

ABSTRACT
Aging is characterized by a cumulative loss of genome integrity, which involves chromatin reorganization, transcriptional dysregulation and the accumulation of DNA damage. Sirtuins participate in the protection against these aging processes by promoting genome homeostasis in response to cellular stress. We recently reported that SirT7−/− mice suffer from partial embryonic lethality and a progeroid like phenotype. At the cellular level, SIRT7 depletion results in the impaired repair of DNA double-strand breaks (DSBs), one the most dangerous DNA lesions, leading to genome instability. SIRT7 is recruited to DSBs, where it specifically deacetylates histone H3 at lysine 18 and affects the focal accumulation of the DNA damage response factor 53BP1, thus influencing the efficiency of repair. Here, we integrate our findings with the current knowledge on the mode of action of other sirtuin family members in DNA repair. We emphasize their capacity to regulate chromatin structure as a response to DNA damage within the constraints imposed by cellular status.
KEYWORDS: DNA damage, DNA double strand breaks, histone acetylation, sirtuins, SIRT7
Sirtuins: Guardians of genome homeostasis
Sirtuins are proteins with multiple enzymatic activities, including NAD+-dependent deacetylation. Mammals have 7 sirtuins, denoted SIRT1 to 7. The NAD+ requirement for their enzymatic activity makes them sensors of the redox and metabolic state of the cell and organism.1 Thus, sirtuins are major effectors in the cellular response to metabolic, oxidative and genotoxic stress, acting to modulate cellular physiology under these conditions. A key function of nuclear sirtuins is the regulation of genome homeostasis under these forms of stress. The loss of sirtuin function is associated with genome instability and compromised organism viability. The majority of SirT1−/− mice die in utero due to severe developmental defects.2-4 SIRT6-deficient mice are born at normal Mendelian ratios but die within the first month after birth with clear signs of accelerated aging.5,6 A substantial proportion of SirT7−/− embryos die at late stages of embryonic development or during the first month after birth,7 and those that survive to adulthood suffer from a shortened lifespan.7, 8 SirT7−/− mice also show phenotypic and molecular signs of accelerated aging.7 These signs include a premature kyphosis, reduced weight and fat content, compromised haematopoietic stem cell function and leukopenia, reduced levels of circulating IGF-1 protein, and increased p16INK4 expression. In addition, SIRT7 depletion is correlated with multiple organ dysfunctions7-11 (Fig. 1).
Figure 1.

Summary of SIRT7 knockout phenotypes and the proposed molecular pathways implicated.
The phenotypic consequences of SIRT7 depletion in mammalian cells could be explained by the functional link of SIRT7 with the maintenance of genome integrity. SIRT7 depletion is associated with an increased mutation rate, sensitivity to different DNA damaging agents and abnormal rates of apoptosis. Indeed, SIRT7 regulates a range of processes that converge on genome stability: transcriptional regulation, DNA replication and the DNA damage response.
SIRT7 is a complex transcriptional regulator acting both as an activator and repressor. This is best exemplified by the SIRT7-mediated regulation of ribosomal biogenesis: SIRT7 promotes the expression of rDNA12 but represses ribosomal protein gene transcription.11 In addition, SIRT7 acts as a transcriptional activator of nuclear-encoded mitochondrial genes.10 Conversely, SIRT7-mediated transcriptional repression via H3K18Ac deacetylation is linked to oncogenic maintenance.13
Similar to SirT2−/− cells,14-16 SIRT7 deficiency leads to replication stress, an important source of endogenous DNA damage. What the relative contribution of these processes is to the observed SirT7−/− phenotype and how these processes are functionally connected are still open questions. Nevertheless, it is broadly accepted that the accumulation of mutations and genomic rearrangements arises from the impaired repair of DNA damage, resulting in devastating consequences for cellular fitness and cumulatively leading to organism aging.17 Importantly, SIRT7 contributes to the maintenance of genome stability by participating in the repair of one of the most dangerous DNA insults, double-strand breaks (DSBs), stressing the importance of nuclear sirtuins in DNA damage repair.
