis to limit mutation induction. Conceivably this function is shared by FANCJ partners, such as Bloom’s syndrome helicase (BLM), or the Fanconi anemia pathway, explaining its link to the UV response and checkpoints that limit genomic instability (59-62). It has been long proposed that ATR-BLM and Fanconi anemia pathway interactions maintain genomic stability by restoring productive replication following replication stress (63-65).
The 2-step mobilization of FANCJ to UV-induced lesions, localization by MMR and further accumulation after NER-dependent postincision could ensure pathway coordination. Indeed, the combined loss of NER and MMR enhances UV-induced mutagenesis (10). Although MMR and NER proteins have been shown to have overlapping substrates (66), it remains to be determined whether they bind the same or a distinct type of UV lesion. FANCJ loading by MLH1 would be reminiscent of the requirement of the bacterial MutL, homologous to the MutLα complex, for loading helicase II (UvrD) onto DNA (67). Helicase II functions with DNA polymerase I to release oligonucleotide fragments containing UV photoproducts (68). In contrast to Helicase II, our data do not support a role for FANCJ or MMR in gap repair (Figs. 4A, B, and F–H and Supplementary Fig. S4D and S4E). However, these findings do not exclude the possibility that MMR and FANCJ contribute to the fidelity of NER-dependent gap filling. Alternatively, loading of FANCJ by MMR factors could unwind and disrupt secondary DNA structures that impede NER processing. Indeed, MMR factors bind secondary structures such as G-4 quadruplex DNA that FANCJ unwinds (69, 70). FANCJ also depends on MLH1 for localization to sites of DNA interstrand crosslinks (29).
Collectively, the data presented in this manuscript provide a framework for understanding the contributions of distinct DNA repair pathways to the DNA damage response to UV irradiation in human cells. The identification of a novel function for MMR in localizing FANCJ to sites of UV-induced damage could be useful for several reasons. First, it could help in the discrimination between missense and pathogenic MMR variants. Loss of FANCJ localization and function could be uniquely disrupted by MMR gene mutations as found in tumors in which canonical MMR is intact. Second, the MMR-FANCJ pathway could represent a unique tumor suppression pathway that provides opportunities for selective therapy in effected tumors. In melanoma, loss of FANCJ function or expression could be a consequence of not only FANCJ mutations (Supplementary Fig. S7A), but also MMR mutations. Indeed, ~5.7% of tumors are affected by FANCJ mutations, which did not co-segregate with MMR gene mutations (51, 52). Associated skin tumors may be selectively sensitive to interstrand crosslink-inducing agents, which is a hallmark of FA-J patient cells. In light of the recent finding that XPF is the Fanconi anemia gene, FANCQ (30), it will be important to determine if the Fanconi anemia pathway has a more fundamental role in the response to UV irradiation and/or in reducing the emergence of disease.
Supplementary Material
Acknowledgments
The authors thank Dr. C. Heinen (University of Connecticut Health Center) for comments on the manuscript, Dr. J. Hays (Oregon State University) for helpful discussions, and B. Morehouse, C. Brown, and N. Patil for technical assistance and quantification of experiments.
