

To gain insights into erythromelalgia disease pathophysiology, this study elucidated changes in peripheral axonal excitability and influences of temperature and mexiletine on axonal function.
Keywords: Erythromelalgia, Pain, SCN9A, Sodium channel, Excitability, Mexiletine
Abstract
Erythromelalgia (EM) is a rare neurovascular disorder characterized by intermittent severe burning pain, erythema, and warmth in the extremities on heat stimuli. To investigate the underlying pathophysiology, peripheral axonal excitability studies were performed and changes with heating and therapy explored. Multiple excitability indices (stimulus–response curve, strength–duration time constant (SDTC), threshold electrotonus, and recovery cycle) were investigated in 23 (9 EMSCN9A+ and 14 EMSCN9A−) genetically characterized patients with EM stimulating median motor and sensory axons at the wrist. At rest, patients with EM showed a higher threshold and rheobase (P < 0.001) compared with controls. Threshold electrotonus and current–voltage relationships demonstrated greater changes of thresholds in both depolarizing and hyperpolarizing preconditioning electrotonus in both EM cohorts compared with controls in sensory axons (P < 0.005). When average temperature was raised from 31.5°C to 36.3°C in EMSCN9A+ patients, excitability changes showed depolarization, specifically SDTC significantly increased, in contrast to the effects of temperature previously established in healthy subjects (P < 0.05). With treatment, 4 EMSCN9A+ patients (4/9) reported improvement with mexiletine, associated with reduction in SDTC in motor and sensory axons. This is the first study of primary EM using threshold tracking techniques to demonstrate alterations in peripheral axonal membrane function. Taken together, these changes may be attributed to systemic neurovascular abnormalities in EM, with chronic postischaemic resting membrane potential hyperpolarization due to Na+/K+ pump overactivity. With heating, a trigger of acute symptoms, axonal depolarization developed, corresponding to acute axonal ischaemia. This study has provided novel insights into EM pathophysiology.
1. Introduction
Erythromelalgia (EM) is a rare neurovascular disorder characterized by recurrent severe pain associated with erythema, swelling, and warmth in the feet or hands. Symptoms are induced by warmth or exercise and eased by cooling. Mutations in the SCN9A gene, encoding the α-subunit of Nav1.7, result in a gain of function and are responsible for primary EM19,23,83
Functional characterization of mutant Nav1.7 channels using patch clamp techniques has established hyperpolarizing shifts in activation curve, increases in ramp current, slowed deactivation, resurgent currents during repolarization, and increased firing frequency with stimulation.13,14,38,69,72 Nav1.7 channels are predominantly expressed on dorsal root ganglia and sympathetic neurons77; however, the functional effects of a mutation are cell background dependent.69 The selective presence of Nav1.8, which is relatively resistant to inactivation by depolarisation, in sensory but not sympathetic neurons, renders dorsal root ganglion (DRG) neurons hyperexcitable.1,18 By contrast, sympathetic ganglion depolarization produces resting inactivation of all the voltage gated sodium channel (VGSC) isoforms and hypoexcitability. These findings provide a molecular basis for clinical manifestations in EM. The hyperexcitability of DRG results in neuropathic pain while dysfunction of the sympathetic tone disturbs vasomotor regulation and skin perfusion.
In terms of understanding the disturbed sympathetic control of vascular tone in EM, previous clinical studies have established the coexistence of marked increases in skin blood flow associated with decreased oxygenation during attacks presenting with sudden swelling, redness, severe pain, and increased temperature.16,22,59 It has been postulated that microvascular arteriovenous shunting may induce a maldistribution of blood flow, resulting in hypoxia, ischaemia, and pain, similar to diabetic neuropathy.76 The warmer temperature may be expected to increase cellular metabolism and modulate VGSC properties, thereby establishing a vicious cycle to sustain attacks. Despite these clinical and pathophysiological insights, there has not been an effective therapeutic armamentarium for EM, with up to 84 medications prescribed, including sodium channel and sympathetic nerve blockers.21 Patients may go to extreme measures to alleviate their symptoms, such as avoiding footwear, using fans, and immersing affected areas with cold water and ice, highlighting the profound impact of temperature on clinical manifestations.
Of relevance to gaining insight into EM pathophysiology, protocols for testing of axonal membrane function have recently been developed for clinical assessment.43 These techniques have been applied to various genetic channelopathies and have demonstrated that nerve impulse conduction along the peripheral axons may be altered, even when the principal channel is not expressed at the site of assessment.45,78,79 The purpose of this study was to evaluate whether EM has changes in axonal excitability and the influence of temperature and mexiletine on axonal excitability.
2. Methods
Clinical manifestations were combined with conventional and specialized neurophysiological assessments in patients with primary EM. The coding region of the human SCN9A gene was sequenced in 23 patients. Authentication of a pathological mutation was performed by screening 200 controls and family segregation analysis. Patients with EM were divided into 2 subgroups according to the presence or absence of a pathogenic mutation in SCN9A: SCN9A-mutated EM (EMSCN9A+) or EM not related to the mutation of SCN9A (EMSCN9A−), respectively. In the latter group secondary causes of EM resulting from an underlying organic disease were excluded, including Fabry disease, diabetes mellitus, medications, toxins, alcoholism, HIV infection, and Lyme disease. All the patients provided informed consent or parental consent to the procedures, which were approved by the South Eastern Sydney and Illawarra Area Health Service Human Research Ethics Committee or the National Taiwan University Hospital Ethics Committee.
Axonal excitability studies used the threshold tracking technique, which measures changes in the strength of a test stimulus needed to produce a target potential. This is set to 40% of the maximum (located in the steepest slope of the stimulus response curve). Different conditioning-test paradigms, including altering the stimulus duration and polarization, were applied to produce changes in excitability8,11,43 and provide indirect information about axonal ion channel function, resting membrane potential, and passive membrane properties.48 In vitro experiments and modelling studies have provided evidence for the relationship of specific ion conductances with cohesive changes in multiple excitability parameters.2,4,6,10,42,70,71
Axonal excitability studies were undertaken stimulating the median nerve at the wrist and recording the compound muscle action potentials (CMAPs) from the abductor pollicis brevis muscle or sensory nerve action potentials (SNAPs) from index finger using the rapid automated threshold tracking protocol known as “TROND” (named after a 3-day training symposium held in 1999 at Trondheim, Norway) within QTRAC software (Institute of Neurology, University College London).43,46 Skin temperature was measured at the site of stimulation and varied by less than 1° during the recordings. A number of assessments were incorporated in excitability testing that measured the nodal and internodal properties for analysis.
Initially, a stimulus–response curve was obtained. This curve was used to determine the threshold for a CMAP or SNAP around 40% of maximum; this was the target response and threshold was the stimulus required to obtain this potential. The strength–duration time constant (SDTC) of the nerve was then determined by adjusting the intensity of stimuli of different durations so that the target response was maintained. To measure threshold electrotonus (TE), prolonged 100-millisecond subthreshold currents were delivered and their strength was set to be +40% (depolarizing, TEd) and −40% (hyperpolarizing, TEh) of the control threshold current. Test pulses were delivered at varying intervals during and after the conditioning currents, and the change in threshold required to produce the target 40% response was recorded. Subsequently the current–threshold relationship (I/V) was measured, reflecting the rectifying properties of the axons.8,43 The I/V relationship was assessed by measuring the change in threshold at the end of polarizing currents of 200-millisecond duration, the strength of which was altered in 10% steps from +50% (depolarizing) to −100% (hyperpolarizing) of the control threshold. Finally, the recovery cycle (RC) was tested, which determines the change in threshold following a supramaximal stimulus at 18 conditioning-test intervals from 2 to 200 milliseconds. The RC consists of a relative refractory period at short interstimulus intervals, a superexcitable period, when a smaller stimulus is necessary to produce the test response, and a late subexcitable period, during which a greater stimulus than control is required.