Sirtuins and double-strand break repair: What we learn from SIRT7
Sirtuins participate in the DNA damage response (DDR) by regulating cell cycle progression and DNA repair (for an extended review of the different DNA repair pathways in which sirtuins are involved see18,19). Evidence in different organisms strongly suggests that nuclear sirtuins are particularly relevant in the signaling pathways that repair DSBs: non-homologous end joining (NHEJ) and homologous recombination (HR)19 (Fig. 2). The cascade of events that follow the sensing of a DSB include the activation of cell cycle checkpoints, the remodeling and modification of the chromatin surrounding the DNA break, and the recruitment of repair proteins.20,21 In mammalian cells, DSBs are primarily sensed by the MRN (MRE11-RAD50-NBS1) complex, which recruits phosphoinositide 3-kinase-related kinases, including ataxia telangiectasia mutated (ATM). ATM plays a major role in the activation of the DNA damage checkpoint, leading to cell cycle arrest or apoptosis, and in the initiation of DNA repair. ATM phosphorylates a myriad of downstream effectors, including the histone H2A variant H2AX to generate γH2AX, which spreads in the chromatin around breaks, forming distinct foci that can be visualized by immunofluorescence. γH2AX is in turn recognized by mediator of DNA damage checkpoint protein 1 (MDC1), another ATM target protein, which initiates a cascade of events that ends with the ubiquitination of histone H2A at Lys13 and/or Lys15 (H2AK13ub and H2AK15ub). H2AK15ub, together with endogenous H4K20me2, participates in the recruitment of 53BP1, one of the drivers that directs DSB repair toward NHEJ by competing with the HR-related protein breast cancer 1 (BRCA1). The commitment to either the NHEJ or HR pathway is marked at the broken DNA ends by the presence of the corresponding limiting factors: the NHEJ kinase catalytic subunit DNA-PKcs and the HR recombinase RAD51.22
Figure 2.
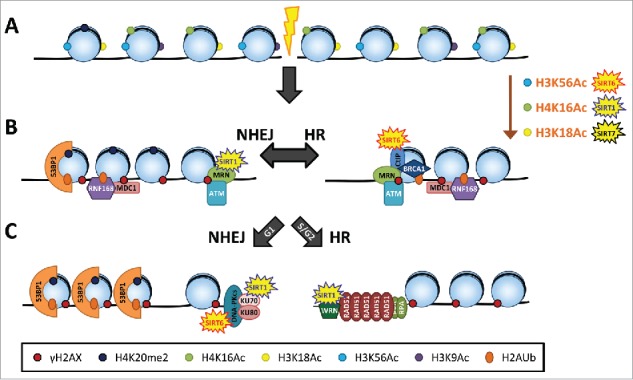
Scheme of the canonical DNA double-strand break (lightning bolt) repair pathways: non-homologous end joining (NHEJ) and homologous recombination (HR). Sirtuins participate at 3 levels within the DNA repair cascade: (A) Chromatin remodeling; (B) Chromatin-based recruitment of the DNA repair machinery; (C) DNA repair at the broken ends. The contribution of each sirtuin family member in each of these processes is highlighted. See text for more details.
The prevalence of one or another pathway of repair is determined by multiple factors. There are cell cycle constrictions, as NHEJ functions throughout the cell cycle, while HR is restricted to late S/G2 phase, where a sister chromatid is available for recombinational repair. The nature of the inflicted DNA damage and the chromatin environment surrounding the damaged site also influence the repair dynamics and the extent of resection of the broken DNA ends, which is a major factor in the choice of repair pathway.23
SIRT1, SIRT6 and SIRT7 have been shown to be directly involved in DSB repair. These sirtuins appear to mostly work at 2 different levels in mediating DNA repair: promoting chromatin structure alterations that facilitate the access and recruitment of repair proteins to the DNA damage sites and directly modulating repair protein activity. However, the relative contribution of these sirtuins in the different DSB repair pathways and the cooperation and/or redundant capabilities between them in the DNA repair process is not clear. The evidence discussed here suggests that the function of sirtuins in DNA repair may be modulated by cell- and development-specific proliferation rates, which in turn impacts the cell cycle profile, the type of genotoxic insult and amount of DNA damage, and the chromatin organization at the DNA damage site.