Grant Support
This work was supported by the NIH (RO1 CA129514-01A1) and charitable contributions from Mr. and Mrs. E.T. Vitone Jr.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors’ Contributions
Conception and design: S.B. Cantor
Development of methodology: S. Guillemette, M. Peng, S.B. Cantor
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): S. Guillemette, A. Branagan, O.D. Schärer
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): S. Guillemette, A. Dhruva, S.B. Cantor
Writing, review, and/or revision of the manuscript: S. Guillemette, A. Branagan, O.D. Schärer, S.B. Cantor
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): M. Peng, A. Dhruva, S.B. Cantor
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Cleaver JE. Defective repair replication of DNA in xeroderma pigmentosum. Nature. 1968;218:652–6. doi: 10.1038/218652a0. [DOI] [PubMed] [Google Scholar]
- 2.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–74. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 3.Gillet LCJ, Scharer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev. 2006;106:253–76. doi: 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
- 4.Bomgarden RD, Lupardus PJ, Soni DV, Yee MC, Ford JM, Cimprich KA. Opposing effects of the UV lesion repair protein XPA and UV bypass polymerase eta on ATR checkpoint signaling. EMBO J. 2006;25:2605–14. doi: 10.1038/sj.emboj.7601123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilljam KM, Muller R, Liabakk NB, Otterlei M. Nucleotide excision repair is associated with the replisome and its efficiency depends on a direct interaction between XPA and PCNA. PLoS ONE. 2012;7:e49199. doi: 10.1371/journal.pone.0049199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- 7.Byun TS, Pacek M, Yee M-C, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–52. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cortez D. Unwind and slow down: checkpoint activation by helicase and polymerase uncoupling. Genes Dev. 2005;19:1007–12. doi: 10.1101/gad.1316905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pathania S, Nguyen J, Hill SJ, Scully R, Adelmant GO, Marto JA, et al. BRCA1 is required for postreplication repair after UV-induced DNA damage. Mol Cell. 2011;44:235–51. doi: 10.1016/j.molcel.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nara K, Nagashima F, Yasui A. Highly elevated ultraviolet-induced mutation frequency in isolated Chinese hamster cell lines defective in nucleotide excision repair and mismatch repair proteins. Cancer Res. 2001;61:50–2. [PubMed] [Google Scholar]
- 11.Meira LB, Cheo DL, Reis AM, Claij N, Burns DK, te Riele H, et al. Mice defective in the mismatch repair gene Msh2 show increased predisposition to UVB radiation-induced skin cancer. DNA Repair. 2002;1:929–34. doi: 10.1016/s1568-7864(02)00143-x. [DOI] [PubMed] [Google Scholar]
- 12.Yoshino M, Nakatsu Y, te Riele H, Hirota S, Kitamura Y, Tanaka K. Additive roles of XPA and MSH2 genes in UVB-induced skin tumorigenesis in mice. DNA Repair. 2002;1:935–40. doi: 10.1016/s1568-7864(02)00144-1. [DOI] [PubMed] [Google Scholar]
- 13.Seifert M, Scherer SJ, Edelmann W, Bohm M, Meineke V, Lobrich M, et al. The DNA-mismatch repair enzyme hMSH2 modulates UV-B-induced cell cycle arrest and apoptosis in melanoma cells. J Invest Dermatol. 2008;128:203–13. doi: 10.1038/sj.jid.5700941. [DOI] [PubMed] [Google Scholar]
- 14.Borgdorff V, Pauw B, van Hees-Stuivenberg S, de Wind N. DNA mismatch repair mediates protection from mutagenesis induced by short-wave ultraviolet light. DNA Repair. 2006;5:1364–72. doi: 10.1016/j.dnarep.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 15.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 16.Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7:335–46. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 17.Kaina B, Christmann M, Naumann S, Roos WP. MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair (Amst) 2007;6:1079–99. doi: 10.1016/j.dnarep.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 18.Mu D, Tursun M, Duckett DR, Drummond JT, Modrich P, Sancar A. Recognition and repair of compound DNA lesions (base damage and mismatch) by human mismatch repair and excision repair systems. Mol Cell Biol. 1997;17:760–9. doi: 10.1128/mcb.17.2.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H, Lawrence CW, Li GM, Hays JB. Specific binding of human MSH2.MSH6 mismatch-repair protein heterodimers to DNA incorporating thymine- or uracil-containing UV light photoproducts opposite mismatched bases. J Biol Chem. 1999;274:16894–900. doi: 10.1074/jbc.274.24.16894. [DOI] [PubMed] [Google Scholar]