Because warmth can trigger and exacerbate symptoms in otherwise asymptomatic areas,56 the influence of temperature on axonal excitability was examined in 6 SCN9A+ mutated patients with EM. The arm was covered with warm towels and heated gradually up to where the subject can tolerate (average skin surface temperature 5° higher than at rest).
Measurements of excitability indices have previously been used for noninvasive assessment and monitoring of the effects of mexiletine, which nonselectively partially blocks VGSCs.49,63 These studies included patients with neuropathic pain or muscle cramp or fasciculation and demonstrated with mexiletine therapy suppression of persistent Na+ currents in human motor axons associated with improvements in clinical phenotype.40,41,50,73 To identify an effective treatment, we assessed the alterations in axonal excitability before and 3 months after mexiletine at a dose of 300 mg daily, a selective sodium channel blocker in 4 SCN9A+mutated patients with EM.
2.1. Data analysis
Peripheral nerve excitability parameters in EMSCN9A+ patients were compared to EMSCN9A− patients and 28 age- and temperature-matched normal controls (17 men; mean age 33.1 ± 1.6 years, range 7-67 years; temperature 33.2 ± 0.2°C).28 Excitability parameters were tested for normality of distribution before performing statistical tests. The one-way analysis of variance was used to compare mean differences between groups for excitability indices that showed the Gaussian distribution. The Kruskal–Wallis (nonparametric one-way analysis of variance) was used for parameters that did not show the normative distribution. All results were expressed as mean ± SEM. A P value of <0.05 was considered statistically significant. The paired t test was used to compare patients' parameters before and after heating and treatment for statistical analysis.
2.2. Modelling
To assess the possible underlying physiological mechanisms of the nerve excitability changes in patients with EM, a model of the human sensory axon was used to undertake mathematical simulations using MEMFIT software within QtracP.5,7,37 Initially, the model was adjusted to closely match the normal sensory control group data. Subsequently, the mechanisms underlying the differences between the excitability of EM and normal control were explored. MEMFIT software uses an iterative approach to minimize the difference between the simulated and the target group (EM) data. The difference between the simulated and group data was calculated using the sum of the squares at each data point with calculations made during the various excitability components (TE, RC, current–threshold relationship, and SDTC) given equal weighting.
3. Results
3.1. Clinical features
The clinical and genetic features for 23 patients with EM are summarized in Table 1. The study population included 9 EMSCN9A+ patients (mean age 23.8 ± 4.5, range 6-54 years, 5 men) and 14 EMSCN9A− patients (mean age 53.6 ± 4.6 years, range 18-77 years, 7 men). By definition, all patients with EM experienced intermittent bouts of heat, erythema, and severe burning pain, with symptoms located in the extremities of 22 patients and isolated to the face in 1 patient. Physical exertion, hot weather, tight shoes, and stress were frequent exacerbating factors. Patients preferred not to wear socks or closed shoes, even in winter. Physical examination between episodes frequently demonstrated erythematous feet, cool limbs with a reticular cutaneous pattern, and excoriated skin. To relieve pain, patients frequently immersed their hands and feet in ice buckets with cold water. Some medications had been prescribed with variable effect, including aspirin, mexiletine, carbamazepine, clonidine, propranolol, gabapentin, and imipramine. Conventional neurophysiological assessments demonstrated no significant changes in distal latencies, conduction velocities, and amplitudes for CMAP and SNAP (median CMAP 11.0 ± 1.1 mV, distal motor latency 3.6 ± 0.1 ms, motor conduction velocity 56.7 ± 1.1 m/s, SNAP 32.6 ± 6.4 µV, and sensory conduction velocity 59.5 ± 1.7 m/s). The patients did not demonstrate any abnormal quantitative skin response such as temperature or vibration sensations.
Table 1.
Clinical and genetic details for 23 patients with inherited erythromelalgia.

To provide an understanding of the therapeutic approach and challenges in EM, the case of patient 1 is described in detail. Symptoms of severe EM began at 2 years of age. Sequencing of the SCN9A gene identified a heterozygous de novo missense variant c.2623C>G which results in a p.Q875E mutation. The biophysical properties of this mutation predict an early-onset severe phenotype, with a large hyperpolarizing shift in voltage dependence of activation of Nav1.7, slowed deactivation, and shortened time to peak current.74 She has severe, persistent pain and itch in her feet, exacerbated by heat and relieved by cooling with ice packs. She has badly excoriated feet (Fig. 1) and markedly disturbed sleep, as do her parents. Multiple treatments were used including Na+ channel blockers, modulators of vascular tone, anaesthetic agents, antihistamines, sedatives, and physical therapy. The effects were evaluated by Faces pain score,34 parental report, and observations of skin. Carbamazepine, gabapentin, pregabalin, amitriptyline, clonidine, various antihistamines, and low-dose aspirin provided no benefit. Mexiletine and phenoxybenzamine were partially helpful (before treatment, Faces pain rating scale [or visual analogue scale] is 10; on mexiletine, is 8; with both mexiletine and phenoxybenzamine, is 7). At severe attacks, ketamine infusion was partly beneficial. Because medical modulation of vascular tone had a positive effect, a transient percutaneous lumbar sympathectomy was performed with some utility. Subsequently, radioablation of lumbar sympathectomy was undertaken; however, pain persistently worsened. Subsequently, topical 0.2% midodrine ointment20 transiently ameliorated the symptoms following sympathectomy. Topical and environmental cooling with air-conditioning to modify chronic pain has partly alleviated the symptoms.
Figure 1.

Lower limbs of a patient with erythromelalgia. (A) During a painful episode, the feet are erythematous, hot, and swollen. Ice packs are being used to relieve the pain. (B) Between episodes, the limbs have a reticular cutaneous pattern. (C) The same patient has excoriated and broken skin resulting from scratching and repetitive submersion in cool water.
3.2. Excitability differences between SCN9A mutations, without SCN9A mutations, and healthy controls
The threshold currents required to elicit a 50% response were greater in both EM patient cohorts compared with controls (motor: EMSCN9A+, 6.9 ± 1.2 mA; EMSCN9A−, 9.5 ± 1.1 mA; controls, 4.0 ± 1.1 mA, F = 20.5, P < 0.001. Sensory: EMSCN9A+, 4.9 ± 1.5 mA; EMSCN9A−, 10.2 ± 1.2 mA; controls, 3.0 ± 1.1 mA, F = 26.8, P < 0.001, Fig. 2A, B and Table 2). In agreement with this, rheobase measurements were significantly greater in patients with EM than in controls (motor: EMSCN9A+, 4.3 ± 1.3 mA; EMSCN9A−, 6.4 ± 1.2 mA; controls, 2.7 ± 1.1 mA, F = 17.9, P < 0.001. Sensory: EMSCN9A+, 2.0 ± 1.5 mA; EMSCN9A−, 4.8 ± 1.2 mA; controls, 1.3 ± 1.1 mA, F = 24.9, P < 0.001, Table 2).
