

Abstract
MicroRNAs (miRNAs) are post-transcriptional regulators of gene expression in animals and plants. Comparative genomic computational methods have been developed to predict new miRNAs in worms, flies, and humans. Here, we present a novel single genome approach for the detection of miRNAs in Arabidopsis thaliana. This was initiated by producing a candidate miRNA-target data set using an algorithm called findMiRNA, which predicts potential miRNAs within candidate precursor sequences that have corresponding target sites within transcripts. From this data set, we used a characteristic divergence pattern of miRNA precursor families to select 13 potential new miRNAs for experimental verification, and found that corresponding small RNAs could be detected for at least eight of the candidate miRNAs. Expression of some of these miRNAs appears to be under developmental control. Our results are consistent with the idea that targets of miRNAs encompass a wide range of transcripts, including those for F-box factors, ubiquitin conjugases, Leucine-rich repeat proteins, and metabolic enzymes, and that regulation by miRNAs might be widespread in the genome. The entire set of annotated transcripts in the Arabidopsis genome has been run through findMiRNA to yield a data set that will enable identification of potential miRNAs directed against any target gene.
Small RNA molecules are now accepted as playing a general role in the regulation of genes, as well as in host defense through the mechanism of RNA interference (RNAi). MicroRNAs (miRNAs) are small RNAs that are processed from hairpin RNA precursors encoded within the genome (for review, see Bartel 2004; Mallory and Vaucheret 2004) and are believed to play significant roles in development within most multicellular organisms by regulating the effective level of developmentally important transcripts (Bergmann and Lane 2003; Emery et al. 2003; Palatnik et al. 2003; Chen 2004; Sempere et al. 2004). In plants, miRNAs tend to show greater complementarity to their targets than do animal miRNAs, and there are a number of examples of mRNA target cleavage for corresponding miRNAs in plants (Llave et al. 2002; Palatnik et al. 2003; Tang et al. 2003). The potential importance of miRNAs in developmental processes is evident in the mutant phenotypes associated with miRNA expression mutants (Hipfner et al. 2002; Brennecke et al. 2003; Palatnik et al. 2003; Chen 2004).
Direct cloning has enabled the identification of many miRNAs; however, significant variation in their expression levels has made it difficult to clone low abundance miRNAs (Lai et al. 2003; Lim et al. 2003b). For this reason, computational approaches have been sought by a number of laboratories to complement such efforts (Grad et al. 2003; Lai et al. 2003; Lim et al. 2003a,b). Methods to date have focused on a number of different characteristics of miRNA genes, but all rely on interspecies comparative genomics at an early stage for identification. Despite the short length of miRNA sequences, the specificity of the interaction between miRNAs and their mRNA targets has demanded considerable conservation of the miRNA target sequences during evolution, but less conservation of the non-miRNA stem sequence and, less still, loop region sequences. This pattern of divergence has been exploited in one algorithm involving the comparison of two or more genomes, and has resulted in the estimation of 110 miRNA genes within Drosophila (Lai et al. 2003). A different approach that emphasizes the statistics of miRNA precursor anatomy, such as characteristic base pairing and nucleotide bias has resulted in the prediction of 120 miRNA genes in Caenorhabditis elegans (Lim et al. 2003b), and 200–255 miRNA genes in human (Lim et al. 2003a).
Here, we describe a computational approach, implemented by an algorithm called findMiRNA, which relies on the more rigid complementarity existing between plant miRNAs and their targets to identify initial miRNA candidates. The algorithm then analyzes these candidates further for adjacent sequence complementarity that would enable stem-loop formation in an RNA molecule consistent with the known structures of miRNA precursors. Analysis of the entire Arabidopsis genome set of 29,399 transcript records, which correspond to 27,987 unique loci, was performed using the findMiRNA algorithm to identify potential miRNA precursors within the intergenic regions. Candidates for experimental verification were selected based on the existence of families of precursors for predicted miRNAs, as well as the conservation of the miRNAs in rice. While this work was under review, algorithms based on comparative genomics have been reported for computational prediction of miRNAs in Arabidopsis (Bonnet et al. 2004; Jones-Rhoades and Bartel 2004), as well as an experimental search for new miRNAs (Sunkar and Zhu 2004), providing independent validation for some of the predicted miRNAs reported here. The results of our analysis have been used to establish a database that can be searched for putative miRNAs directed against any annotated target transcript in the Arabidopsis genome, and represents a resource of potential miRNA target sites, their respective potential miRNAs, and the corresponding precursors.
Results
Exploratory scan of 5701 transcripts by the findMiRNA algorithm
The findMiRNA algorithm uses annotated transcripts to search the intergenic regions for candidate miRNAs directed against the transcripts (see Methods). An initial exploratory set of transcripts was used for a preliminary assessment of the findMiRNA algorithm for its efficiency to detect known miRNA precursors, their miRNAs, and their respective targets. This exploratory set was also used to choose candidates for further analysis. The selected set contained only 5701 of the total of 29,399 transcript records for the Arabidopsis genome. This set of transcripts included transcription factors and other transcripts that showed potential for targeting by families of precursors as determined from a less sensitive full-genome analysis of an older version of the algorithm.
At the time the algorithm was run, the miRNA Registry held at Rfam in 2004 (http://www.sanger.ac.uk/Sofware/Rfam/mirna/; Griffiths-Jones et al. 2003; Griffiths-Jones 2004) contained 43 Arabidopsis miRNA precursors that could be grouped into 15 sequence-related precursor families (SRPF) as follows: MIR156/157, MIR158, MIR159/319/JAW, MIR160, MIR161, MIR162, MIR163, MIR164, MIR165/166, MIR167, MIR168, MIR169, MIR170/171, MIR172, and MIR173. An SRPF is a family of precursor sequences that fall within the same group according to sequence homology rather than Rfam groupings. The mRNA transcript sequences that may be targets of these miRNAs have previously been suggested (Rhoades et al. 2002; Palatnik et al. 2003) and some confirmed experimentally (Llave et al. 2002; Aukerman and Sakai 2003). Of the 62 suggested or confirmed targets previously reported for the 43 precursor genes (Park et al. 2002; Rhoades et al. 2002; Palatnik et al. 2003; Xie et al. 2003), 54 were present in the exploratory set of 5701 transcripts. Of the 15 SRPFs, 11 were predicted while correctly identifying the miRNA sequence within 2 nt of the known miRNA. The MIR162 SRPF, which has previously been reported to target dicer (At1g01040) (Xie et al. 2003), was not detected due to the requirement for gapped alignments to allow for the presence of a single-stranded bulge near the center of the target site in the dicer transcript when aligned with the miR162 sequence. For the SRPFs MIR163, MIR168, and MIR173, the target transcripts were not present in the 5701 exploratory set. Hence, all 11 SRPFs that were included within the data set could be detected. The findMiRNA algorithm therefore appears to have the sensitivity required to identify most of the known miRNAs using previously predicted targets.
Identification of potential miRNA precursor families by using the characteristic pattern of precursor family sequence divergence
It has been previously observed that miRNA precursor families have a characteristic pattern of sequence divergence (Pasquinelli et al. 2000). We decided to use this divergence pattern to rank families of sequences on the presumption that this would enrich for true miRNA precursor families. Candidate miRNA precursors that are predicted to target the same nucleotides of a transcript, and for which the predicted miRNA is on the same side of the central loop, were considered to belong to a family of sequences. A group of findMiRNA records containing such a family of sequences with its associated target sequence has been designated as a cluster. A family of predicted precursor sequences that targets more than one transcript will therefore give rise to more than one cluster, one for each target. Each of these clusters may or may not contain all of the same predicted precursor candidates, since some predicted miRNA sequences may match some transcripts better than others do as a result of, among other things, the existence of G-U pairing.
A set of 1599 candidate precursor clusters from the exploratory scan were ranked using an algorithm that relies on the difference between the combined sequence identities for predicted miRNA/miRNA* sequences (within aligned candidate precursors) and percent identity of the interstice sequence, i.e., the sequence which lies between miRNA sequence and the complementary miRNA* sequence in the precursor hairpin (see Methods and Supplemental material). Within these ranked clusters, the correct miRNA sequence of the correct strand was identified for 10 SRPFs, and these tended to be present in clusters that were high on the ranking (Fig. 1A). Some SRPFs are represented by more than one sense strand cluster, which might be either due to the precursor families being detected as two families by the program, and/or the same precursor sequence being detected by findMiRNA as two different length versions of the same sequence.
Figure 1.

Ranked clusters of predicted candidate miRNA precursor sequences derived from two different findMiRNA experiments. Crosses (×) mark clusters containing previously reported miRNA precursors, filled triangles (▴) show clusters containing candidate miRNA precursors that were analyzed further, dots (·) indicate clusters of other candidate sequences. Clusters containing antisense sequences of previously reported miRNA precursors, for which the predicted target is erroneous, are shown in gray type. (A) Preliminary scan of 5701 transcripts. (Inset) All 1599 ranked clusters, with the magnified region delineated by gray borders. Clusters containing aligned sequences with 100% sequence identity across the entire alignment have a score of 0.0. (Magnified) Shows top-ranking clusters down to the cluster containing the cmr214 candidate precursor sequences. (B) Whole-genome scan, showing only ranked clusters that contain exact matches for the predicted miRNA sequence in rice. (Inset) All 236 ranked clusters, with the magnified region delineated by gray borders. (Magnified) Open circles (○) show candidates additional to those shown for preliminary scan. Small crosses (×) represent clusters containing some sequences that are also present in superior clusters (×) of previously reported miRNA precursors. Clusters containing aligned sequences with 100% sequence identity across the entire alignment have a score of 4.0.