An important clue about the role of sirtuins in DSB repair comes from the distinct dynamics of the recruitment of nuclear sirtuins to the DNA damage site. SIRT6 and SIRT1 are recruited with fast kinetics to DSBs, supporting an initiating role of these sirtuins in the DDR,24, 25 while SIRT7 is recruited with slower kinetics, pointing to a more distal role in the DNA repair process.7 Consistent with this, the depletion of SIRT1 and SIRT6 affects the initial steps of the DDR cascade such as ATM-dependent H2AX phosphorylation,3, 24 while the phosphorylation of these proteins is not affected by SIRT7 depletion.7
A second clue comes from the proteins that recruit sirtuins to the DNA damage sites. ATM seems to be crucial for the recruitment of SIRT1 to damage sites in several cell types, including mouse embryonic stem (ES) cells26 and primary neurons.25 In primary neurons, SIRT1 further regulates ATM autophosphorylation and recruitment to an I-PpoI-generated DSB. This is plausibly facilitated by the interaction of SIRT1 with the MRN complex member NBS1, as the recruitment of NBS1 to DSBs is also reduced in SIRT1-depleted neurons.25 Indeed, SIRT1-mediated deacetylation of NBS1 is necessary for irradiation-induced NBS1 phosphorylation,27 and NBS1 is a known recruiter and activator of ATM.28 The reciprocal regulation of SIRT1/ATM does not seem to occur in MEFs, because ATM phosphorylation levels do not change at irradiation-induced foci.3 Despite the limitations of comparing different experimental approaches, it is interesting to note that SIRT1 activity might be specified by the cell type, which also specifies the prevalent DNA repair pathway. Rapidly dividing cells such as undifferentiated embryonic cells rely more on HR repair,29 while in non-proliferative cells such as neurons, NHEJ might be more prevalent. The recruitment of SIRT7 to chromatin does not seem to be affected by ATM depletion.7 How SIRT6 is recruited to DNA damaged sites is not clear. It is known, however, that lamin A binds to SIRT6 and stimulates its accumulation on chromatin upon DNA damage in addition to enhancing the DNA repair activities of SIRT6.30
The recruitment of SIRT7 to DNA-damaged chromatin is dependent on poly[adenosine diphosphate (ADP)–ribose] polymerase (PARP).7 Consistent with this, a direct interaction between SIRT7 and PARP1 has been recently reported.31 PARP proteins are recruited to DNA damage sites where they promote DNA repair by facilitating the recruitment of chromatin remodelers such as SNF2H and therefore enhance chromatin accessibility. PARP proteins also poly-ADP-ribosylate other proteins around DNA damage sites, facilitating the recruitment of DNA repair factors.32 The interplay between sirtuins and PARPs has been long documented, as both families are major consumers of cellular NAD+33. Under mechanical stress conditions, the SIRT1-mediated deacetylation of PARP increases cell survival, although this effect does not seem to be related to the association of PARP with DNA.34 As stated above, the question of how SIRT6 is recruited to DSBs is not known. However, SIRT6-mediated ADP-ribosylation of PARP1 stimulates PARP1 activity and DSB repair under oxidative stress conditions.35 Consistent with this, SIRT6 is able to rescue HR deficiency in a PARP-dependent manner in senescent cells.36 However, if the action of SIRT6 in DNA repair occurred exclusively by modifying PARP activity, we would expect that PARP activation would change in SIRT6-depleted cells, which does not always seem to be the case.24 Therefore, the SIRT6-PARP axis may be more relevant under oxidative stress conditions or in senescent or aged cells, in which PARP activation is increased due to chronic DNA damage.37 Increased PARP activity could lead to NAD+ depletion and mitochondrial dysfunction and therefore increased oxidative stress.33 The activation and autoPARylation of PARP-1 also results in its release from chromatin to enable downstream DNA repair to occur.38 An interesting parallel comes from a mouse model of Cockayne Syndrome group B (CSB). In this model, the lack of the CSB protein is correlated with increased DNA damage due to the lack of CSB-mediated displacement of active PARP from chromatin. Remarkably, the protein levels of all sirtuins are decreased in CSB-depleted cells, with the exception of SIRT6, which is increased.37 It will be interesting to investigate whether the SIRT6-PARP interplay is in fact related to the removal of PARP from chromatin under oxidative stress conditions and whether this interplay is extended to other sirtuins such as SIRT7. However, it remains to be determined whether SIRT7 might regulate PARP activity.