Figure 2.
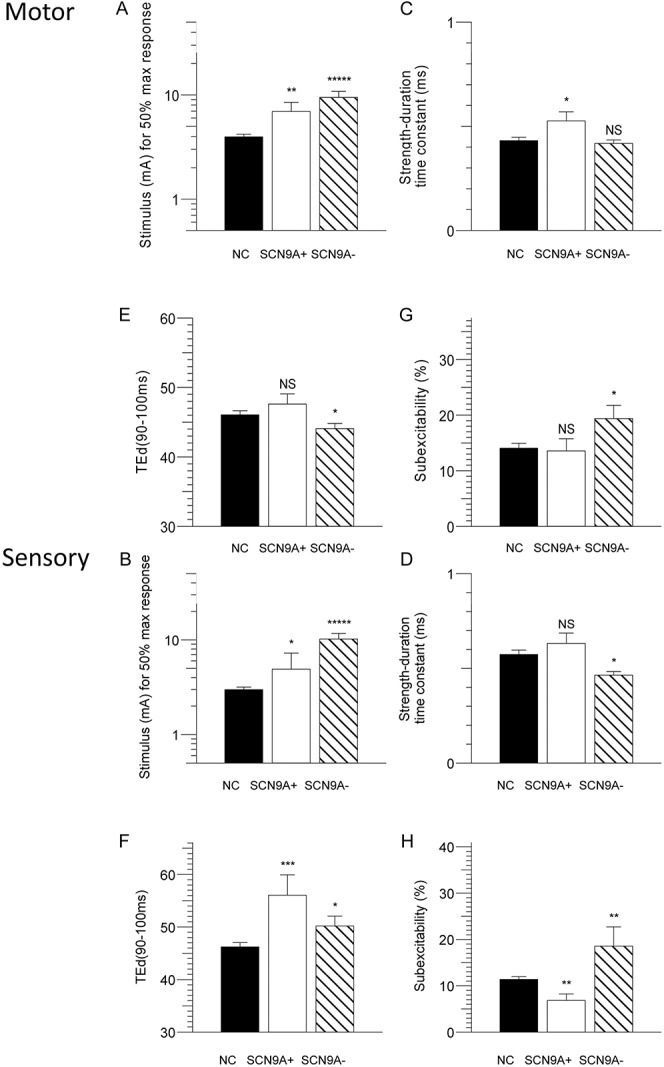
Comparison of multiple measurements of axonal excitability in motor (top panel) and sensory axons (bottom panel) in patients with erythromelalgia (EMSCN9A+, open; EMSCN9A−, hatched) and normal controls (filled) plotted as mean and SEM of motor and sensory fibres. (A, B) Stimulus for 50% maximum response. (C, D) Strength–duration time constant. (E, F) TEd (90-100 ms). (G, H) Subexcitability.
Table 2.
Indices in multiple excitability measurements: mean ± SEM.
There were significant alterations in the strength–duration properties in patients with EM compared with healthy controls in both motor and sensory axons, which provide an indirect measurement of nodal persistent Na+ conductances.9 Strength–duration time constant was significantly prolonged in EMSCN9A+ patients compared with EMSCN9A− patients and healthy controls (motor: SCN9A+, 0.53 ± 0.04 ms; SCN9A−, 0.42 ± 0.02 ms; controls, 0.43 ± 0.02 ms, F = 4.7, P = 0.01; sensory: SCN9A+, 0.63 ± 0.05 ms; SCN9A−, 0.46 ± 0.02 ms; controls, 0.58 ± 0.02 ms, F = 3.6, P < 0.05, Fig. 2C, D).
Threshold electrotonus demonstrated greater changes in response to depolarizing and hyperpolarizing currents in both EM cohorts compared with controls in sensory axons. This was accompanied by similar changes in the current–voltage relationship. Threshold electrotonus and current voltage relationships were similar between patients with EM and controls in motor fibres (Fig. 2E, F).
The RC showed variable changes in subexcitability in EM SCN9A+ and SCN9A− patients compared with controls, predominantly in sensory fibres (motor: EMSCN9A+, 14.5 ± 1.6%; EMSCN9A−, 19.3 ± 2.4%; controls, 14.1 ± 0.9%, F = 3.7, P < 0.05. Sensory: EMSCN9A+, 6.8 ± 1.4%; EMSCN9A−, 18.6. ± 4.2%; controls, 11.4 ± 0.6.%, F = 7.3, P < 0.05, Fig. 2G, H).
3.3. Nerve excitability changes with temperature
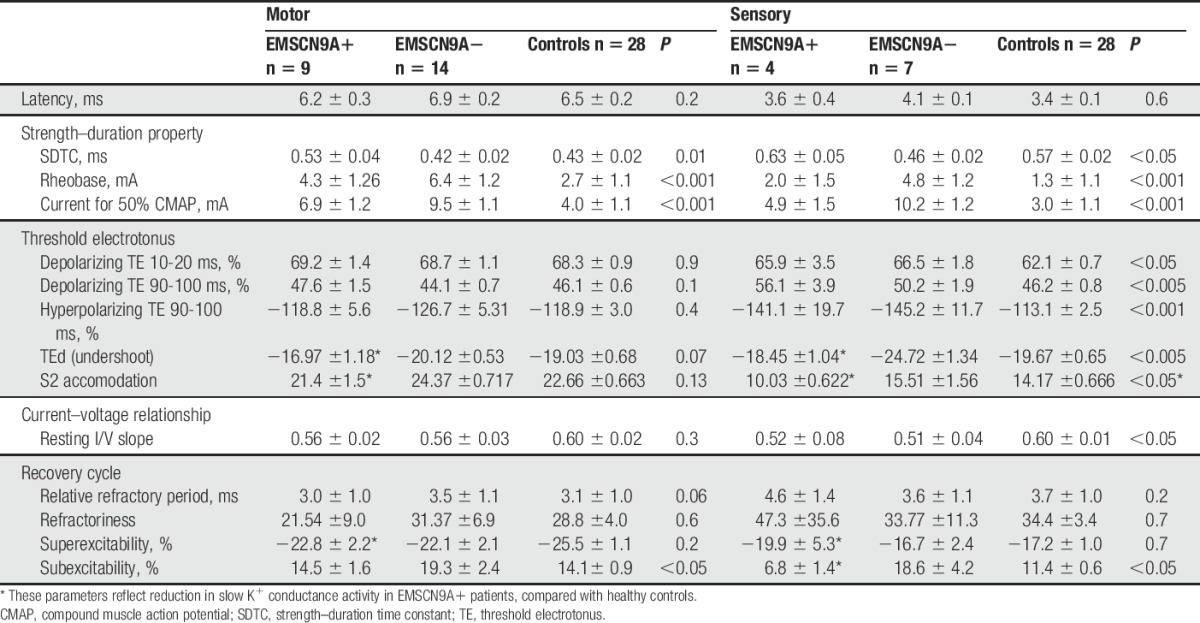
The pathophysiological mechanisms leading to the clinical attacks with increasing temperature in EM raise the importance of investigating their effects on axonal dysfunction. As such, the changes in nerve excitability parameters with different temperature were investigated in 6 EMSCN9A+ patients. Figure 3 demonstrates the changes in a SCN9A+ patient with heating where both motor and sensory excitability were recorded simultaneously to document the gradual changes as the temperature increases. Similar to the effects of temperature previously established in healthy subjects over the range of 29°C to 35°C,44 heating reduced the relative refractory period (P < 0.05) and quickened the accommodation to depolarising currents (P = 0.01) in both motor and sensory fibres. By contrast, SDTC in EMSCN9A+ patients increased with increased temperature (motor: preheating, 0.49 ± 0.07 ms vs postheating, 0.54 ± 0.07 ms, P < 0.05; preheating temperature, 31.5 ± 1.2°C, postheating temperature, 36.3 ± 0.5°C, P < 0.005. Sensory: preheating, 0.62 ± 0.05 ms, postheating, 0.75 ± 0.04 ms, P < 0.05; preheating temperature, 29.7 ± 1.3°C, postheating temperature, 35.2 ± 2.1°C, P < 0.05; Fig. 4A, B).