Analysis of the 1599 ranked clusters from the exploratory scan for candidate novel miRNAs was performed for the top ∼200 clusters down to cmr214 (candidate microRNA 214; Fig. 1A). The analysis consisted of inspection for suggestive sequence features in Arabidopsis, as well as a comparison with the available rice genome sequence to search for predicted miRNAs that might be conserved in rice using the program prec_extract (see Methods). Ten candidate precursor families were selected for further analysis (cmr3, cmr5, cmr6, cmr7, cmr75, cmr84, cmr165, cmr195, cmr201, and cmr214; Fig. 1A). Some of these candidates appeared very promising on the basis of their structure and conservation in rice, while others were selected somewhat arbitrarily. For example, the cmr214 (candidate microRNA 214) miRNA sequence had a predicted target sequence that was conserved across four Arabidopsis transcripts, and the cmr6 candidate miRNA sequence had five target sites within the untranslated leader of the target transcript within Arabidopsis. These sequences are unlikely to occur by chance across this many diverged transcript sequences targeted by cmr214, or within the relatively short sequence of a 5′ UTR targeted by cmr6, suggesting functional conservation as miRNA targets.
Whole-genome scan and alignment of predicted miRNA sequences to rice genome
Using the current revised version of the findMiRNA algorithm, a scan of the entire 29,399 transcript set using a result limit of 500 candidate precursors per transcript was performed (see Methods). This resulted in the production of 2,382,269 candidate-miRNA-target records, from which a subset of records was extracted that could be grouped into clusters. The candidate miRNAs contained in these clusters were used to search the rice genome with the program hashedPrecExtract (see Methods) to identify equivalent miRNA candidate sequences that have the potential to form suitable hairpin structures. Using a stringent zero mismatch criterion for the predicted miRNA sequence, 236 candidate-miRNA-target clusters contained predicted miRNA sequences that had potential orthologs in rice. These clusters were ranked using a method that is similar to that used for the selection of the initial 10 candidates described above (Fig. 1B). Precursors belonging to nine of the known SRPFs were present in 25 clusters, among a total of 64, that ranked higher than score 4.0 (the level at which the cluster has 100% identity across the entire alignment for this version of the ranking algorithm). Of the remaining 43 clusters, six contained predicted precursors for three of the candidates previously predicted in the exploratory ranked clusters. These were cmr3, cmr6, and cmr214. The remaining 37 clusters were examined for likely bona fide miRNA and precursor sequences, and an additional three candidates were chosen for further analysis; these are designated as cand1, cand2, and cand3.
The predicted candidate miRNA precursors resulting from the preliminary screen of the 5701 transcripts and the whole-genome rice matching screen, are included in the Supplemental material as aligned sequences and as hairpin structure diagrams.
Detection of small RNAs corresponding to predicted miRNA candidates
RNA gel blot analysis was used to determine whether any of the 13 candidate miRNAs accumulated to detectable levels in either wild-type Columbia or in Columbia transgenic Arabidopsis plants expressing the viral suppressor of RNA silencing, P1/HC-Pro. Because most miRNAs have previously been shown to accumulate to elevated levels in P1/HC-Pro plants (Mallory et al. 2002; Kasschau et al. 2003), we reasoned that the use of RNA isolated from a P1/HC-Pro transgenic line would facilitate the detection of miRNAs that might be present below the level of detection in wild-type plants. Total RNA was isolated from rosette leaves, stems, and flowers of wild-type and P1/HC-Pro transgenic plants. This RNA was then enriched for small RNAs and analyzed by RNA gel-blot analysis using radiolabeled oligonucleotide probes complementary to each potential miRNA. Clear signals for small RNAs were detected for five of the 13 candidates tested, cand1, cmr214, cmr6, cmr3, and cand2 (Fig. 2A). The RNAs are similar in size to previously reported miRNAs (∼21–22 nt) and accumulated to detectable levels in one or more tissues (Fig. 2A). Similar to previously reported miRNAs, four of these five predicted miRNAs accumulated to elevated levels in the P1/HC-Pro transgenic plants. Cand1 and cmr214 were detected at about the same level in all three tested tissues, and at higher levels in the equivalent P1/HC-Pro tissues. Consistent with it being a true miRNA, the signal for cand1 was very strong, and was enhanced in the RNA from the HC-Pro plants, similar to the miR169 control (Fig. 2A). Potential precursor intermediates were also detected for cand1 (Fig. 2C). For cmr6 and cmr3, signals were generally weaker and were only detected for P1/HC-Pro samples and accumulated in a tissue-specific manner. Thus, cmr6 was found preferentially in the leaves, and cmr3 was found exclusively in the stems of P1/HC-Pro plants (Fig. 2A). Unlike all other miRNAs reported to date, the small RNA corresponding to cand2 accumulated to higher levels in wild-type plants than in the P1/HC-Pro transgenic plants.
Figure 2.

RNA gel-blot analysis of size-selected small RNAs from three tissues, leaves, stems, and flowers, from both wild-type Columbia plants and transgenic Columbia plants expressing the HC-Pro gene under the control of the 35S promoter. Signals were detected for 8 of the 13 selected miRNA candidates following hybridization of specific end-labeled DNA oligonucleotides. (Bottom) (EtBr) show representative RNA loadings for the set of replicate blots above.(A) Signals corresponding to ∼21 nt small RNAs. The candidates, cand1 and cmr214, show the expected increase in expression in the HC-Pro samples. Expression is detected only in HC-Pro lanes for cmr6 and cmr3, with higher expression in RNA isolated from leaf and stem tissue, respectively. For cand2, lower expression was observed in HC-Pro than the Columbia wild-type samples. Arrows indicate the position of a 23-nt DNA size marker that corresponds to RNAs of ∼20 nt. (B) StarFire probes were used to improve weaker signals corresponding to ∼24 nt small RNAs. Probes to the antisense sequences were also used to help identify whether the correct genomic strand was identified by findMiRNA (prefix `as-'). (C) Signals corresponding to end-labeled probes specific for miR167 and the cand1 candidate miRNA for RNA isolated from HC-Pro samples. An RNA size marker shown on the left edge has values shown in nucleotides. Larger bands that may represent precursor intermediates are evident for cand1. Two size classes of small RNA, ∼22 nt and ∼24 nt, are observed for the control, miR167 (arrowheads).
Using simple end labeled probes, small RNAs corresponding to the remaining six candidate miRNAs (cand3, cmr7, cmr195, cmr75, cmr5, and cmr84) were barely detectable or undetectable, even in P1/HC-Pro plants (data not shown). RNA gel-blot hybridizations were repeated using StarFire probes for those candidates that gave weak signals with simple end labeled probes. In addition, the recently reported miRNA, miR398 (Jones-Rhoades and Bartel 2004; Sunkar and Zhu 2004), has a large overlap with the antisense sequence of cand3 and is derived from the opposite strand in the genome, suggesting that the signal arising from the cand3 probe is coming from miR398*, and that the wrong strand was initially identified. In order to confirm this possibility, Star-Fire-labeled probes specific to the sense and antisense sequences overlapping cand3 and miR398 were used to probe fresh blots. In case the weak signals for cmr7, cmr75, and cmr84 also arose from hybridization of the probe to miRNA* molecules that might be derived from the opposite strand in the genome, StarFire probes for sense and antisense sequences were also used for these candidate miRNAs. For cand3, both probes gave distinct signals, but the probe against the antisense cand3 (miR398) gave the strongest signal, supporting the conclusion that the signal for cand3 corresponds to the miRNA* molecule, miR398* (Fig. 2B). Signals were also obtained for both cmr7 and anti-cmr7; however, the signal was stronger for the cmr7 probe (Fig. 2B). It should be noted that differences in the signal intensity observed between sense and antisense miRNA probes are not necessarily quantitative indicators of the actual differences in small RNA levels, as there will be differences in the efficiency of hybridization to the potential miRNA and miRNA* molecules, as well as differences in labeling efficiency and hybridization conditions. The cmr7 sense-specific probe detected the presence of two RNAs that appear to differ in length by one nucleotide. Similarly for cmr84, distinct signals were obtained for two small RNA species also appearing to differ in length by one nucleotide. The anti-cmr84 probe gave a very weak signal for the HC-Pro tissues only. For cmr5 and cmr75, unambiguous signals could not be obtained (data not shown).
While this work was under review, Rfam numbers were assigned for five of the above candidates, (see Table 2, below) following reports by other labs (Jones-Rhoades and Bartel 2004; Sunkar and Zhu 2004).