In addition to their role in the initiation of the DDR, sirtuins have the capacity to directly interact with and regulate the activity of downstream repair proteins. SIRT6 supports different types of DNA damage repair, including NHEJ by interacting with and stabilizing the DNA-PKcs protein at break sites,39 and HR by deacetylating and enhancing the activity of CtIP,40 an essential protein for the resection of broken DNA ends. Whether SIRT6 is able to simultaneously regulate either pathway or whether its function has cell type and cell cycle restrictions need further investigation. Interestingly, the overexpression of SIRT6 in DNA-PKcs-null MEFs stimulates microhomology end-joining (MHEJ) repair, which is also PARP1-dependent,35 and suggests the ability of SIRT6 to regulate alternative repair pathways in the absence of canonical DSB repair. Similarly, SIRT1 directly binds to and deacetylates members of core DNA repair complexes, such as the NHEJ mediator Ku7041 and members of the RecQ DNA and RNA helicase family (WRN, Werner Syndrome), the exonuclease and helicase activity of which is necessary for efficient HR repair.42 Thus, SIRT1 depletion is correlated with impaired HR and NHEJ in ES cells and MEFs from the different SIRT1 KO mouse models.3,26 However, there are inconsistent reports about the involvement of SIRT1 in the DDR in immortalized cell lines. siRNA-mediated SIRT1 depletion does not impair damage signaling in U2OS cells after camptothecin (CPT) treatment40 and, similarly, using an I-SCEI-induced DNA DSB reporter system, the overexpression of SIRT1 does not enhance HR or NHEJ repair in human immortalized fibroblasts after paraquat treatment.35 It is not clear whether this indicates that the regulation of DNA repair, mostly of HR, by SIRT1 is most prominent in undifferentiated cells such as ES cells and MEFs. Interestingly, in immortalized cells, SIRT1 has the capacity to promote alternative recombinational pathways such as Rad51-independent single-strand annealing (SSA),43 in which the SIRT1-interacting protein WRN is involved. SIRT7 modulates NHEJ activity by facilitating the recruitment of 53BP1 to DSBs.7 A previous report suggested that SIRT7 may regulate HR repair.35 However, HR does not seem to be affected in irradiated primary cells from the SirT7−/− mice.7 Whether this disparity might be related to the cell type or type of cellular stress inflicted needs further investigation.
Histone acetylation and DNA repair: SIRT7 and the DSB pathway choice
Chromatin modification at and around the DNA damaged sites seems to be an important factor in the DDR.20 Strikingly, upon DNA damage, there is an increase of chromatin mobility that parallels the recruitment of chromatin remodelers and nucleosome eviction. This mobility could facilitate the encounter of recombinational partners and the anchoring of broken ends to avoid translocations. This is accompanied by the modification of chromatin landscapes, including histone acetylation profiles.44 Several HDACs and HATs are recruited to chromatin in response to DNA damage.20 Indeed, the biphasic regulation of histone acetylation as a response to DNA damage has long been documented and is initiated by a rapid (detectable within minutes) acetylation increase.45 Histone acetylation, mostly in the form of H4K16Ac, has been correlated with an open and more accessible chromatin.18 Intuitively, histone acetylation might favor the accessibility of the DNA damage site to repair proteins, while at the pan-nuclear level, it might facilitate the observed increased of chromatin mobility and remodeling that follows DNA damage. In apparent contradiction to this view, the global deacetylation of H3K56, H3K9 and H4K16 is an early event in the DNA damage response.39,46 Whether the acetylation wave precedes the deacetylation one or vice versa is not clear, and this warrants further investigation at high temporal and spatial resolution in living cells. Similarly, the functional implications of pan-nuclear acetylation signals also need further investigation.