- 20.Mathiak M, Rutten A, Mangold E, Fischer H-P, Ruzicka T, Friedl W, et al. Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations: establishment of immunohistochemical analysis as a screening test. Am J Surg Pathol. 2002;26:338–43. doi: 10.1097/00000478-200203000-00007. [DOI] [PubMed] [Google Scholar]
- 21.Kruse R, Rutten A, Lamberti C, Hosseiny-Malayeri HR, Wang Y, Ruelfs C, et al. Muir-Torre phenotype has a frequency of DNA mismatch-repair-gene mutations similar to that in hereditary nonpolyposis colorectal cancer families defined by the Amsterdam criteria. Am J Hum Genet. 1998;63:63–70. doi: 10.1086/301926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suspiro A, Fidalgo P, Cravo M, Albuquerque C, Ramalho E, Leitao CN, et al. The Muir-Torre syndrome: a rare variant of hereditary nonpolyposis colorectal cancer associated with hMSH2 mutation. Am J Gastroenterol. 1998;93:1572–4. doi: 10.1111/j.1572-0241.1998.00487.x. [DOI] [PubMed] [Google Scholar]
- 23.Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105:149–60. doi: 10.1016/s0092-8674(01)00304-x. [DOI] [PubMed] [Google Scholar]
- 24.Peng M, Litman R, Xie J, Sharma S, Brosh RM, Jr, Cantor SB. The FANCJ/MutLα interaction is required for correction of the cross-link response in FA-J cells. EMBO J. 2007;26:3238– 49. doi: 10.1038/sj.emboj.7601754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong Z, Kim JE, Leung CC, Glover JN, Chen J. BACH1/FANCJ acts with TopBP1 and participates early in DNA replication checkpoint control. Mol Cell. 2010;37:438–46. doi: 10.1016/j.molcel.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cotta-Ramusino C, McDonald ER, 3rd, Hurov K, Sowa ME, Harper JW, Elledge SJ. A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science. 2011;332:1313–7. doi: 10.1126/science.1203430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie J, Peng M, Guillemette S, Quan S, Maniatis S, Wu Y, et al. FANCJ/BACH1 acetylation at lysine 1249 regulates the DNA damage response. PLoS Genet. 2012;8:e1002786. doi: 10.1371/journal.pgen.1002786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie J, Litman R, Wang S, Peng M, Guillemette S, Rooney T, et al. Targeting the FANCJ-BRCA1 interaction promotes a switch from recombination to polη-dependent bypass. Oncogene. 2010;29:2499–508. doi: 10.1038/onc.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suhasini AN, Sommers JA, Muniandy PA, Coulombe Y, Cantor SB, Masson JY, et al. Fanconi anemia group J helicase MRE11 nuclease interact to facilitate the DNA damage response. Mol Cell Biol. 2013;33:2212–27. doi: 10.1128/MCB.01256-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet. 2013;92:800–6. doi: 10.1016/j.ajhg.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kashiyama K, Nakazawa Y, Pilz DT, Guo C, Shimada M, Sasaki K, et al. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes cockayne syndrome, xeroderma pigmentosum, and fanconi anemia. Am J Hum Genet. 2013;92:807–19. doi: 10.1016/j.ajhg.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakatani Y, Ogryzko V. Immunoaffinity purification of mammalian protein complexes. Methods Enzymol. 2003;370:430–44. doi: 10.1016/S0076-6879(03)70037-8. [DOI] [PubMed] [Google Scholar]