Table 2.
miRNA candidates confirmed at the molecular level
| Candidate | Family and locusa | Intergenic regionb | Predicted miRNA sequencec | Predicted target transcriptsd | |||
|---|---|---|---|---|---|---|---|
| cmr3 | miR395JR | a | At1g26990 | 5.3 kb | CUGAAGUGUUUGGGGGAACUC | At5g43780JR | APS4 |
| b | At1g26990 | 4.0 kb | CUGAAGUGUUUGGGGGGACUC | At3g49430 | SF2 (SR1) | ||
| c | At1g26990 | 1.4 kb | CUGAAGUGUUUGGGGGGACUC | At2g28780 | exp | ||
| d | At1g69800 | 4.4 kb | CUGAAGUGUUUGGGGGAACUC | At5g10180 | Sulfate Transporter | ||
| e | At1g69800 | 1.6 kb | CUGAAGUGUUUGGGGGAACUC | ||||
| f | AT1g69800 | 0.5 kb | CUGAAGUGUUUGGGGGGACUCu | ||||
| cmr6 | miR399JR,S | a | At1g29270 | UGCCAAAGGAGAUUUGCCCug | At2g33770S | UBC (607 627) | |
| d | At2g34210 | 2.9 kb | UGCCAAAGGAGAUUUGCCC | (740 760) | |||
| e | At2g34210 | 2.3 kb | UGCCAAAGGAGAUUUGCCU | (830 849) | |||
| f | At2g34210 | 0.9 kb | UGCCAAAGGAGAUUUGCCC | (887 906) | |||
| b | At1g63010 | UGCCAAAGGAGAGUUGCCC | (943 963) | ||||
| c | At5g62165 | UGCCAAAGGAGAGUUGCCC | At4g09730* | DEAD/DEAHboxhelicase | |||
| cmr7 | At5g35340 | uaaUAACCGAACCCGAACCGAUuug | At2g31700 | hyp (186 210) | |||
| At5g35360 | uaaUACCCGAACCCGAACCGAUacc | (207 231) | |||||
| At1g75980 | exp | ||||||
| At1g28540 | exp | ||||||
| At3g16760 | TPR | ||||||
| At5g05250 | exp | ||||||
| At1g13100 | Cytochrome P450 | ||||||
| cmr214 | miR393JR,S | a | At2g39890 | aagAUCCAAAGGGAUCGCAUUG | At3g62980JR,S | TIR1 | |
| b | At3g55740 | AUCCAAAGGGAUCGCAUUG | At3g26810JR,S | ||||
| At1g12820JR,S | |||||||
| At4g03190JR,S | |||||||
| At4g19780 | copia | ||||||
| At4g17450 | copia | ||||||
| cand1 | miR390S,C | b | At5g58470 | agCUCAGGAGGGAUAGCGCCau | At1g30040* | GA2OX2 | |
| a | At2g38330 | CUCAGGAGGGAUAGCGCC | At5g53470 | acyl-CoA BP | |||
| At4g13430 | Aconitase | ||||||
| At5g14210 | LRR-kinase | ||||||
| At4g08850* | LRR-kin (964 985) | ||||||
| (1612 1633) | |||||||
| At4g03205 | copro-ox | ||||||
| At1g03475 | copro-ox | ||||||
| At5g07280 | LRR-kinase | ||||||
| At1g60720 | hyp | ||||||
| At1g56130 | LRR-kinase | ||||||
| At1g61520 | CAB | ||||||
| cand2 | miR394JR | a | At1g20380 | UUUGGCAUUCUGUCCACCUCCu | At1g27340JR | F-box | |
| b | At1g76140 | UUUGGCAUUCUGUCCACCUCC | At1g10200 | LIM | |||
| At4g24850/60* | AAA-ATPase/MSP1 | ||||||
| At5g20580 | exp | ||||||
| anti-cand3 | miR398JR,S | a | At2g03450 | uguGUUCUCAGGUCACCCCUU | At3g15640JR,S | Cyt C oxidase VB | |
| b | At5g14550 | ugugUUCUCAGGUCACCCCUGCUG | At3g06370* | NHX3 putative | |||
| c | At5g14570 | uguguucucaggucaccccug | At1g08830*JR | CSD1 | |||
Rfam IDs for miRNAs that overlap the predicted candidates. For cand3, the antisense strand that corresponds to miR398 was found in the findMiRNA records. (JR) Jones-Rhoades and Bartel (2004); (S) Sunkar and Zhu (2004); (C) Carrington lab.—Arabidopsis thaliana Small RNA project—http://gac.bcc.orst.edu/smallRNA/
Intergenic regions containing precursor loci as defined by TAIR (intergenic regions are North of loci corresponding to AGI locus indentifiers). Where more than one locus occurs in an intergenic region, the position of the precursor relative to the AGI locus is indicated in kilobase pairs
Predicted miRNA sequence identified by findMiRNA is in uppercase. Underlined sequence was used with predicted target mRNAs to estimate miRNA-target stabilities (see Supplemental material). In the case of miR398 (anti-cand3), the predicted miRNA sequence was extended to fit a previously cloned miRNA, miR398b (Jones-Rhoades and Bartel 2004; Carrington J. lab. Arabidopsis thaliana Small RNA project, http://gac.bcc.orst.edu/smallRNA/)
Potential target transcripts were identified with a demand for an alignment with no single-strand bulges. Numbers refer to AGI locus identifiers and are ordered from top to bottom with decreasing thermal stability with predicted miRNA (also see Supplementary material). Those in bold type or underlined have respectively conserved or potentially conserved target sites in related transcripts in other plant species. When more than one target site occurs within the transcript, the corresponding nucleotides are indicated in the parentheses. Transcript IDs with asterisks correspond to targets that were manually identified and were not identified by findMiRNA at the 500 result limit. At4g24850/60 represent a pair of overlapping loci. (JR) Jones-Rhoades and Bartel (2004); (S) Sunkar and Zhu (2004) have suggested targets and these are underlined where cleavage products were detected by 5′ RACE. (exp) Expressed; (hyp) hypothetical protein; (SP2) pre-mRNA splicing factor 2, (TPR) tetratricopeptide repeat containing protein; (LRR-kin) leucine rich repeat kinase; (copro-ox) coproporthyrin III oxidase; (CAB) chlorophyll A-B binding protein; (MSP1) intramitochondrial sorting protein 1; (NHX3) putative sodium proton exchanger; (CSD1) copper/zinc superoxide dismutase
Diversity and potential multiplicity of miRNA targets
For the five RNA-blot-validated miRNAs, further potential targets to those identified by findMiRNA were added using relaxed search parameters with BLAST and the original ungapped version of PATMATCH using manually adjusted predicted miRNA sequences to the Arabidopsis transcripts. The miRNA-target pairs were then reduced to a conservative set (see Table 2, below, and Supplement to Table 2), which have minimum-free energies for the miRNA-target duplexes that are as good or better than the free energy of -28.2 kcal mol-1 as estimated using Mfold 3.1 (http://www.bioinfo.rpi.edu/applications/mfold/; Zuker 2003) (see Discussion).
For cmr3, the candidate miRNA sequence was predicted to target at least three Arabidopsis transcripts. One of these transcripts, At5g43780 (APS4), was recently reported as a potential target along with two other members of the ATP sulfurylase precursor protein family, APS1 and APS3 (Jones-Rhoades and Bartel 2004). The findMiRNA algorithm and BLAST searches do not identify the latter two members of the family as potential targets (see Discussion). Searches in other species revealed similar putative target sites in APS gene homologs in maize, Allium cepa and Brassica juncea. In addition to members of the APS gene family, we identified a potential target site within the 5′ end of the transcript for At5g10180, which encodes a sulfate transporter protein. By searching in other species, we found an equivalent target site in the 5′ UTR of a rice transcript (AK111395) that encodes a protein that is at least 59% identical to the Arabidopsis sulfate transporter. These putative target sites had 19 and 20 exactly complementary nucleotides with the cmr3 candidate miRNA, respectively, while the immediately surrounding sequence was significantly diverged. The sulfate transporter proteins are not related at the sequence level to the ATP sulfurylase precursor protein family, but may also be involved in processes related to sulfate assimilation. We identified two other potential targets, At2g28780 and At3g49430, encoding an unknown protein and a splicing factor, respectively, but these did not have conserved target sites in transcripts from other species. The conservation of target sites between species for transcripts belonging to two different gene families, i.e., encoding ATP sulfurylase precursors and sulfate transporters, supports the conclusion that this miRNA has more than one target gene family.
The predicted miRNA for candidate cmr6 is a predicted miRNA sequence that has five highly conserved target sequences within the 5′ UTR of the gene At2g33770 that encodes an ubiquitin conjugase (UBC) (Table 2, below). This target was not predicted by Jones-Rhoades and Bartel (2004). By searching in other plant species, five target sites were also found in a similarly spaced region from the 5′ end of a UBC gene in rice on chromosome 5 and Medicago (data not shown).
The miRNA precursor family cmr214 has a conserved target site that is present within four Arabidopsis transcripts and three rice transcripts (Table 2, below). Among these targets is the TIR1 gene (At3g62980) that encodes an F-box protein involved in the negative regulation of AUX/IAA proteins, repressors of auxin response, and in the regulation of lateral root formation (Gray et al. 1999). Three relatively close homologs of TIR1 that are also clear targets of cmr214 are At1g12820, At3g26810, and At4g03190. The presumptive target site in each of these transcript sequences was almost identical, despite neighboring sequences differing to a significant extent, indicating that this site was highly conserved. The presence of similar precursor-like sequences and an equivalent conserved target sequence across three transcripts in rice made it extremely likely, if not certain, that these candidate sequences represented bona fide miRNA precursor sequences. Other unrelated transcripts, At4g19780 and At4g17450, also contained sequences that have the potential to be targets of cmr214 (Table 2, below); however, these are both members of a copia-like retrotransposon family.