A pivotal role of sirtuins in the DDR is the regulation of discrete histone acetylation levels.7,24,47 Specifically, studies have demonstrated SIRT1-dependent deacetylation of H4K16Ac,47 SIRT6-dependent deacetylation of H3K9Ac and H3K56Ac,24,39 and SIRT7-dependent deacetylation of H3K18Ac.7 Nevertheless, the role of sirtuin-dependent histone deacetylation in the DDR is not known for most cases. At the pan-nuclear level, the SIRT7 deacetylase activity seems to counterbalance a first wave of H3K18Ac that occurs soon (minutes) after the induction of DNA damage. Four hours after ionizing radiation exposure, most of the DNA damage has been repaired and the levels of H3K18Ac have already been restored to near steady-state levels.7 At this time point, SIRT7 is sequestered in the cytoplasm in a manner dependent on the ribonuclease III enzyme Dicer, with a concomitant decrease of SIRT7 and an increase of H3K18Ac in the chromatin fraction.48 Overall, these results indicate that the regulation of H3K18Ac levels is fine-tuned in response to DNA damage.
Genome-wide transcriptional silencing is a cellular response to DNA damage, and the transcriptional profiles seem to be restored upon DNA repair.49 Similar to the role of SIRT1 in transcriptional repression and heterochromatin formation,50 SIRT1 mediates gene silencing at DNA damage-flanking genes.47 The exact role of SIRT6- and SIRT7-dependent pan-nuclear deacetylation of H3K9Ac, H3K56Ac and H3K18Ac is not known. This deacetylation might also be associated with the DDR-related transcriptional repression and/or the restoration of transcriptional profiles, considering that these histone modifications are enriched at transcriptional regulatory regions.51
SIRT7-mediated deacetylation at the DNA damage site has a very precise role in DNA repair of facilitating the recruitment of the 53BP1 protein.7 Indeed, the importance of histone modifications in DNA damage repair is exemplified by their role in the recruitment of 53BP1 to damaged sites.21 53BP1 forms foci at damaged chromatin through the bivalent recognition of H2AK15ub and H4K20me2 marks. Upon DNA damage, H2AK15 is ubiquitinated by the DNA damage-specific RNF168 ubiquitin ligase. The H4K20me2 levels increase at damage sites in an MMSET7-dependent manner. In addition, factors that compete with DNA repair proteins for H4K20me2 binding are ubiquitinated by the ubiquitin ligases RNF8 and RNF168 and are subsequently degraded, facilitating the accessibility of preexisting H4K20me2. 53BP1 then accumulates at foci through the recognition of H4K20me2 and H2AK15ub by its tudor and UDR domains, respectively. Histone acetylation appears to counteract the positive effect of these histone modifications by adversely affecting the binding of 53BP1 to damaged chromatin.52-54 For instance, the acetylation of H4K16 on H4K20me2-marked histones disrupts a salt bridge between H4K16 and Glu1551 in the 53BP1 tudor domain, reducing its affinity for H4K20me253. H2AK15Ac has also been proposed to counteract RNF168-dependent H2AK15ub and therefore 53BP1 occupancy at damage sites.54 In agreement with these observations, SIRT7-mediated H3K18 deacetylation is important for 53BP1 stabilization at break sites and, conversely, H3K18 acetylation disrupts the recruitment of 53BP1 to chromatin.7 Given the role of 53BP1 in inhibiting DNA end resection, it appears that histone acetylation regulates DSB pathway choice by releasing 53BP1 from chromatin, thus favoring HR repair over NHEJ. Although this model seems to hold true for H4K16ac and H2AK15ac,53, 54 H3K18 acetylation does not seem to be associated with DSB pathway choice, because SIRT7 depletion does not correlate with an increase in HR repair.7 Interestingly, H3K18 acetylation levels can modulate the extent of 53BP1 focal accumulation at DNA damaged sites. This observation is reminiscent of a previous report using a mutant 53BP1 protein lacking the “oligomerization” domain, which impaired NHEJ but was still able to repress CtIP-dependent end resection at dysfunctional telomeres and therefore HR repair.55 It remains to be determined whether H3K18 interacts directly with 53BP1, and if so, what is the domain involved. Since the tudor domain does not interact with the unmodified H3 tail,56 other domains including the oligomerization domain could be responsible for the H3K18–53BP1 interaction. Additionally, a high-throughput proteomics screen identified 53BP1 as a direct SIRT7 binding partner,57 underscoring the importance of the SIRT7-H3K18–53BP1 axes in the DDR. Further research will shed light into the SIRT7-mediated regulation of 53BP1 recruitment to DNA damage sites, which might involve both epigenetic regulation and protein-protein functional interactions.