Figure 3.
Nerve excitability changes in motor (top panel) and sensory axons (bottom panel) with increase temperature in EMSCN9A+ patient. (A, E) Stimulus response curve. (B, F) Strength–duration time constant. (C, G) Threshold electrotonus waveforms. (D, H) Recordings were made at wrist temperatures of 32ºC (blue), 33.5ºC (green), and 37.8ºC (red) by heating the arm proximal to the wrist.
Figure 4.

Changes of strength–duration time constant (SDTC) by increasing of temperature and treatment of mexiletine. Upper panels: effects of heating: changes in SDTC of motor (left panel) and sensory axons (right panel) by increasing temperature in EMSCN9A+ patients. Bottom panels: effects of mexiletine on SDTC parameters in motor and sensory axons in patients with erythromelalgia (n = 4). Our results have demonstrated that these patients may have normal SDTC parameters at rest, they developed abnormal changes in SDTC with heating reflecting an increase in persistent sodium channel activity, and this can be partially reversed by sodium channel blocker such as Mexiletine; our results also demonstrated that after mexiletine treatment, the SDTC was reduced (C, D). Arrow depicts direction of change heating normal controls36 and with mexiletine treatment.50
3.4. Nerve excitability changes with mexiletine
Among our SCN9A+ patient cohorts, only 4 patients reported clinical improvement with mexiletine, accompanied by a 1- or 2- point reduction in Faces pain rating scale. These patients were studied using nerve excitability tests before or after medication, and the results are shown in Figures 4C and D; there is a reduction in SDTC in both motor and sensory axons while other excitability measures were not altered (motor SDTC: pre-mexiletine, 0.46 ± 0.03 ms vs on mexiletine, 0.42 ± 0.04 ms; sensory SDTC: pre-mexiletine, 0.61 ± 0.00 ms vs on mexiletine, 0.51 ± 0.08 ms). These results are in agreement with previous reports on the effects of mexiletine on excitability parameters.15,40,41,50,73
3.5. Mathematical modeling of abnormal excitability properties
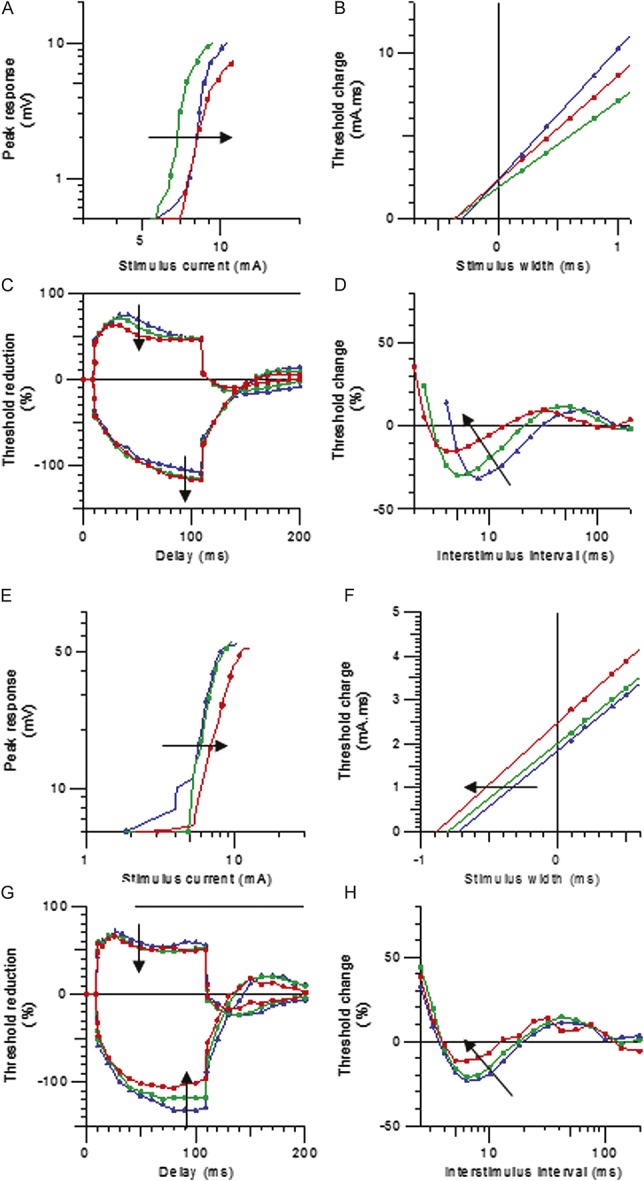
A mathematical model of the human sensory axon was used to assist in the interpretation of the changes measured in clinical nerve excitability in patients with EM. Membrane hyperpolarization best explained the differences between the EM SCN9A+ and EMSCN9A− patients and the sensory controls. Specifically, hyperpolarization of the resting membrane potential by 2.8 mV accounted for 89.4% of the discrepancy between the EMSCN9A+ and sensory control data. Similarly, a 1.6-mV hyperpolarization of resting membrane potential accounted for 83.6% of the discrepancy between the EMSCN9A− patients and sensory controls. Internodal shortening was also included in the modelling and reduced the discrepancy by 25%. It was not considered the most plausible explanation for the findings. The model did not show any further improvement with additional changes in Na+ channel expression or the fraction of Na+ channels operating in a persistent mode. The effect of temperature on nerve excitability changes in EM is shown in Figure 5. The 5° warming in 1 subject was modelled well by a 2-mV depolarization of resting membrane potential from the EM model.
Figure 5.
Mathematical model of axonal excitability changes with heating in patients with erythromelalgia. (A) Threshold electrotonus in a single subject during heating: 33.4°C, red; 35.1°C, green; 37, blue; 38.4°C, grey. (B) Modelled depolarization of resting membrane potential in threshold electrotonus, with resting membrane potentials (RMPs): −79.5 mV, red; −78.8 mV green, −78.2 mV, blue; −77.5 mV grey. C. Recovery cycles during heating in the same subject (temperatures and colours as in A). D. Modelled depolarization of recovery cycle during heating (RMPs and colours are the same as in B).