The candidate miRNA cand1 has the potential to be directed against a variety of targets. These include the transcripts of at least two members of a family of LRR kinases (At4g08850, At5g14210, At5g07280, and At1g56130). Other potential targets include two proteins from the coporporphyrin III oxidase family (At4g03205 and At1g03475), as well as transcripts encoding an aconitate hydratase (At4g13430), a gibberellin 2-oxidase (At1g30040), and an acyl-CoA-binding protein (At5g53470) (Table 2, below). Since a conserved miRNA precursor family was found in rice for cand1, searches were performed for conserved target sites in transcripts of other plant species. Potential target sites were also found in transcripts that encode proteins from the same family of LRR kinase from Oryza sativa (gi-34904253, MSP1 – gi-33383177) and Glycine max (RLK3 – gi-9651944). However, it could not be determined whether any of these sites were the result of sequence conservation, or were coincidental and due to the large size of the LRR kinase gene family in plants.
For cand2, a variety of potential targets were identified and included genes encoding an F-box (At1g27340), an AAA-ATPase (MSP1 – At4g24860 [formerly At4g24850]) a transcription factor LIM (At1g10200), a TPR repeat protein (At1g01320), and a zinc-finger protein (At4g22820).
Additional members of precursor families for known miRNAs
The findMiRNA algorithm identified additional precursors that belong to miRNA precursor families already reported to be in Arabidopsis. For five SRPFs (MIR156/157, MIR158, MIR159, MIR167, and MIR172) a single new precursor locus was identified. An additional two loci were found for two other SRPFs (MIR164 and MIR170/171). The largest number of newly identified precursor loci was seen for the MIR169 family for which the algorithm found an additional 12 precursor genes (Table 1). The detection of additional loci for known precursor families is in support of the sensitivity of the findMiRNA algorithm.
Table 1.
New loci related to previously reported miRNA precursors
| Precursor loci (Rfam ID)a | Intergenic regionb | miRNA sequencec |
|---|---|---|
| MIR157a | At1g66790 | |
| MIR157new (MIR156h) | At5g55840 | UUGACAGAAGAAAGAGAGCAC |
| MIR158 | At1g55600 | |
| MIR158new (MIR158a) | At3g10750 | CCAAAUGUAGACAAAGCA |
| MIR159a | At1g18080 | |
| MIR159new | At2g46260 | UUGGAUUGAAGGGAGCUCC |
| MIR164a | At2g47590 | UGGAGAAGCAGGGCACGUGC |
| MIR164new (MIR164c) | At5g27810 | UGGAGAAGCAGGGCACGUGC |
| MIR164new | At3g01210 | UUCGAGAAGCAGGGCACG |
| MIR167a | At3g22890 | |
| MIR167new (MIR167c) | At3g04770 | AAGCUGCCAGCAUGAUCU |
| MIR169 | At3g13410 | |
| MIR169new (MIR169b) | At5g24830 | AGCCAAGGAUGACUUGCC |
| MIR169new (MIR169d) | At1g53690 3.1 kb | AGCCAAGGAUGACUUGCC |
| MIR169new (MIR169e) | At1g53690 1.1 kb | AGCCAAGGAUGACUUGCC |
| MIR169new (MIR169f) | At3g14390 | AGCCAAGGAUGACUUGCC |
| MIR169new (MIR169g) | At4g21600 | AGCCAAGGAUGACUUGCC |
| MIR169new (MIR169h) | At1g19380 | AGCCAAGGAUGACUUGCCUG |
| MIR169new (MIR169i) | At3g26820 9.0 kb | AGCCAAGGAUGACUUGCCUG |
| MIR169new (MIR169j) | At3g26820 8.6 kb | AGCCAAGGAUGACUUGCCUG |
| MIR169new (MIR169k) | At3g26820 5.4 kb | AGCCAAGGAUGACUUGCCUG |
| MIR169new (MIR169l) | At3g26820 5.1 kb | AGCCAAGGAUGACUUGCCUG |
| MIR169new (MIR169m) | At3g26820 2.8 kb | AGCCAAGGAUGACUUGCCUG |
| MIR169new (MIR169n) | At3g26820 2.4 kb | AGCCAAGGAUGACUUGCCUG |
| MIR170 | At5g66050 | UGAUUGAGCCGUGUCAAUAUC |
| MIR170new (MIR171b) | At1g62040 | UGAUUGAGCCGUGCCAAUAUC |
| MIR170new (MIR171c) | At1g11740 | GAUUGAGCCGUGCCAAUAUC |
| MIR171 | AT3g51380 | UGAUUGAGCCGCGCCAAUAUC |
| MIR172a1 | At2g28060 | |
| MIR172new (MIR172d) | At3g55520 | UGAGAAUCUUGAUGAUGCUGCAG |
miRNA name as registered in Rfam (http://www.sanger.ac.uk/Software/Rfam/mirna/). Loci that were found by findMiRNA pior to recent reports are indicated with a superscript `new'
Intergenic regions North of indicated AGI pseudogenes that contain the precursor locus (loci). Each MIR169 precursor locus of intergenic region At2g26820 are distinguished by their locations on the 10.7-kb intergenic region North of At2g26820 relative to the ATG start codon of At2g26820 in kilobase pairs
Bolded nucleotides indicate the known miRNA sequences. Gray-bolded nucleotides were not indicated by the findMiRNA algorithm
It is of note that a number of the precursors detected in this study were found within the same intergenic region. Among the known miRNA families, six of the 13 loci of MIR169 occur as a tandem array within the intergenic region North of locus At3g26820 (Table 1). Two of the new miRNA precursor families reported here also occur as an array of more than one precursor within a single intergenic region. The cmr3 candidate has three loci in the intergenic region North of At1g26990 and three loci North of At1g69800, and the cmr6 candidate has three of its six loci within the intergenic region North of At2g34210 (Table 2).
An online resource of candidate miRNA-target pairs
The potential for miRNA precursors to be missed by any comparative genomics approach and the difficultly in identifying all likely bona fide miRNAs from among the miRNA candidates found by the findMiRNA algorithm prompted us to produce an interactive database. This database, which can be found at http://sundarlab.ucdavis.edu/mirna/ will facilitate the investigation of miRNA regulation by the research community by enabling easy access to the candidate miRNA data corresponding to their genes of interest. The database of candidate miRNA-target pairs provides an interface (Fig. 3A) for users to enter a transcript name with the same naming convention as The Arabidopsis Information Resource (TAIR) (Huala et al. 2001; Rhee et al. 2003). Information provided by a query returns all important information related to a candidate precursor, such as the precursor and miRNA sequence, the region of the transcript targeted with its sequence, hashedPrecExtract results in Oryza sativa, and links to relevant information at TAIR (http://www.arabidopsis.org) for a given transcript. Using a result limit of 500, the whole data set is comprised of ∼2.4 million records. In order to produce a useful database for the research community, a number of filters have been developed to reduce the data to a more manageable quantity, and to provide control over the values for these filters. The criteria for these filters include (1) a defined range for precursor stability estimated as the length normalized precursor minimum-free energy, (2) the requirement of a 5′ U residue in the predicted miRNA, (3) a defined range for the GC content of the predicted miRNA, and (4) a lower limit for miRNA-target duplex stability. Using the default settings (length normalized precursor MFE -0.3 to -0.56 kcal mol-1, miRNA GC content of 36%–67%, and miRNA-target Δ G threshold of -28 kcal mol-1), the data is reduced by 98%, and generates an average hit number of two candidates per transcript. The output of the Web site is shown for an example transcript (Fig. 3B).
Figure 3.

(A) Web site entry panel for query transcript At1g10200, with default precursor and miRNA parameter options. (B) Output from above query. (Graphic) Shows a map of the query transcript, below which is marked the positions of potential target sites (filled squares). These are labeled with the intergenic regions containing the precursor candidate loci from which the predicted miRNA candidates originate. The two labels on the right of this map, At1g20380 and At1g76140, correspond to the two cand2 loci that have Rfam names MIR394a and MIR394b, respectively. (Text) The predicted miRNA sequences are shown (white text on black), and, for comparison in this figure, the known mature miRNAs are indicated above the predicted precursors. When the `predicted orthologs in rice' box for the query options is also checked, only the At1g76140 cand2 candidate (MIR394b) record is returned. The At1g20380 cand2 candidate (MIR394a) record is not returned because the predicted miRNA for this record is 2 nt longer and does not have an exact match in rice. Boxes on the left can be checked and the precursor candidates aligned with ClustalW or downloaded in fasta format using the option shown below the listed candidate records.
The data returned by the Web site is similar to that for many other bioinformatics tools, e.g., BLAST, in that it should be interpreted carefully along with gene-specific information already in hand. For example, this might involve searching for target-site conservation or other significant evidence, and only after this, would one embark upon experimental verification. The Web interface allows the selection of records that contain potential orthologs in rice, as well as records that have overlapping target sites on the transcript, a property of precursor families; both factors can be utilized to identify particularly promising candidates. In progress, but not yet incorporated, is an additional filter to screen out records for which the miRNA/miRNA* Δ G asymmetry would suggest that the predicted miRNA is actually a miRNA*. The entire unfiltered prediction data set is available for download to enable researchers to compare or integrate with future prediction algorithms, along with the source code for both findMiRNA and hashedPrecExtract.
Discussion
Computational prediction of miRNAs
The small size of miRNAs means that they present special challenges for identification. At the experimental level, their identification has generally been accomplished by cloning and sequencing of size-selected RNA. However, the outcome of this direct approach is that miRNAs that are expressed at very high levels tend to be the dominant representatives of the libraries, and those that may be expressed at low levels or in a limited population of cells within an organism will be underrepresented and evade detection. In the absence of prior experimental data, their identification will rest upon computational prediction, which is tedious, if not impossible, using the current sequence-matching or alignment software, due to the small size of miRNAs and their targets and the sequence structures of miRNA precursors. Hence, there is clearly a need for new algorithms to be implemented as publicly available software to assist in the identification of sequences that may represent miRNAs or miRNA precursors.