Concluding remarks
In light of the new discovery of SIRT7-mediated regulation of NHEJ repair, it is becoming evident that an important role of sirtuins in the maintenance of genome integrity is the promotion of DNA damage repair. The different reports discussed here emphasized that sirtuins can act at different levels on the DDR, but our knowledge of how their relative contributions are regulated in vivo is in its infancy. Some of the remaining questions include: What is the level of redundancy and/or synergy between the different sirtuins in the DDR? What are the limiting factors that dictate the involvement of a particular sirtuin in one or more DNA repair pathways? Is the remodeling of chromatin by sirtuins required for its interaction with downstream repair proteins? If so, it might imply that sirtuin DNA repair activity in vivo depends on the pre-existing state of chromatin, such as histone acetylation levels, which is determined by cell type, developmental stage and cell fitness. Most importantly, what are the consequences of sirtuin activity in DNA repair versus other forms of sirtuin-mediated genome regulation, specifically transcriptional regulation? In this regard, it is known that the genome-wide redistribution of SIRT1 that occurs in response to genotoxic stress is associated with the activation of heterochromatic repetitive DNA as well as of individual genes.26 Moreover, the re-localization of SIRT7 from the nucleolus to DNA damage sites affects the SIRT7-mediated regulation of ribosomal gene expression.7,12 This suggests that under chronic DNA damage situations, sirtuin-mediated DNA repair might have genome-wide transcriptional consequences. On the other hand, sirtuin-mediated chromatin regulation in the DDR is correlated with acute transcriptional repression47 and plausibly also with the restoration of transcriptional profiles following DNA repair. Overall, the modulation of sirtuins in different cellular contexts can have a range of outcomes with potentially distinct mutagenic consequences, which should be taken into consideration before any therapeutic intervention involving the modification of sirtuin activity.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
We thank Dr. Jay Tischfield director of the Human Genetics Institute of New Jersey (HGINJ) for helpful discussion. This study was supported by a grant from the HGINJ (to L.S). B.N.V. was supported by postdoctoral fellowships from the Spanish Ministry of Education, Culture, and Sports (EX-2010-278) and the HGINJ.