4. Discussion
This is the first study of primary EM using threshold tracking techniques to evaluate peripheral motor and sensory axonal function and the excitability changes related to temperature and mexiletine treatment. The techniques applied in this study allowed noninvasive in vivo assessment of the biophysical properties of axonal membranes, although Nav1.7 is not known to be expressed on large alpha-myelinated motor fibres and may only be expressed in some of these sensory fibres.3,24 Accordingly, this approach differs from previous electrophysiological studies of EM, which are focused on the short-term effects on single cells by voltage and current clamp directly or microneurography at the site of abnormal Nav1.7 expression.60 Notably, there were significant differences in sensory and motor excitability parameters between EM patients with and without SCN9A pathogenic variants compared with the healthy controls in this study. These findings reveal alterations in peripheral axonal membrane potential and conductances and reflect systemic abnormalities in EM. A paradoxical pattern of changes in Na+ channel-dependent parameters with heating was evident in SCN9A mutant patients in comparison to normal controls. There was variable and partial clinical benefit or change in neurophysiological parameters with pain medications, revealing challenges in therapy.
4.1. Peripheral axonal function in patients with erythromelalgia: alterations in membrane potential and conductances
Significant reductions in sensory axonal excitability were seen in both groups of EM patients, with a “fanning out” of responses during TE, increases in threshold and rheobase, similar to the effects established with membrane hyperpolarization.42 A similar but lesser pattern of changes occurred in motor nerves of patients with EM.35 While these changes are subclinical, in part related to Nav1.7 not being expressed in these neurons, they are important in comprehensively understanding EM pathophysiology and systemic effects. In the same way, subclinical changes in axonal membrane function have been demonstrated with temperature changes or maturation paralleling alterations in conduction velocity and in many neuropathies.28,44,51,61 Associated with greater membrane hyperpolarization in EMSCN9A+ patients, there was also evidence of reduction in slow K+ currents, with decreased subexcitability, S2 accommodation, TE undershoot, and greater superexcitability. The mathematical modelling suggests that these changes are secondary to the changes in membrane potential, as approximately 35% of slow K+ channels are open at resting membrane potential.66
Nav1.7-mediated disturbances in vascular regulation may underlie the distinct electrophysiological changes observed in myelinated motor and cutaneous afferents in this study, leading to membrane hyperpolarization following nerve ischemia. This contrasts with previously established hyperexcitability and membrane depolarisation in smaller diameter nociceptive fibers directly associated mutant Nav1.7 activity.13,14,38,60,69,72 While the effects of Nav1.7 signaling are cell background dependent,69 intriguingly in each cell type activation of Nav1.7 promotes vasodilatation64 and ensuing abnormal arteriole–venule shunting with decreased oxygenation may establish a vicious cycle of pain, redness, swelling, and warmth or chronic mild ischaemia.59 Similar clinical vascular changes are observed in EMSCN9A− patients, although the pathophysiological mechanisms are unclear, further supporting the secondary effects of EM on peripheral axons demonstrated in this study. Previous cellular models of nerve hypoxia and in vivo models of nerve ischaemia have demonstrated increases in persistent Na+ currents.33,39,75 The model was unable to resolve a small increase in persistent Na+ currents in this case, but this may be due in part to the fact that the measurements were made interictally. A small increase in resting persistent Na+ currents could lead to increased intracellular Na+ concentration, thereby stimulating the Na+/K+ pump. Consequently, in the postischaemic state, the Na+/K+ pump is overactive to maintain electrochemical gradients, exporting 3 Na+ ions in exchange for 2 K+ ions, producing a chronic state of axonal hyperpolarization. Furthermore, axonal ischaemic vulnerability differs between types of nerve fibres, with sensory more susceptible than motor axons and greater vulnerability in large myelinated fibres compared with small unmyelinated fibres.31,35,37,55,62 This is consistent with the pattern of changes in this study. Separately, increased levels of intracellular Na+ have been observed in dorsal root ganglion neurons expressing Nav1.7 gain-of-function mutations.25 Interestingly, sodium channel blockers have provided an in vitro protective benefit against the adverse long-term effects of anoxia on axons.30,80
Altered ion channel expression or gating properties may provide an alternative explanation for alterations in membrane potential.67 While this was not supported by changes in multiple excitability parameters or mathematical modelling, this may be complicated by interneuronal heterogeneity in the dynamics of Na+ conductances that reflect differences in the structure of the underlying channel molecule in the neuronal membrane.65 Alterations in passive cable properties (myelin thickness and internodal length) may also change membrane potential or ion channel conductances, for example, shorter internodes in regenerated nerve and an increased number of nodes provide a greater intra-axonal Na+ load to drive the Na+/K+ pump.58 This concept is unlikely as latency and conduction velocities were maintained and mathematical modeling did not support this as the most plausible explanation.
We deliberately chose to test the median nerve at the wrist, remote from the ongoing clinical symptomatology, to avoid possible secondary changes associated with EM (eg, cutaneous thickening or swelling) that may have influenced nerve excitability testing and thereby ensuring changes were constitutional.
4.2. Heat hyperalgesia: pathophysiological mechanisms
The most striking result was the significant increases in SDTC in EMSCN9A+ patients, an indirect measure of the activity of nodal persistent Na+ current with heating,9 the opposite of controls.36 Similar to controls, increases of skin temperature reduced superexcitability and shortened accommodation to depolarizing current, related to activation of slow K+ channels during hyperthermia. Heating typically triggers acute EM symptoms, such that the excitability changes with increased temperature are likely to measure acute modulations in membrane potential and conductances. Mathematical modelling indicated that the acute excitability changes with heating in patients with EM correspond to axonal ischaemic depolarisation and supporting the concept of vascular dysregulation.42 This will produce an intra-axonal Na+ load to chronically drive the Na+/K+ pump. Recent in vitro studies have demonstrated that heating selectively increases excitability in gain-of-function mutant Nav1.7 channels, in comparison to wild-type Nav1.7 channels,.12,82 Together with increases in skin metabolism and oxygen consumption, the resultant neurovascular changes would exacerbate tissue hypoxia and worsen cutaneous arteriovenous shunting in EM, creating a vicious cycle to evoke the heat hyperalgesia.59 It is possible that these paradoxical changes may also be caused by structural membrane changes as a function of temperature, disturbing the temperature dependence of Na+ channel conductance and gating kinetics.68 Tissue oedema from skin hyperaemia and release of inflammatory peptides may also influence the membrane structure and Na+ channel activity. Importantly, increases in persistent Na+ conductances in peripheral axons have been linked to a number of common painful neuropathies,17,52,53 participating in the generation of ectopic neuropathic symptoms.
4.3. Challenges in therapy
Concerning the episodic excruciating pain in patients with EM, a frequently used treatment of this disorder are the sodium channel blockers such as lidocaine and mexiletine. This nonselective partial block of voltage-gated Na+ channels targets segment 6 of domain IV of the alpha subunit.63 Clinical response varies and suggests that genotype may be important.12,81 In agreement with the previous study by Kuwabara et al.,50 the effect of mexiletine on axonal excitability in EMSCN9A+ patients demonstrates small reductions in SDTC. Even so, SDTC remained increased such that it may be expected to have an ongoing part in neuropathic symptoms. There is an urgent clinical need to develop more effective Na+ channel blockers for the treatment of neuropathic pain, particularly the selective and potent Nav1.7 channel blockers, with initial results showing promise in EM.12
The variability of pain between and within subjects poses challenges in assessing response to treatment and highlights the need to develop sensitive and relevant biomarkers to facilitate establishing novel disease modifying therapies. Recently, molecular modelling, thermodynamic analysis, and functional profiling were able to predict the responsiveness to treatment and may provide a platform for personalized medicine in the future with a preclinical assessment of potential efficacy.32
5. Conclusions
Excitability studies are now being used widely in the clinical investigation of patients with neurological disease to provide functional information about peripheral nerves, with changes in measurements of excitability being interpreted in terms of direct and/or remote effects of altered ion channel function.47 These have provided novel insights into disease mechanisms and treatment strategies.26,27,29,49,54,57,73 This study advances this possibility by exploring the variations in excitability properties along with temperature in patients with EM. Specific indices of nerve excitability responded very differently to increases in temperature and these effects can be related to the indirect behaviour of mutant Nav1.7 channels systemically, producing a vicious cycle of pain. These preliminary findings support the use of excitability techniques as a biomarker to assess abnormalities of axonal function and monitor changes with the development of novel therapies in vivo.