Here, we present a target-based algorithm, findMiRNA, that starts with transcript sequences and looks for corresponding 18–25-nt sequences embedded in intergenic- or intron-hairpins that have the potential to target any part of these transcripts from their 5′ to their 3′ ends. Unlike comparative genomics methods that compare two or more genomes at an early stage to obtain their initial data sets, the findMiRNA algorithm produces its data set from a single genome, and should, therefore, also contain data for species-specific miRNAs. Comparative genomics methods result in a highly specific data set, but at the cost of removing species-specific candidates.
There is a considerable number of sequences within the Arabidopsis genome that have the characteristics of hairpin sequences. Because of the small size of miRNAs, and the fact that many known miRNA-target pairs involve mismatches as well as GU pairs, the selection of candidate miRNA-target pairs was necessarily relaxed. For this reason, the processing of the whole genome resulted in a large data set of 2.4 million candidate miRNA-target pairs. There are many ways to extract useful predictions from this data set. For the end users of the online resource, the data set is reduced by 98% with the default filters to an average of two candidates per transcript (see last section of Results). Promising candidates for experimental verification can be selected by additional criteria, e.g., the occurrence of a precursor family that targets the same region of the transcript, or the conservation of the candidate precursor(s) in rice.
In this study, for the purpose of obtaining candidates for experimental validation independent of the choice of target, we ranked families of related candidate precursors using the characteristic divergence pattern seen for known miRNA precursor families. This process resulted in the successful extraction of candidate miRNA-target pairs corresponding to previously reported miRNA precursor families. In addition, by focusing on a subset of targets and by paying attention to the sequence family-alignment pattern, we were able to predict additional small RNAs with only limited use of sequence comparison with other genomes.
The use of ranking algorithms to extract meaningful data from the entire findMiRNA data set, as used in this study, is only suitable for miRNA precursors that are members of precursor families. Three known miRNAs (miR161, miR163, and miR173), are defined by singleton precursors in the Arabidopsis genome, and therefore, not identified by the cluster ranking method. Interestingly, these miRNAs also lack corresponding precursors in the rice genome. Therefore, the identification of these miRNA precursors using comparative genomics approaches between rice and Arabidopsis would have failed, as they would be eliminated from the initial data set. In contrast, the findMiRNA retains the miR161 and miR173 precursors in the data set, and they can be identified through searches using the target transcripts. For instance, the transcript for At1g06580 corresponds to a RACE-confirmed target of miR161. When this transcript is used as a query with the default filters set on the Web interface, two miRNA candidate records are returned—one for each of the intergenic regions At1g48270 and REV_COM_At1g32585, with the former region containing the MIR161 precursor locus.
As discussed above, the results returned by the Web interface for a given query can be further narrowed by various methods, such as the existence of a precursor family or conservation of the precursor in other species such as rice. For a singleton non-conserved precursor like miR161, an alternative approach is to look for the presence of conserved target sites within related transcripts within the Arabidopsis genome. If we use the above transcript, At1g06580, to perform a BLASTN search of all the transcripts in the Arabidopsis genome, the best hits consist of transcripts encoding a family of pentatricopeptide repeat proteins. When entered as queries on the miRNA candidate Web site and filtered using the default Web site parameters, the miR161 precursor is returned as a candidate for nine of the most closely related transcripts. Although an average of two to three additional candidate precursors unrelated to miR161 are also returned for each transcript, none of these are predicted to target more than two of the nine transcripts. This is consistent with the idea that only the target sites for the real miRNA molecules (those derived from the MIR161 precursor) have been conserved, thus enabling the identification of miR161 from among these other candidates in the Arabidopsis findMiRNA data set.
The results from the two recent comparative genomics-based predictions of Jones-Rhoades and Bartel (2004) and Bonnet et al. (2004) are in general agreement with the predictions from findMiRNA, but several differences can be found, some of which can be clearly ascribed to the different limitations of these algorithms. Whereas the comparative genomic-based miRNA prediction algorithms look for conservation of miRNA sequences embedded in precursor sequences across species, the findMiRNA algorithm identifies initial miRNA-target sequence pairs for scoring, by using a 7-nt word (7mer) index of transcripts and intergenic regions (see Methods). The size of 7 is an adjustable parameter in the findMiRNA program that proved to sufficiently balance run time and sensitivity. In cases where there are four or more evenly spaced mismatches (GU pairs or other mismatches) in the ∼21-nt miRNA-target pair, a contiguous stretch of at least seven or more complementary base pairs will not occur, and hence, scoring of these miRNA-target pairs will not be initiated, because a 7-nt match will not be found. Such miRNA-target pairs will therefore evade detection by findMiRNA. It is for this reason that some members of the APS family, APS1 and APS3, were not identified as targets of cmr3, whereas the APS4 target was successfully predicted. Improvements could be made to the find-MiRNA algorithm to decrease the likelihood of missing such miRNA-target pairs, but because these modifications would likely require significantly more computing power, they can only be considered for future versions of the algorithm. Another limitation of findMiRNA is that miRNA-target pairs containing bulges in the miRNA-target duplex will be missed, for example, miR162a/b-DCL1. In order to detect these, the algorithm would have required the use of gapped alignments in the initial miRNA-target scoring process. Implementing the use of gapped alignments in this stage of the algorithm would have significantly increased the processing time to scan the entire genome. We note that most currently known plant miRNAs do not involve bulges during pairing with their targets. Even if bulge-containing miRNA-target pairs are not detected, this does not preclude the detection of other miRNA-target pairs for the same miRNA that do not require gapped alignments.
The miRNA cand1 was not included in the predictions from Jones-Rhoades and Bartel (2004), but is in agreement with a miRNA referred to as `miR26' predicted by a comparative genomics computational approach (Bonnet et al. 2004), although not experimentally confirmed. A cloned small RNA, miR390a.2, in the recent report by Sunkar and Zhu (2004), corresponds to a sequence that overlaps the miRNA* sequence within the predicted cand1 precursor. This report indicates that the `miR390a.2' molecule could not be detected on miRNA Northerns. By comparison, we obtained a very strong signal for cand1, which is further enhanced in the HC Pro plants. Further support for the idea that cand1, and not miR390a.2, is the main product of processing from this precursor, is the presence of a greater number of cand1-related clones than cand1*-related clones that are referred to as miR390 and miR390*, respectively, within the Arabidopsis small RNA project database (http://gac.bcc.orst.edu/smallRNA/), the latter being defined as miR390a.2 by Sunkar and Zhu (2004).
As discussed above, the findMiRNA algorithm can successfully detect singleton miRNA precursors. However, the singleton precursor corresponding to miR163 is missed, because the precursor sequence, as defined by the distance between the miRNA and miRNA* sequences, is larger than the 200-nt range that was set for the algorithm. Again, this adjustable parameter can be changed to allow for larger precursors in future runs. Hence, two of these limitations in our predictions could be precluded by further parameter optimization with longer run times. Since the source code is being made publicly available, the algorithm may also be customized for specific precursor properties that might be found in different species.
Variations in the size of the small RNAs
The small RNA gel blots performed in this study revealed the presence of potential miRNAs of two size classes (21 and 24 nt). For about half of the miRNA candidates experimentally tested in this study, only ∼24 nt RNAs could be detected. Two hybridizing bands could also be detected within the same lane for the control miRNA miR167 (Fig. 2C), as has also been observed for previously reported miRNAs, but not commented on (Kasschau et al. 2003). This variation in band size may result from different processing of precursors derived from different loci or from alternative processing pathways for single precursor RNAs in a manner that is similar to that previously reported for the production of two size classes of siRNAs (Hamilton et al. 2002; Mallory et al. 2002; Nicolas et al. 2003) that are the products of a more general RNA interference pathway. Larger siRNAs that are ∼24 nt have been correlated with the methylation of DNA and with the systemic spread of RNA interference (Hamilton et al. 2002; Zilberman et al. 2003), whereas the 21-nt RNA class is known to act on mRNA transcripts, whether these are siRNAs or miRNAs, and has been shown in at least one case not to be connected with methylation of DNA or systemic spread of RNAi (Mallory et al. 2001).
The miRNA candidates have the potential to target a diverse set of transcripts
One approach toward validating potential target transcripts is by using 5′ RACE to detect the products resulting from miRNA-directed cleavage. However, results from 5′ RACE may not be biologically meaningful, since the PCR assay is sensitive enough to detect the presence of low levels of cleavage products that are developmentally or physiologically neutral. A good example is the regulation of AP2 and some other members of this family by miR172. In this case, the use of 5′ RACE enabled the detection of mRNA cleavage products (Aukerman and Sakai 2003). However, a reduction in the level of the full-target transcripts was not observed using RNA gel-blot analysis even in plants overexpressing the miR172 RNA, and the actual regulation was found to be at the level of translation (Aukerman and Sakai 2003; Chen 2004).
Despite the necessary caution in the use of 5′ RACE in assessing the biological impact of a miRNA, a positive result from RACE is suggestive of an interaction between the miRNA and the suspected target. As shown in the Supplemental material for Table 2, all of the potential target transcripts that were predicted for the miRNA candidates have estimated Δ G values, for the miRNA-target pairs, that are better than -28.2 kcal mol-1 as determined by Mfold 3.1. This value corresponds to the duplex between miR398b and At3g15640, which is one of the least stable miRNA-target pairs giving a positive 5′ RACE result in the list compiled recently by Jones-Rhoades and Bartel (2004).