References
- [1].Guarente L. Calorie restriction and sirtuins revisited. Genes Dev 2013; 27:2072-85; PMID:24115767; http://dx.doi.org/ 10.1101/gad.227439.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A 2003; 100:10794-9; PMID:12960381; http://dx.doi.org/ 10.1073/pnas.1934713100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wang RH, Sengupta K, Li C, Kim HS, Cao L, Xiao C, Kim S, Xu X, Zheng Y, Chilton B, et al.. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 2008; 14:312-23; PMID:18835033; http://dx.doi.org/ 10.1016/j.ccr.2008.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].McBurney MW, Yang X, Jardine K, Hixon M, Boekelheide K, Webb JR, Lansdorp PM, Lemieux M. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol 2003; 23:38-54; PMID:12482959; http://dx.doi.org/ 10.1128/MCB.23.1.38-54.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, et al.. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006; 124:315-29; PMID:16439206; http://dx.doi.org/ 10.1016/j.cell.2005.11.044 [DOI] [PubMed] [Google Scholar]
- [6].Zhong L, D'Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, et al.. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 2010; 140:280-93; PMID:20141841; http://dx.doi.org/ 10.1016/j.cell.2009.12.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vazquez BN, Thackray JK, Simonet NG, Kane-Goldsmith N, Martinez-Redondo P, Nguyen T, Bunting S, Vaquero A, Tischfield JA, Serrano L. SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. EMBO J 2016; 35:1483-595; PMID:27225932; http://dx.doi.org/ 10.15252/embj.201593499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Vakhrusheva O, Smolka C, Gajawada P, Kostin S, Boettger T, Kubin T, Braun T, Bober E. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ Res 2008; 102:703-10; PMID:18239138; http://dx.doi.org/ 10.1161/CIRCRESAHA.107.164558 [DOI] [PubMed] [Google Scholar]
- [9].Yoshizawa T, Karim MF, Sato Y, Senokuchi T, Miyata K, Fukuda T, Go C, Tasaki M, Uchimura K, Kadomatsu T, et al.. SIRT7 controls hepatic lipid metabolism by regulating the ubiquitin-proteasome pathway. Cell Metab 2014; 19:712-21; PMID:24703702; http://dx.doi.org/ 10.1016/j.cmet.2014.03.006 [DOI] [PubMed] [Google Scholar]
- [10].Ryu D, Jo YS, Lo Sasso G, Stein S, Zhang H, Perino A, Lee JU, Zeviani M, Romand R, Hottiger MO, et al.. A SIRT7-dependent acetylation switch of GABPbeta1 controls mitochondrial function. Cell Metab 2014; 20:856-69; PMID:25200183; http://dx.doi.org/ 10.1016/j.cmet.2014.08.001 [DOI] [PubMed] [Google Scholar]
- [11].Shin J, He M, Liu Y, Paredes S, Villanova L, Brown K, Qiu X, Nabavi N, Mohrin M, Wojnoonski K, et al.. SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell Rep 2013; 5:654-65; PMID:24210820; http://dx.doi.org/ 10.1016/j.celrep.2013.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ford E, Voit R, Liszt G, Magin C, Grummt I, Guarente L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev 2006; 20:1075-80; PMID:16618798; http://dx.doi.org/ 10.1101/gad.1399706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Barber MF, Michishita-Kioi E, Xi Y, Tasselli L, Kioi M, Moqtaderi Z, Tennen RI, Paredes S, Young NL, Chen K, et al.. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 2012; 487:114-8; PMID:22722849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang H, Park SH, Pantazides BG, Karpiuk O, Warren MD, Hardy CW, Duong DM, Park SJ, Kim HS, Vassilopoulos A, et al.. SIRT2 directs the replication stress response through CDK9 deacetylation. Proc Natl Acad Sci U S A 2013; 110:13546-51; PMID:23898190; http://dx.doi.org/ 10.1073/pnas.1301463110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang H, Head PE, Daddacha W, Park SH, Li X, Pan Y, Madden MZ, Duong DM, Xie M, Yu B, et al.. ATRIP deacetylation by SIRT2 drives ATR checkpoint activation by promoting binding to RPA-ssDNA. Cell Rep 2016; 14:1435-47; PMID:26854234; http://dx.doi.org/ 10.1016/j.celrep.2016.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhang H, Head PE, Yu DS. SIRT2 orchestrates the DNA damage response. Cell Cycle 2016; 15:2089-90; PMID:27153288; http://dx.doi.org/ 10.1080/15384101.2016.1184517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013; 153:1194-217; PMID:23746838; http://dx.doi.org/ 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bosch-Presegue L, Vaquero A. Sirtuin-dependent epigenetic regulation in the maintenance of genome integrity. FEBS J 2015; 282:1745-67; PMID:25223884; http://dx.doi.org/ 10.1111/febs.13053 [DOI] [PubMed] [Google Scholar]