Conflict of interest statement
M.-J. Lee was supported by the grants from National Taiwan University Hospital (NTUH 105-003153) and the Ministry of Science and Technology, Taiwan, R.O.C. (MOST 104-2314-B-002-061).
M. A. Farrar and M.-J. Lee contributed equally to this article.
Acknowledgements
M.-J. Lee thank the assistance from the second and third common laboratory, National Taiwan University Hospital, Taipei, Taiwan.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
References
- [1].Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature 1996;379:257–62. [DOI] [PubMed] [Google Scholar]
- [2].Baker M, Bostock H, Grafe P, Martius P. Function and distribution of three types of rectifying channel in rat spinal root myelinated axons. J Physiol 1987;383:45–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Black JA, Frezel N, Dib-Hajj SD, Waxman SG. Expression of Nav1.7 in DRG neurons extends from peripheral terminals in the skin to central preterminal branches and terminals in the dorsal horn. Mol Pain 2012;8:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bostock H. The strength-duration relationship for excitation of myelinated nerve: computed dependence on membrane parameters. J Physiol 1983;341:59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bostock H. FC28.1 MEMFIT: a computer program to aid interpretation of multiple excitability measurements on human motor axons. Clin Neurophysiol 2006;117(suppl 1):1.16318927 [Google Scholar]
- [6].Bostock H, Baker M. Evidence for two types of potassium channel in human motor axons in vivo. Brain Res 1988;462:354–8. [DOI] [PubMed] [Google Scholar]
- [7].Bostock H, Baker M, Reid G. Changes in excitability of human motor axons underlying post-ischaemic fasciculations: evidence for two stable states. J Physiol 1991;441:537–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bostock H, Cikurel K, Burke D. Threshold tracking techniques in the study of human peripheral nerve. Muscle Nerve 1998;21:137–58. [DOI] [PubMed] [Google Scholar]
- [9].Bostock H, Rothwell JC. Latent addition in motor and sensory fibres of human peripheral nerve. J Physiol 1997;498(pt 1):277–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bostock H, Sears TA, Sherratt RM. The spatial distribution of excitability and membrane current in normal and demyelinated mammalian nerve fibres. J Physiol 1983;341:41–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Burke D, Kiernan MC, Bostock H. Excitability of human axons. Clin Neurophysiol 2001;112:1575–85. [DOI] [PubMed] [Google Scholar]
- [12].Cao L, McDonnell A, Nitzsche A, Alexandrou A, Saintot PP, Loucif AJ, Brown AR, Young G, Mis M, Randall A, Waxman SG, Stanley P, Kirby S, Tarabar S, Gutteridge A, Butt R, McKernan RM, Whiting P, Ali Z, Bilsland J, Stevens EB. Pharmacological reversal of a pain phenotype in iPSC-derived sensory neurons and patients with inherited erythromelalgia. Sci Transl Med 2016;8:335ra356. [DOI] [PubMed] [Google Scholar]
- [13].Cheng X, Dib-Hajj SD, Tyrrell L, Waxman SG. Mutation I136V alters electrophysiological properties of the Na(v)1.7 channel in a family with onset of erythromelalgia in the second decade. Mol Pain 2008;4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cheng X, Dib-Hajj SD, Tyrrell L, Wright DA, Fischer TZ, Waxman SG. Mutations at opposite ends of the DIII/S4-S5 linker of sodium channel Na V 1.7 produce distinct pain disorders. Mol Pain 2010;6:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Choi JS, Zhang L, Dib-Hajj SD, Han C, Tyrrell L, Lin Z, Wang X, Yang Y, Waxman SG. Mexiletine-responsive erythromelalgia due to a new Na(v)1.7 mutation showing use-dependent current fall-off. Exp Neurol 2009;216:383–9. [DOI] [PubMed] [Google Scholar]
- [16].Cook-Norris RH, Tollefson MM, Cruz-Inigo AE, Sandroni P, Davis MD, Davis DM. Pediatric erythromelalgia: a retrospective review of 32 cases evaluated at Mayo Clinic over a 37-year period. J Am Acad Dermatol 2012;66:416–23. [DOI] [PubMed] [Google Scholar]
- [17].Craner MJ, Klein JP, Renganathan M, Black JA, Waxman SG. Changes of sodium channel expression in experimental painful diabetic neuropathy. Ann Neurol 2002;52:786–92. [DOI] [PubMed] [Google Scholar]
- [18].Cummins TR, Dib-Hajj SD, Black JA, Akopian AN, Wood JN, Waxman SG. A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J Neurosci 1999;19:RC43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cummins TR, Dib-Hajj SD, Waxman SG. Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J Neurosci 2004;24:8232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Davis MD, Morr CS, Warndahl RA, Sandroni P. Topically applied midodrine, 0.2%, an alpha1-agonist, for the treatment of erythromelalgia. JAMA Dermatol 2015;151:1025–6. [DOI] [PubMed] [Google Scholar]
- [21].Davis MD, O'Fallon WM, Rogers RS, III, Rooke TW. Natural history of erythromelalgia: presentation and outcome in 168 patients. Arch Dermatol 2000;136:330–6. [DOI] [PubMed] [Google Scholar]
- [22].Davis MD, Sandroni P, Rooke TW, Low PA. Erythromelalgia: vasculopathy, neuropathy, or both? A prospective study of vascular and neurophysiologic studies in erythromelalgia. Arch Dermatol 2003;139:1337–43. [DOI] [PubMed] [Google Scholar]
- [23].Dib-Hajj SD, Rush AM, Cummins TR, Hisama FM, Novella S, Tyrrell L, Marshall L, Waxman SG. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain 2005;128(pt 8):1847–54. [DOI] [PubMed] [Google Scholar]
- [24].Djouhri L, Newton R, Levinson SR, Berry CM, Carruthers B, Lawson SN. Sensory and electrophysiological properties of Guinea-pig sensory neurones expressing Nav 1.7 (PN1) Na+ channel alpha subunit protein. J Physiol 2003;546(pt 2):565–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Estacion M, Vohra BP, Liu S, Hoeijmakers J, Faber CG, Merkies IS, Lauria G, Black JA, Waxman SG. Ca2+ toxicity due to reverse Na+/Ca2+ exchange contributes to degeneration of neurites of DRG neurons induced by a neuropathy-associated Nav1.7 mutation. J Neurophysiol 2015;114:1554–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Farrar MA, Lin CS, Krishnan AV, Park SB, Andrews PI, Kiernan MC. Acute, reversible axonal energy failure during stroke-like episodes in MELAS. Pediatrics 2010;126:e734–739. [DOI] [PubMed] [Google Scholar]
- [27].Farrar MA, Park SB, Krishnan A, Kiernan MC, Lin C. Axonal dysfunction and conduction failure in HNPP. Muscle Nerve 2014;49:858–64. [DOI] [PubMed] [Google Scholar]