Previously reported miRNAs in Arabidopsis have targets that are predominantly transcription factors or parts of the RNAi machinery such as Dicer and AGO1 (for review, see Bartel 2004). Here, we show that miRNAs may target transcripts that encode a wide range of proteins. These include components of the ubiquitination pathway, namely TIR1 (At3g62980) and it's closest relatives (At3g26810, At1g12820, and At4g03190) that are F-box proteins involved in the auxin response (Dharmasiri and Estelle 2002), and an uncharacterized protein (At2g33770) that is a member of the ubiquitin-conjugating family of proteins (E2) (Scheffner et al. 1995; Weissman 2001). The possibility that components of the ubiquitylation pathway may be regulated by miRNAs is interesting, since it is well known that the ubiquination is widely used by organisms to modulate the levels of important regulatory proteins such as transcription factors and regulators of the cell cycle.
Another miRNA from this study, cmr3, has the potential to target the transcripts that encode an ATP sulfurylase precursor protein (APS4), and was recently shown to be up-regulated by sulfate starvation (Jones-Rhoades and Bartel 2004). The APS4 protein has a suggested role in the assimilation of sulfate in plants (Rotte and Leustek 2000). In addition to the ATP Sulfurylase genes, we identified a target site that is clearly conserved in the 5′ end of an Arabidopsis and a rice transcript that encode sulfate transporter proteins. This indicates that these transcripts are very likely additional targets of cmr3. Interestingly, these transcripts are not related to the APS gene family, but may be components of a common metabolic pathway.
For the new miRNA cand1, which was determined to be highly abundant from the Northern blot analysis, potential cross-species target sites were observed in a LRR kinase gene family in rice and soybean. Within the Arabidopsis genome, the cand1 miRNA has candidate targets other than the LRR-kinase transcripts that appear as good or better, based on predicted miRNA-target thermal stability. These transcripts encode a giberrellin 2-oxidase (At1g30040), an aconitase (At4g13430), and an acyl-CoA-binding protein (At5g53470). It is possible that cand1 and other miRNAs could target more than one transcript that are apparently unrelated but linked by some common biological process. Alternatively, the potential target sites in these different transcripts might only coincidentally match the miRNA, and the actual expression of the miRNA and the potential target transcripts might not overlap, or they do overlap, but are biologically neutral, a model that has been previously suggested (Bartel 2004).
Tandem arrays of miRNA precursors
In animal systems, the presence of tandem arrays of miRNA genes has been previously reported (Tanzer and Stadler 2004). In Arabidopsis, only the MIR166 family had been found to contain a tandem array of two loci, MIR166c and MIR166d, although not indicated (Reinhart et al. 2002). We detected the presence of miRNA precursor arrays for the MIR169 family and for two of the newly discovered miRNA precursor families reported here. The MIR169 family contains an array of six precursor sequences. This array appears to have resulted from a duplication of an ancestral precursor, resulting in a pair of precursor genes. This pair may then have duplicated to create two pairs of genes, one of which may have duplicated again, creating three pairs of precursor genes. The cmr3 family consists of six precursor genes. These are divided between two intergenic regions. Within each intergenic region, two of the three precursors are in the same orientation. The similarity in the arrangement between the two intergenic regions suggests that the last event to occur was a duplication of a single intergenic region. For cmr6, we report the presence of six precursor loci. Three of these are within the same intergenic region. The anti-cand3 family (MIR398), which consists of three members, has two loci that, although not within the same intergenic region, are relatively close on the same chromosome. It is conceivable that precursors may have become duplicated to nearby regions by the activity of transposable elements.
Screening for more miRNAs in Arabidopsis
In this study, we described the use of an algorithm called find-MiRNA to produce a data set of candidate miRNA-target pairs for the Arabidopsis genome. This algorithm differs from the other algorithms in that it does not use comparative genomics to obtain the initial set of candidate miRNAs and their respective targets. Therefore, the database contains candidate miRNAs directed against any given target gene, irrespective of their conservation in rice. The requirement of only a single genome to produce the initial data set thus provides flexibility for the identification of miRNA precursors in the future. For example, the genome sequence for poplar will soon be officially released, and poplar is a dicot as is Arabidopsis, whereas rice is a monocot. Therefore, Arabidopsis miRNAs conserved with poplar alone can be quickly and easily identified from among the Arabidopsis candidate miRNA data set using the algorithm precExtract. We plan to update the Web interface to include these data as new genome sequences become available with the addition of corresponding new filters. In addition to looking for conserved miRNA sequences, looking for conserved target sites in transcripts is an important alternative and can also be done within a single genome. However, this is not easily automated and is best done on a small scale by individual researchers. It is hoped that by providing a Web interface to our data in a readily accessible form, as a miRNA candidate database, the discovery of potentially elusive miRNAs is possible.
Methods
Computational prediction of precursors and targets
Intergenic regions and mRNA transcripts of Arabidopsis thaliana were downloaded from The Arabidopsis Information Resource, TAIR (Huala et al. 2001; Rhee 2003) with the last BAC sequence release date 4/17/2003. In an effort to preclude pseudogenes, transposons, and unannotated genes from spuriously matching homologous transcripts, known transcripts were aligned to the intergenic region using BLAT (Kent 2002), and regions with an E-Value lower than 10-6 and longer than 35 nt were masked. In addition, any base not labeled as A, T, G, or C was masked as well.
The algorithm, implemented in a program called find-MiRNA, initially indexes each overlapping 7mer (nucleotide sequence of length 7) of the intergenic region if (1) it is not contained in a repeat region of size 2 and length 4, and (2) at least two of the seven bases is a G or C. Each considered transcript is cut into overlapping 7mers, and checked against the indexed 7mers of the intergenic region. For each matching pair, the surrounding areas are aligned without gaps and scored; each Watson-Crick pair receives two points, while a G-U receives one point. All other combinations score no points. The maximum length normalized score of the ungapped alignment with a window size ranging from 18 to 25 is the predicted miRNA length. For miRNAs where mismatches with the target occur at the ends, this procedure underestimates the length of the miRNA. This implies the predicted miRNA may be less than or equal to the actual length of the miRNA. If the length-normalized score exceeds 1.55 with at least 35 total points, the score is recorded, and a 400-nt region centered at the miRNA is inspected for hairpin potential.
A dynamic programming algorithm specific for overlap matches (Durbin et al. 1998) aligns the miRNA to the 400-nt region with a weight matrix giving two points for Watson-Crick pairs, one point for G-U pairs, and minus one point for each gap. Conceptually, overlap matches between two sequences is a global alignment, but without penalties for overhangs. This alignment choice is ideal for finding embedded sequences, in this case, an embedded sequence, miRNA*, in the 400 nt that is complementary to the miRNA. The predicted miRNA and its corresponding miRNA* sequence are recorded and queued if their length-normalized overlap match score (overlap match alignment score/miRNA length) is greater than one.
After processing all overlapping 7mers of a transcript, all miRNAs and corresponding miRNA* sequences are ranked by combined descending (1) miRNA-target score, and (2) miRNA-miRNA* overlap score. The precursor is temporarily defined as the region delimited by the miRNA and its miRNA* sequence. An arbitrary integer, the Result Limit (RL), represents the number of regions that are subsequently passed to RNAfold for structural analysis, and is reset to zero for the scan of the complement strand of the genome. Almost all previously reported miRNAs/precursors appear in the ranked results within the bounds of the result limit of 100, and therefore, almost all are passed on to RNAfold. RNAfold calculates the minimum free energy (MFE) of the region delimited by the miRNA and the predicted miRNA* sequence, and regions not satisfying the following equation are dropped.
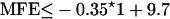 |
where l is the length of the predicted precursor.
This threshold of MFE as a function of precursor length was determined by comparing the distribution of length normalized MFE values of random intergenic regions with lengths equal to previously reported precursors (delimited by the miRNA and its corresponding miRNA*), to the length normalized MFE values of the previously reported precursors. Finally, RNAfold must predict at least 70% of the miRNA sequence to align with its previously predicted miRNA* sequence for the RNA fold structure to be considered to be in agreement with the formally predicted miRNA–miRNA* overlap.
In summary, the core of the miRNA and target-detection algorithm is essentially predicated on four factors as follows: (1) significant ungapped complementarity between the miRNA and its target transcript, which allows only a few mismatches, (2) the presence of a complementary region, miRNA*, upstream or downstream of the miRNA to indicate the potential of an RNA to form a hairpin, (3) a minimum-free energy threshold as a function of the predicted precursor length, which is delimited by the miRNA and its predicted miRNA* sequence, (4) at least 70% of the miRNA and its previously predicted miRNA* must be bound in the precursor as subsequently predicted by RNAfold.
Rice alignments
Given the remote homology within families of precursors in the same genome, a companion program, precExtract, was designed to locate precursors in other genomes given a set of hypothetical miRNAs, in this case, the miRNAs predicted from processing all transcripts in Arabidopsis. precExtract works by scanning a genome to match provided miRNA sequences with a certain number of mismatches, and predicts the region of optimal complementary, miRNA*, using the same dynamic programming algorithm described previously (Durbin et al. 1998). The region delimited by the miRNA and its miRNA* is excised from the genome, and filtered with the same criteria as the findMiRNA implementation. Another program, hashedPrecExtract, operates similarly to precExtract, except that it indexes for exact matches to query miRNAs for all overlapping Kmers in the provided genome, where a Kmer is the size of any provided miRNA. Hashed-PrecExtract is a memory-intensive implementation that can handle hundreds of thousands of queries, while precExtract is a CPU-intensive implementation that can handle a few thousand queries. An additional criterion was added for post-processing the alignments; the orientation of the potential heterologous precursor homolog (i.e., miRNA on 5′ end or 3′ end of precursor) must be the same as that found for Arabidopsis precursor candidate.