- 33.Kannouche P, Broughton BC, Volker M, Hanaoka F, Mullenders LH, Lehmann AR. Domain structure, localization, and function of DNA polymerase η, defective in xeroderma pigmentosum variant cells. Genes Dev. 2001;15:158–72. doi: 10.1101/gad.187501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M. Rad18 guides polη to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004;23:3886–96. doi: 10.1038/sj.emboj.7600383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mone MJ, Volker M, Nikaido O, Mullenders LH, van Zeeland AA, Verschure PJ, et al. Local UV-induced DNA damage in cell nuclei results in local transcription inhibition. EMBO Rep. 2001;2:1013–7. doi: 10.1093/embo-reports/kve224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiu RK, Brun J, Ramaekers C, Theys J, Weng L, Lambin P, et al. Lysine 63-polyubiquitination guards against translesion synthesis-induced mutations. PLoS Genet. 2006;2:e116. doi: 10.1371/journal.pgen.0020116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Limsirichaikul S, Niimi A, Fawcett H, Lehmann A, Yamashita S, Ogi T. A rapid non-radioactive technique for measurement of repair synthesis in primary human fibroblasts by incorporation of ethynyl deoxyuridine (EdU) Nucleic Acids Res. 2009;37:e31. doi: 10.1093/nar/gkp023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hidaka M, Takagi Y, Takano TY, Sekiguchi M. PCNA-MutSα-mediated binding of MutLα to replicative DNA with mismatched bases to induce apoptosis in human cells. Nucleic Acids Res. 2005;33:5703–12. doi: 10.1093/nar/gki878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cantor S, Drapkin R, Zhang F, Lin Y, Han J, Pamidi S, et al. The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc Natl Acad Sci U S A. 2004;101:2357–62. doi: 10.1073/pnas.0308717101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Volker M, Mone MJ, Karmakar P, van Hoffen A, Schul W, Vermeulen W, et al. Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell. 2001;8:213–24. doi: 10.1016/s1097-2765(01)00281-7. [DOI] [PubMed] [Google Scholar]
- 41.Staresincic L, Fagbemi AF, Enzlin JH, Gourdin AM, Wijgers N, Dunand-Sauthier I, et al. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 2009;28:1111–20. doi: 10.1038/emboj.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sertic S, Pizzi S, Cloney R, Lehmann AR, Marini F, Plevani P, et al. Human exonuclease 1 connects nucleotide excision repair (NER) processing with checkpoint activation in response to UV irradiation. Proc Natl Acad Sci U S A. 2011;108:1, 3647–52. doi: 10.1073/pnas.1108547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Young LC, Hays JB, Tron VA, Andrew SE. DNA mismatch repair proteins: potential guardians against genomic instability and tumorigenesis induced by ultraviolet photoproducts. J Invest Dermatol. 2003;121:435–40. doi: 10.1046/j.1523-1747.2003.12450.x. [DOI] [PubMed] [Google Scholar]
- 44.Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006;20:34–46. doi: 10.1101/gad.1381306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–42. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 46.Kaufmann WK. The human intra-S checkpoint response to UVC-induced DNA damage. Carcinogenesis. 2010;31:751–65. doi: 10.1093/carcin/bgp230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carty MP, Zernik-Kobak M, McGrath S, Dixon K. UV light-induced DNA synthesis arrest in HeLa cells is associated with changes in phosphorylation of human single-stranded DNA-binding protein. EMBO J. 1994;13:2114–23. doi: 10.1002/j.1460-2075.1994.tb06487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Niu H, Erdjument-Bromage H, Pan ZQ, Lee SH, Tempst P, Hurwitz J. Mapping of amino acid residues in the p34 subunit of human single-stranded DNA-binding protein phosphorylated by DNA-dependent protein kinase and Cdc2 kinase in vitro. J Biol Chem. 1997;272:12634–41. doi: 10.1074/jbc.272.19.12634. [DOI] [PubMed] [Google Scholar]
- 49.Zernik-Kobak M, Vasunia K, Connelly M, Anderson CW, Dixon K. Sites of UV-induced phosphorylation of the p34 subunit of replication protein A from HeLa cells. J Biol Chem. 1997;272:23896–904. doi: 10.1074/jbc.272.38.23896. [DOI] [PubMed] [Google Scholar]
- 50.Auclair Y, Rouget R, Affar el B, Drobetsky EA. ATR kinase is required for global genomic nucleotide excision repair exclusively during S phase in human cells. Proc Natl Acad Sci U S A. 2008;105:17896–901. doi: 10.1073/pnas.0801585105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:p11. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–50. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cantor SB, Guillemette S. Hereditary breast cancer and the BRCA1-associated FANCJ/BACH1/BRIP1. Future Oncol. 2011;7:253–61. doi: 10.2217/fon.10.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gupta R, Sharma S, Sommers JA, Jin Z, Cantor SB, Brosh RM., Jr Analysis of the DNA substrate specificity of the human BACH1 helicase associated with breast cancer. J Biol Chem. 2005;280:25450–60. doi: 10.1074/jbc.M501995200. [DOI] [PubMed] [Google Scholar]