- [28].Farrar MA, Park SB, Lin CSY, Kiernan M. Evolution of peripheral nerve function in humans: novel insights from motor nerve excitability. J Physiol 2013;591:273–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Farrar MA, Vucic S, Lin CS, Park SB, Johnston HM, du Sart D, Bostock H, Kiernan MC. Dysfunction of axonal membrane conductances in adolescents and young adults with spinal muscular atrophy. Brain 2011;134(pt 11):3185–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fern R, Ransom BR, Stys PK, Waxman SG. Pharmacological protection of CNS white matter during anoxia: actions of phenytoin, carbamazepine and diazepam. J Pharmacol Exp Ther 1993;266:1549–55. [PubMed] [Google Scholar]
- [31].Fink BR, Cairns AM. A bioenergetic basis for peripheral nerve fiber dissociation. PAIN 1982;12:307–17. [DOI] [PubMed] [Google Scholar]
- [32].Geha P, Yang Y, Estacion M, Schulman BR, Tokuno H, Apkarian AV, Dib-Hajj SD, Waxman SG. Pharmacotherapy for pain in a family with inherited erythromelalgia guided by genomic analysis and functional profiling. JAMA Neurol 2016;73:659–67. [DOI] [PubMed] [Google Scholar]
- [33].Hammarstrom AK, Gage PW. Inhibition of oxidative metabolism increases persistent sodium current in rat CA1 hippocampal neurons. J Physiol 1998;510(pt 3):735–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hicks CL, von Baeyer CL, Spafford PA, van Korlaar I, Goodenough B. The Faces Pain Scale-Revised: toward a common metric in pediatric pain measurement. PAIN 2001;93:173–83. [DOI] [PubMed] [Google Scholar]
- [35].Hofmeijer J, Franssen H, van Schelven LJ, van Putten MJ. Why are sensory axons more vulnerable for ischemia than motor axons? PLoS One 2013;8:e67113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Howells J, Czesnik D, Trevillion L, Burke D. Excitability and the safety margin in human axons during hyperthermia. J Physiol 2013;591:3063–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Howells J, Trevillion L, Bostock H, Burke D. The voltage dependence of I(h) in human myelinated axons. J Physiol 2012;590:1625–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Huang CW, Lai HJ, Huang PY, Lee MJ, Kuo CC. The biophysical basis underlying gating changes in the p.V1316A mutant Nav1.7 channel and the molecular pathogenesis of inherited erythromelalgia. PLoS Biol 2016;14:e1002561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hyllienmark L, Brismar T. Effect of hypoxia on membrane potential and resting conductance in rat hippocampal neurons. Neuroscience 1999;91:511–17. [DOI] [PubMed] [Google Scholar]
- [40].Isose S, Misawa S, Sakurai K, Kanai K, Shibuya K, Sekiguchi Y, Nasu S, Noto Y, Fujimaki Y, Yokote K, Kuwabara S. Mexiletine suppresses nodal persistent sodium currents in sensory axons of patients with neuropathic pain. Clin Neurophysiol 2010;121:719–24. [DOI] [PubMed] [Google Scholar]
- [41].Kanai K, Kuwabara S, Arai K, Sung JY, Ogawara K, Hattori T. Muscle cramp in Machado-Joseph disease: altered motor axonal excitability properties and mexiletine treatment. Brain 2003;126(pt 4):965–73. [DOI] [PubMed] [Google Scholar]
- [42].Kiernan MC, Bostock H. Effects of membrane polarization and ischaemia on the excitability properties of human motor axons. Brain 2000;123(pt 12):2542–51. [DOI] [PubMed] [Google Scholar]
- [43].Kiernan MC, Burke D, Andersen KV, Bostock H. Multiple measures of axonal excitability: a new approach in clinical testing. Muscle Nerve 2000;23:399–409. [DOI] [PubMed] [Google Scholar]
- [44].Kiernan MC, Cikurel K, Bostock H. Effects of temperature on the excitability properties of human motor axons. Brain 2001;124(pt 4):816–25. [DOI] [PubMed] [Google Scholar]
- [45].Kiernan MC, Krishnan AV, Lin CS, Burke D, Berkovic SF. Mutation in the Na+ channel subunit SCN1B produces paradoxical changes in peripheral nerve excitability. Brain 2005;128(pt 8):1841–6. [DOI] [PubMed] [Google Scholar]
- [46].Kiernan MC, Lin CS, Andersen KV, Murray NM, Bostock H. Clinical evaluation of excitability measures in sensory nerve. Muscle Nerve 2001;24:883–92. [DOI] [PubMed] [Google Scholar]
- [47].Kiernan MC, Lin CSY. Nerve excitability: a clinical translation A2-Aminoff. Chapter 15 In: Michael J. Aminoff's Electrodiagnosis in Clinical Neurology. 6th ed London: W.B. Saunders, 2012. p. 345–65. [Google Scholar]
- [48].Krishnan AV, Lin CS, Park SB, Kiernan MC. Axonal ion channels from bench to bedside: a translational neuroscience perspective. Prog Neurobiol 2009;89:288–313. [DOI] [PubMed] [Google Scholar]
- [49].Kuwabara S, Misawa S. Pharmacologic intervention in axonal excitability: in vivo assessment of nodal persistent sodium currents in human neuropathies. Curr Mol Pharmacol 2008;1:61–7. [DOI] [PubMed] [Google Scholar]
- [50].Kuwabara S, Misawa S, Tamura N, Kanai K, Hiraga A, Ogawara K, Nakata M, Hattori T. The effects of mexiletine on excitability properties of human median motor axons. Clin Neurophysiol 2005;116:284–9. [DOI] [PubMed] [Google Scholar]
- [51].Kwai NC, Arnold R, Poynten AM, Howells J, Kiernan MC, Lin CS, Krishnan AV. In vivo evidence of reduced nodal and paranodal conductances in type 1 diabetes. Clin Neurophysiol 2016;127:1700–6. [DOI] [PubMed] [Google Scholar]
- [52].Kwai NC, Arnold R, Wickremaarachchi C, Lin CS, Poynten AM, Kiernan MC, Krishnan AV. Effects of axonal ion channel dysfunction on quality of life in type 2 diabetes. Diabetes Care 2013;36:1272–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lin CS, Farrar MA. No gain—no pain? J Neurol Neurosurg Psychiatry 2013;84:364. [DOI] [PubMed] [Google Scholar]
- [54].Lin CS, Krishnan AV, Lee MJ, Zagami AS, You HL, Yang CC, Bostock H, Kiernan MC. Nerve function and dysfunction in acute intermittent porphyria. Brain 2008;131(pt 9):2510–19. [DOI] [PubMed] [Google Scholar]
- [55].Lin CS, Kuwabara S, Cappelen-Smith C, Burke D. Responses of human sensory and motor axons to the release of ischaemia and to hyperpolarizing currents. J Physiol 2002;541(pt 3):1025–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].McDonnell A, Schulman B, Ali Z, Dib-Hajj SD, Brock F, Cobain S, Mainka T, Vollert J, Tarabar S, Waxman SG. Inherited erythromelalgia due to mutations in SCN9A: natural history, clinical phenotype and somatosensory profile. Brain 2016;139(pt 4):1052–65. [DOI] [PubMed] [Google Scholar]