HashedPrecExtract was used to align all predicted miRNAs from Arabidopsis to Rice with zero mismatches for the miRNA. Predicted target pairs without identical miRNAs in Rice were filtered, clustered, and ranked by differential sequence conservation. The Oryza sativa genome (10/20/2003) was downloaded from Gramene (Jaiswal et al. 2002; Ware et al. 2002a,b) at http://www.gramene.org.
Ranking precursors based on differential sequence conservation
Although the algorithm provides the sensitivity to detect miRNAs, it does not provide the specificity required to exclude sequences that are not miRNA precursors. One approach is to rank precursors in clusters, based on conservation of the miRNA and its miRNA*, and less conservation in the interstices.
All precursors targeting the same region of a transcript ±2 bp were clustered and ranked according to the following criteria: (1) percent identity of the miRNAs in the cluster, (2) percent identity of its miRNA* in the precursor, and (3) a reduced percent identity in the interstices. These identities were generated from excising the respective regions from the precursors and aligning the regions separately. A scoring system was developed with these criteria using the multiple-sequence alignment program T-Coffee (Notredame et al. 2000) in conjunction with the Bioperl utilities (Stajich et al. 2002). For this algorithm, the three scores are integrated into a single score:
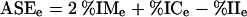 |
1 |
where ASEe is the alignment score of the excised region, %IMe is the percent identity of the excised miRNA sequences, %ICe is the percent identity of the excised complementary miRNA* sequences, and %IIe is the percent identity of the excised interstice sequences. In this case, all sequences were taken before performing the multiple-sequence alignment.
An additional alignment was made in a global context, all precursors in the cluster were aligned using T-Coffee, and the same three regions (miRNAs, miRNA*s, and interstices) were inferred from their average positions in the alignments, and scored in this context using a similar equation 1:
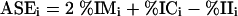 |
2 |
where ASEi is the alignment score of the inferred regions, %IMi is the percent identity of the inferred miRNA sequences, %ICi is the percent identity of the inferred miRNA* sequences, and %IIi is the percent identity of the inferred interstice sequences. The percent identity for a particular region, %IMi, %ICi, or %IIi, was inferred by taking the percent identity of the average location of that region in the multiple sequence alignment.
The two scores from equations 1 and 2 were summed to give a final score for that cluster:
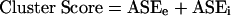 |
3 |
(Note: for the old method, the Cluster Score = ASEe * ASEi with weights [1,1,2] instead of [2,1,1] This method produces clusters where each cluster represents a set of two or more miRNAs targeting a given region of a transcript. This produces redundancies in the clusters, since the same miRNAs may target more than one transcript. A second stage in the clustering process integrates members from different clusters that have identical or overlapping miRNAs. An optimization procedure was implemented to search parameter space for improved rankings, but the rankings produced were not significantly different from the rankings using the parameters shown above (data not shown).
Web site parameters
See the Web site at http://sundarlab.ucdavis.edu/mirna/ for more details on the precursor and miRNA parameters that are used to enrich for likely miRNA candidates during end-user query submission.
HC-Pro transgenic Arabidopsis line
The Arabidopsis thaliana line (Columbia ecotype) expressing Turnip mosaic virus (TuMV) P1/HC-Pro under the control of the cauliflower mosaic virus 35S promoter was generated by agro-bacterium-mediated transformation with binary vector PCX305 containing the P1/HC-Pro coding region from TuMV isolate 1.
Low molecular-weight RNA isolation and gel-blot analysis
Plant material from 30-d-old plants was frozen in liquid nitrogen, ground to a fine powder, and vortexed in a mixture of 5 vol/gram tissue of extraction buffer (100 mM Tris-HCl at pH 8.0, 100 mM LiCl, 10 mM EDTA, and 1% SDS) and an equal volume of phenol. Two volumes of chloroform per volume of phenol were added, the solution mixed intermittently for 5 min, and phases separated by centrifugation. An equal volume of 4 M LiCl, 10 mM EDTA (pH 7.0), was added to the aqueous phase, incubated overnight at 4°C, and centrifuged at 12,000g for 15 min to pellet high molecular-weight RNA. The supernatant, containing DNA and low molecular-weight RNAs, was made 10% in polyethylene glycol 8000 and 0.5 M NaCl, and incubated for 30 min on ice to precipitate the DNA. After centrifugation to remove DNA, low molecular-weight RNA was concentrated by ethanol precipitation and resuspended in deionized formamide.
A total of 10 μg per lane of low molecular-weight RNAs were separated by 20% denaturing polyacrylamide gel electrophoresis and blotted onto a nylon membrane (Hybond NX, Amersham Biosciences) as described earlier (Mallory et al. 2001). Specific miRNA probes were prepared by end-labeling antisense oligonucleotides with [γ-32P]ATP and T4 polynucleotide kinase (New England Biolabs) (Mallory et al. 2002). The most predominant ethidium bromide-stained species of low molecular-weight RNA, running at ∼200 nt on a 1% nondenaturing agarose gel, was used as a loading control. For the StarFire-labeled probings, EtBr-stained gels were photographed to affirm that the RNA loadings were comparable. DNA oligonucleotides of known sizes were end-labeled with [γ-32P]ATP and used as size markers for miRNAs. The sizes of the miRNAs were further verified using the Ambion mirVana RNA marker system prepared according to the manufacturer's protocol. For end-labeled probes, DNA oligonucleotides were ordered from Operon, while StarFire probes were ordered from IDT and labeled using the associated StarFire kit. The sequences for the probes were as follows: end-labeled probes;— cmr3, GAGTCCCCCCAAACACTTCAGT; cmr6, GGGCAAATCTCCTTTGGCA; cmr5, ATGGTTCGGGCATTGACTTCTA; cmr7, GGTATCGGTTCGGGTTCGGGTA; cmr84, GCCCAGTGGCGGATCTAGAAACA; cmr75, AAAACGACGTCGTTTTGATAG; cmr165, ATCCAGGATCCGAGATCCGATC; cmr195, ACTGTTTGATACGTACACTTAGATT; cmr201, TCACTCAATTAGAGAGATCTGAA; cmr214, AATGCGATCCCTTTGGATCCT; cand1, at GGCGCTATCCCTCCTGAGcttta; cand2, GGAGGTGGACAGAATGCCAAA; cand3, TGTTCTCAGGTCACCCCTTTga, and StarFire probes—cmr5, ATGGTTCGGGCATTGACTTCTAxxxxxx; cmr7, GGTATCGGTTCGGGTTCGGGTAxxxxxx; as-cmr7, TACCCGAACCCGAACCGATACCxxxxxx; as-cmr75, CTATCAAAACGACGTCGTTTTxxxxxx; cmr75, AAAACGACGTCGTTTTGATAGxxxxxx; cmr84, CCCAGTGGCGGATCTAGAAACAxxxxxx; as-cmr84, TGTTTCTAGATCCGCCACTGGGxxxxxx; cand3, TGT GTTCTCAGGTCACCCCTTTGAxxxxxx; as-cand3, TCAAAGGGGTGACCTGAGAACACAxxxxxx
Note added in proof
Wang et al. (2004) have reported another computational study for the prediction of miRNAs in Arabidopsis thaliana. Using a comparative genomics method, they have also predicted many of the miRNAs described here and by others (Jones-Rhoades et al. 2004; Sunkar and Zhu 2004).
Acknowledgments
We thank Dr. Edward Marcotte of the University of Texas for providing Web space for the database of candidate miRNAs and the University of California–Davis for funding. We would also thank Andrej Sali of the University of California San Francisco for providing access to critical computational resources for the most recent runs of findMiRNA.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2908205.
Footnotes
[Supplemental material is available online at www.genome.org. All programs are freely available, and the miRNA candidate data is available through a Web interface at http://sundarlab.ucdavis.edu/mirna/.]
References
- Aukerman, M.J. and Sakai, H. 2003. Regulation of flowering time and floral organ identity by a MicroRNA and its APETALA2-like target genes. Plant Cell 15: 2730-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel, D.P. 2004. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116: 281-297. [DOI] [PubMed] [Google Scholar]
- Bergmann, A. and Lane, M.E. 2003. HIDden targets of microRNAs for growth control. Trends Biochem. Sci. 28: 461-463. [DOI] [PubMed] [Google Scholar]
- Bonnet, E., Wuyts, J., Rouze, P., and Van de Peer, Y. 2004. Detection of 91 potential conserved plant miRNAs in Arabidopsis thaliana and Oryza sativa identifies important target genes. Proc. Natl. Acad. Sci. 101: 11511-11516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke, J., Hipfner, D.R., Stark, A., Russell, R.B., and Cohen, S.M. 2003. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 113: 25-36. [DOI] [PubMed] [Google Scholar]
- Chen, X. 2004. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 303: 2022-2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmasiri, S. and Estelle, M. 2002. The role of regulated protein degradation in auxin response. Plant Mol. Biol. 49: 401-409. [PubMed] [Google Scholar]
- Durbin, R., Eddy, S., Krogh, A., and Mitchison, G. 1998. Biological sequence analysis: Probabilistic models of proteins and nucleic acids. Cambridge University Press, Cambridge, UK.