- 56.Dumaz N, Drougard C, Sarasin A, Daya-Grosjean L. Specific UV-induced mutation spectrum in the p53 gene of skin tumors from DNA-repair-deficient xeroderma pigmentosum patients. Proc Natl Acad Sci U S A. 1993;90:10529–33. doi: 10.1073/pnas.90.22.10529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Oosten M, Stout GJ, Backendorf C, Rebel H, de Wind N, Darroudi F, et al. Mismatch repair protein Msh2 contributes to UVB-induced cell cycle arrest in epidermal and cultured mouse keratinocytes. DNA Repair. 2005;4:81–9. doi: 10.1016/j.dnarep.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 58.Lv L, Wang F, Ma X, Yang Y, Wang Z, Liu HL, et al. Mismatch repair protein MSH2 regulates translesion DNA synthesis following exposure of cells to UV radiation. Nucleic Acids Res. 2013 Sep 12; doi: 10.1093/nar/gkt793. [Epub ahead of print.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suhasini AN, Rawtani NA, Wu Y, Sommer JA, Sharma S, Mosedale G, et al. Interaction between the helicases genetically linked to Fanconi anemia group J and Bloom’s syndrome. EMBO J. 2011;30:692–705. doi: 10.1038/emboj.2010.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kelsall IR, Langenick J, MacKay C, Patel KJ, Alpi AF. The Fanconi anaemia components UBE2T and FANCM are functionally linked to nucleotide excision repair. PLoS ONE. 2012;7:e36970. doi: 10.1371/journal.pone.0036970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Singh TR, Bakker ST, Agarwal S, Jansen M, Grassman E, Godthelp BC, et al. Impaired FANCD2 monoubiquitination and hypersensitivity to camptothecin uniquely characterize Fanconi anemia complementation group M. Blood. 2009;114:174–80. doi: 10.1182/blood-2009-02-207811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nalepa G, Enzor R, Sun Z, Marchal C, Park SJ, Yang Y, et al. Fanconi anemia signaling network regulates the spindle assembly checkpoint. J Clin Invest. 2013;123:3839–47. doi: 10.1172/JCI67364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davies SL, North PS, Dart A, Lakin ND, Hickson ID. Phosphorylation of the Bloom’s syndrome helicase and its role in recovery from S-phase arrest. Mol Cell Biol. 2004;24:1279–91. doi: 10.1128/MCB.24.3.1279-1291.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Olson E, Nievera CJ, Klimovich V, Fanning E, Wu X. RPA2 is a direct downstream target for ATR to regulate the S-phase checkpoint. J Biol Chem. 2006;281:39517–33. doi: 10.1074/jbc.M605121200. [DOI] [PubMed] [Google Scholar]
- 65.Sobeck A, Stone S, Costanzo V, de Graaf B, Reuter T, de Winter J, et al. Fanconi anemia proteins are required to prevent accumulation of replication-associated DNA double-strand breaks. Mol Cell Biol. 2006;26:425–37. doi: 10.1128/MCB.26.2.425-437.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao J, Jain A, Iyer RR, Modrich PL, Vasquez KM. Mismatch repair and nucleotide excision repair proteins cooperate in the recognition of DNA interstrand crosslinks. Nucleic Acids Res. 2009;37:4420–9. doi: 10.1093/nar/gkp399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mechanic LE, Frankel BA, Matson SW. Escherichia coli MutL loads DNA helicase II onto DNA. J Biol Chem. 2000;275:38337–46. doi: 10.1074/jbc.M006268200. [DOI] [PubMed] [Google Scholar]
- 68.Caron PR, Kushner SR, Grossman L. Involvement of helicase II (UvrD gene product) and DNA polymerase I in excision mediated by the uvrABC protein complex. Proc Natl Acad Sci U S A. 1985;82:4925–9. doi: 10.1073/pnas.82.15.4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Larson ED, Duquette ML, Cummings WJ, Streiff RJ, Maizels N. MutSa binds to and promotes synapsis of transcriptionally activated immunoglobulin switch regions. Curr Biol. 2005;15:470– 4. doi: 10.1016/j.cub.2004.12.077. [DOI] [PubMed] [Google Scholar]
- 70.Wu Y, Shin-Ya K, Brosh RM., Jr FANCJ helicase defective in Fanconia anemia and breast cancer unwinds g-quadruplex DNA to defend genomic stability. Mol Cell Biol. 2008;28:4116–28. doi: 10.1128/MCB.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.