- [57].Menezes MP, Farrar MA, Webster R, Antony J, O'Brien K, Ouvrier R, Kiernan MC, Burns J, Vucic S. Pathophysiology of motor dysfunction in a childhood motor neuron disease caused by mutations in the riboflavin transporter. Clin Neurophysiol 2016;127:911–18. [DOI] [PubMed] [Google Scholar]
- [58].Moldovan M, Alvarez S, Rosberg MR, Krarup C. Persistent alterations in active and passive electrical membrane properties of regenerated nerve fibers of man and mice. Eur J Neurosci 2016;43:388–403. [DOI] [PubMed] [Google Scholar]
- [59].Mork C, Kvernebo K, Asker CL, Salerud EG. Reduced skin capillary density during attacks of erythromelalgia implies arteriovenous shunting as pathogenetic mechanism. J Invest Dermatol 2002;119:949–53. [DOI] [PubMed] [Google Scholar]
- [60].Namer B, Orstavik K, Schmidt R, Kleggetveit IP, Weidner C, Mork C, Kvernebo MS, Kvernebo K, Salter H, Carr TH, Segerdahl M, Quiding H, Waxman SG, Handwerker HO, Torebjork HE, Jorum E, Schmelz M. Specific changes in conduction velocity recovery cycles of single nociceptors in a patient with erythromelalgia with the I848T gain-of-function mutation of Nav1.7. PAIN 2015;156:1637–46. [DOI] [PubMed] [Google Scholar]
- [61].Park SB, Lin CS, Krishnan AV, Goldstein D, Friedlander ML, Kiernan MC. Long-term neuropathy after oxaliplatin treatment: challenging the dictum of reversibility. Oncologist 2011;16:708–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Parry GJ, Brown MJ. Selective fiber vulnerability in acute ischemic neuropathy. Ann Neurol 1982;11:147–54. [DOI] [PubMed] [Google Scholar]
- [63].Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 1994;265:1724–8. [DOI] [PubMed] [Google Scholar]
- [64].Rice FL, Albrecht PJ, Wymer JP, Black JA, Merkies IS, Faber CG, Waxman SG. Sodium channel Nav1.7 in vascular myocytes, endothelium, and innervating axons in human skin. Mol Pain 2015;11:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Rizzo MA, Kocsis JD, Waxman SG. Slow sodium conductances of dorsal root ganglion neurons: intraneuronal homogeneity and interneuronal heterogeneity. J Neurophysiol 1994;72:2796–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Roper J, Schwarz JR. Heterogeneous distribution of fast and slow potassium channels in myelinated rat nerve fibres. J Physiol 1989;416:93–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Rosberg MR, Alvarez S, Klein D, Nielsen FC, Martini R, Levinson SR, Krarup C, Moldovan M. Progression of motor axon dysfunction and ectopic Nav1.8 expression in a mouse model of Charcot-Marie-Tooth disease 1B. Neurobiol Dis 2016;93:201–14. [DOI] [PubMed] [Google Scholar]
- [68].Rosen AD. Nonlinear temperature modulation of sodium channel kinetics in GH(3) cells. Biochim Biophys Acta 2001;1511:391–6. [DOI] [PubMed] [Google Scholar]
- [69].Rush AM, Dib-Hajj SD, Liu S, Cummins TR, Black JA, Waxman SG. A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc Natl Acad Sci U S A 2006;103:8245–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Scholz A, Reid G, Vogel W, Bostock H. Ion channels in human axons. J Neurophysiol 1993;70:1274–9. [DOI] [PubMed] [Google Scholar]
- [71].Schwarz JR, Reid G, Bostock H. Action potentials and membrane currents in the human node of Ranvier. Pflugers Arch 1995;430:283–92. [DOI] [PubMed] [Google Scholar]
- [72].Sheets PL, Jackson JO, II, Waxman SG, Dib-Hajj SD, Cummins TR. A Nav1.7 channel mutation associated with hereditary erythromelalgia contributes to neuronal hyperexcitability and displays reduced lidocaine sensitivity. J Physiol 2007;581(pt 3):1019–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Shibuya K, Misawa S, Kimura H, Noto Y, Sato Y, Sekiguchi Y, Iwai Y, Mitsuma S, Beppu M, Watanabe K, Fujimaki Y, Tsuji Y, Shimizu T, Mizuno T, Nakagawa M, Sawaguchi K, Hanaoka H, Kuwabara S. A single blind randomized controlled clinical trial of mexiletine in amyotrophic lateral sclerosis: efficacy and safety of sodium channel blocker phase II trial. Amyotroph Lateral Scler Frontotemporal Degener 2015;16:353–8. [DOI] [PubMed] [Google Scholar]
- [74].Stadler T, O'Reilly AO, Lampert A. Erythromelalgia mutation Q875E Stabilizes the activated state of sodium channel Nav1.7. J Biol Chem 2015;290:6316–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Stys PK, Lopachin RM. Mechanisms of calcium and sodium fluxes in anoxic myelinated central nervous system axons. Neuroscience 1998;82:21–32. [DOI] [PubMed] [Google Scholar]
- [76].Tesfaye S, Harris N, Jakubowski JJ, Mody C, Wilson RM, Rennie IG, Ward JD. Impaired blood flow and arterio-venous shunting in human diabetic neuropathy: a novel technique of nerve photography and fluorescein angiography. Diabetologia 1993;36:1266–74. [DOI] [PubMed] [Google Scholar]
- [77].Toledo-Aral JJ, Moss BL, He ZJ, Koszowski AG, Whisenand T, Levinson SR, Wolf JJ, Silos-Santiago I, Halegoua S, Mandel G. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci U S A 1997;94:1527–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Tomlinson SE, Bostock H, Grinton B, Hanna MG, Kullmann DM, Kiernan MC, Scheffer IE, Berkovic SF, Burke D. In vivo loss of slow potassium channel activity in individuals with benign familial neonatal epilepsy in remission. Brain 2012;135(pt 10):3144–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Tomlinson SE, Tan SV, Kullmann DM, Griggs RC, Burke D, Hanna MG, Bostock H. Nerve excitability studies characterize Kv1.1 fast potassium channel dysfunction in patients with episodic ataxia type 1. Brain 2010;133(pt 12):3530–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Waxman SG, Black JA, Stys PK, Ransom BR. Ultrastructural concomitants of anoxic injury and early post-anoxic recovery in rat optic nerve. Brain Res 1992;574:105–19. [DOI] [PubMed] [Google Scholar]
- [81].Wu MT, Huang PY, Yen CT, Chen CC, Lee MJ. A novel SCN9A mutation responsible for primary erythromelalgia and is resistant to the treatment of sodium channel blockers. PLoS One 2013;8:e55212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Yang Y, Huang J, Mis MA, Estacion M, Macala L, Shah P, Schulman BR, Horton DB, Dib-Hajj SD, Waxman SG. Nav1.7-A1632G mutation from a family with inherited erythromelalgia: enhanced firing of dorsal root ganglia neurons evoked by thermal stimuli. J Neurosci 2016;36:7511–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, Fan J, Bu D, Liu B, Fan Z, Wu G, Jin J, Ding B, Zhu X, Shen Y. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet 2004;41:171–4. [DOI] [PMC free article] [PubMed] [Google Scholar]