- Emery, J.F., Floyd, S.K., Alvarez, J., Eshed, Y., Hawker, N.P., Izhaki, A., Baum, S.F., and Bowman, J.L. 2003. Radial patterning of Arabidopsis shoots by class III HD-ZIP and KANADI genes. Curr. Biol. 13: 1768-1774. [DOI] [PubMed] [Google Scholar]
- Grad, Y., Aach, J., Hayes, G.D., Reinhart, B.J., Church, G.M., Ruvkun, G., and Kim, J. 2003. Computational and experimental identification of C. elegans microRNAs. Mol. Cell 11: 1253-1263. [DOI] [PubMed] [Google Scholar]
- Gray, W.M., del Pozo, J.C., Walker, L., Hobbie, L., Risseeuw, E., Banks, T., Crosby, W.L., Yang, M., Ma, H., and Estelle, M. 1999. Identification of an SCF ubiquitin-ligase complex required for auxin response in Arabidopsis thaliana. Genes & Dev. 13: 1678-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones, S. 2004. The microRNA Registry. Nucleic Acids Res. 32: D109-D111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones, S., Bateman, A., Marshall, M., Khanna, A., and Eddy, S.R. 2003. Rfam: An RNA family database. Nucleic Acids Res. 31: 439-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton, A., Voinnet, O., Chappell, L., and Baulcombe, D. 2002. Two classes of short interfering RNA in RNA silencing. EMBO J. 21: 4671-4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipfner, D.R., Weigmann, K., and Cohen, S.M. 2002. The bantam gene regulates Drosophila growth. Genetics 161: 1527-1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huala, E., Dickerman, A.W., Garcia-Hernandez, M., Weems, D., Reiser, L., LaFond, F., Hanley, D., Kiphart, D., Zhuang, M., Huang, W., et al. 2001. The Arabidopsis Information Resource (TAIR): A comprehensive database and Web-based information retrieval, analysis, and visualization system for a model plant. Nucleic Acids Res. 29: 102-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal, P., Ware, D., Ni, J., Chang, K., Zhao, W., Schmidt, S., Pan, X., Clark, K., Teytelman, L., Cartinhour, S., et al. 2002. Gramene: Development and integration of trait and gene ontologies for rice. Comp. Funct. Genom. 3: 132-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones-Rhoades, M. and Bartel, D. 2004. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Cell 14: 787-799. [DOI] [PubMed] [Google Scholar]
- Kasschau, K.D., Xie, Z., Allen, E., Llave, C., Chapman, E.J., Krizan, K.A., and Carrington, J.C. 2003. P1/HC-Pro, a viral suppressor of RNA silencing, interferes with Arabidopsis development and miRNA unction. Dev. Cell 4: 205-217. [DOI] [PubMed] [Google Scholar]
- Kent, W.J. 2002. BLAT—the BLAST-like alignment tool. Genome Res. 12: 656-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, E.C., Tomancak, P., Williams, R.W., and Rubin, G.M. 2003. Computational identification of Drosophila microRNA genes. Genome Biol. 4: R42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, L.P., Glasner, M.E., Yekta, S., Burge, C.B., and Bartel, D.P. 2003a. Vertebrate microRNA genes. Science 299: 1540. [DOI] [PubMed] [Google Scholar]
- Lim, L.P., Lau, N.C., Weinstein, E.G., Abdelhakim, A., Yekta, S., Rhoades, M.W., Burge, C.B., and Bartel, D.P. 2003b. The microRNAs of Caenorhabditis elegans. Genes & Dev. 17: 991-1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llave, C., Xie, Z., Kasschau, K.D., and Carrington, J.C. 2002. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science 297: 2053-2056. [DOI] [PubMed] [Google Scholar]
- Mallory, A.C. and Vaucheret, H. 2004. MicroRNAs: Something important between the genes. Curr. Opin. Plant Biol. 7: 120-125. [DOI] [PubMed] [Google Scholar]
- Mallory, A.C., Ely, L., Smith, T.H., Marathe, R., Anandalakshmi, R., Fagard, M., Vaucheret, H., Pruss, G., Bowman, L., and Vance, V.B. 2001. HC-Pro suppression of transgene silencing eliminates the small RNAs but not transgene methylation or the mobile signal. Plant Cell 13: 571-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallory, A.C., Reinhart, B.J., Bartel, D., Vance, V.B., and Bowman, L.H. 2002. A viral suppressor of RNA silencing differentially regulates the accumulation of short interfering RNAs and micro-RNAs in tobacco. Proc. Natl. Acad. Sci. 99: 15228-15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas, F.E., Torres-Martinez, S., and Ruiz-Vazquez, R.M. 2003. Two classes of small antisense RNAs in fungal RNA silencing triggered by non-integrative transgenes. EMBO J. 22: 3983-3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame, C., Higgins, D.G., and Heringa, J. 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302: 205-217. [DOI] [PubMed] [Google Scholar]
- Palatnik, J.F., Allen, E., Wu, X., Schommer, C., Schwab, R., Carrington, J.C., and Weigel, D. 2003. Control of leaf morphogenesis by microRNAs. Nature 425: 257-263. [DOI] [PubMed] [Google Scholar]
- Park, W., Li, J., Song, R., Messing, J., and Chen, X. 2002. CARPEL FACTORY, a Dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr. Biol. 12: 1484-1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquinelli, A.E., Reinhart, B.J., Slack, F., Martindale, M.Q., Kuroda, M.I., Maller, B., Hayward, D.C., Ball, E.E., Degnan, B., Muller, P., et al. 2000. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 408: 86-89. [DOI] [PubMed] [Google Scholar]
- Reinhart, B.J., Weinstein, E.G., Rhoades, M.W., Bartel, B., and Bartel, D.P. 2002. MicroRNAs in plants. Genes & Dev. 16: 1616-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee, S.Y., Beavis, W., Berardini, T.Z., Chen, G., Dixon, D., Doyle, A., Garcia-Hernandez, M., Huala, E., Lander, G., Montoya, M., et al. 2003. The Arabidopsis Information Resource (TAIR): A model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res. 31: 224-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoades, M.W., Reinhart, B.J., Lim, L.P., Burge, C.B., Bartel, B., and Bartel, D.P. 2002. Prediction of plant microRNA targets. Cell 110: 513-520. [DOI] [PubMed] [Google Scholar]
- Rotte, C. and Leustek, T. 2000. Differential subcellular localization and expression of ATP sulfurylase and 5′-adenylylsulfate reductase during ontogenesis of Arabidopsis leaves indicates that cytosolic and plastid forms of ATP sulfurylase may have specialized functions. Plant Physiol. 124: 715-724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner, M., Nuber, U., and Huibregtse, J.M. 1995. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature 373: 81-83. [DOI] [PubMed] [Google Scholar]
- Sempere, L.F., Freemantle, S., Pitha-Rowe, I., Moss, E., Dmitrovsky, E., and Ambros, V. 2004. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 5: R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajich, J.E., Block, D., Boulez, K., Brenner, S.E., Chervitz, S.A., Dagdigian, C., Fuellen, G., Gilbert, J.G., Korf, I., Lapp, H., et al. 2002. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 12: 1611-1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkar, R. and Zhu, J-K. 2004. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 16: 2001-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, G., Reinhart, B.J., Bartel, D.P., and Zamore, P.D. 2003. A biochemical framework for RNA silencing in plants. Genes & Dev. 17: 49-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzer, A. and Stadler, P.F. 2004. Molecular evolution of a microRNA cluster. J. Mol. Biol. 339: 327-335. [DOI] [PubMed] [Google Scholar]
- Wang, X-J., Reyes, J.L., Chua, N-H., and Gaasterland, T. 2004. Prediction and identification of Arabidopsis thaliana microRNAs and their mRNA targets. Genome Biol. 5: R65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware, D., Jaiswal, P., Ni, J., Pan, X., Chang, K., Clark, K., Teytelman, L., Schmidt, S., Zhao, W., Cartinhour, S., et al. 2002a. Gramene: A resource for comparative grass genomics. Nucleic Acids Res. 30: 103-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware, D.H., Jaiswal, P., Ni, J., Yap, I.V., Pan, X., Clark, K.Y., Teytelman, L., Schmidt, S.C., Zhao, W., Chang, K., et al. 2002b. Gramene, a tool for grass genomics. Plant Physiol. 130: 1606-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman, A.M. 2001. Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell. Biol. 2: 169-178. [DOI] [PubMed] [Google Scholar]
- Xie, Z., Kasschau, K.D., and Carrington, J.C. 2003. Negative feedback regulation of Dicer-Like1 in Arabidopsis by microRNA-guided mRNA degradation. Curr. Biol. 13: 784-789. [DOI] [PubMed] [Google Scholar]
- Zilberman, D., Cao, X., and Jacobsen, S.E. 2003. ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science 299: 716-719. [DOI] [PubMed] [Google Scholar]
- Zuker, M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31: 3406-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Web site references
- http://sundarlab.ucdavis.edu/mirna/; This study.
- http://www.arabidopsis.org; The Arabidopsis Information resource (TAIR).
- http://www.sanger.ac.uk/Software/Rfam/mirna/; The miRNA Registry.
- http://www.gramene.org/; Gramene: A comparative mapping resource for grains.
- http://gac.bcc.orst.edu/smallRNA/; Arabidopsis thaliana Small RNA project.
- http://www.bioinfo.rpi.edu/applications/mfold/; Mfold RNA and DNA folding at The Bioinformatics Center at Rensselaer and Wadsworth.