- [19].Roos WP, Krumm A. The multifaceted influence of histone deacetylases on DNA damage signalling and DNA repair. Nucleic Acids Res 2016; 44:10017-30; PMID:27738139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 2011; 25:409-33; PMID:21363960; http://dx.doi.org/ 10.1101/gad.2021311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 2014; 15:7-18; PMID:24326623; http://dx.doi.org/ 10.1038/nrm3719 [DOI] [PubMed] [Google Scholar]
- [22].Daley JM, Sung P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol Cell Biol 2014; 34:1380-8; PMID:24469398; http://dx.doi.org/ 10.1128/MCB.01639-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet 2011; 45:247-71; PMID:21910633; http://dx.doi.org/ 10.1146/annurev-genet-110410-132435 [DOI] [PubMed] [Google Scholar]
- [24].Toiber D, Erdel F, Bouazoune K, Silberman DM, Zhong L, Mulligan P, Sebastian C, Cosentino C, Martinez-Pastor B, Giacosa S, et al.. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol Cell 2013; 51:454-68; PMID:23911928; http://dx.doi.org/ 10.1016/j.molcel.2013.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dobbin MM, Madabhushi R, Pan L, Chen Y, Kim D, Gao J, Ahanonu B, Pao PC, Qiu Y, Zhao Y, et al.. SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat Neurosci 2013; 16:1008-15; PMID:23852118; http://dx.doi.org/ 10.1038/nn.3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, Park SK, Hartlerode A, Stegmuller J, Hafner A, Loerch P, et al.. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 2008; 135:907-18; PMID:19041753; http://dx.doi.org/ 10.1016/j.cell.2008.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yuan Z, Zhang X, Sengupta N, Lane WS, Seto E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol Cell 2007; 27:149-62; PMID:17612497; http://dx.doi.org/ 10.1016/j.molcel.2007.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007; 26:7741-8; PMID:18066086; http://dx.doi.org/ 10.1038/sj.onc.1210872 [DOI] [PubMed] [Google Scholar]
- [29].Serrano L, Liang L, Chang Y, Deng L, Maulion C, Nguyen S, Tischfield JA. Homologous recombination conserves DNA sequence integrity throughout the cell cycle in embryonic stem cells. Stem Cells Dev 2011; 20:363-74; PMID:20491544; http://dx.doi.org/ 10.1089/scd.2010.0159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ghosh S, Liu B, Wang Y, Hao Q, Zhou Z. Lamin A is an endogenous SIRT6 activator and promotes SIRT6-mediated DNA repair. Cell Rep 2015; 13:1396-406; PMID:26549451; http://dx.doi.org/ 10.1016/j.celrep.2015.10.006 [DOI] [PubMed] [Google Scholar]
- [31].Li L, Shi L, Yang S, Yan R, Zhang D, Yang J, He L, Li W, Yi X, Sun L, et al.. SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat Commun 2016; 7:12235; PMID:27436229; http://dx.doi.org/ 10.1038/ncomms12235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev 2012; 26:417-32; PMID:22391446; http://dx.doi.org/ 10.1101/gad.183509.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol 2014; 24:464-71; PMID:24786309; http://dx.doi.org/ 10.1016/j.tcb.2014.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, Hottiger MO, Gupta MP. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol Cell Biol 2009; 29:4116-29; PMID:19470756; http://dx.doi.org/ 10.1128/MCB.00121-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V. SIRT6 promotes DNA repair under stress by activating PARP1. Science 2011; 332:1443-6; PMID:21680843; http://dx.doi.org/ 10.1126/science.1202723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mao Z, Tian X, Van Meter M, Ke Z, Gorbunova V, Seluanov A. Sirtuin 6 (SIRT6) rescues the decline of homologous recombination repair during replicative senescence. Proc Natl Acad Sci U S A 2012; 109:11800-5; PMID:22753495; http://dx.doi.org/ 10.1073/pnas.1200583109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Scheibye-Knudsen M, Mitchell SJ, Fang EF, Iyama T, Ward T, Wang J, Dunn CA, Singh N, Veith S, Hasan-Olive MM, et al.. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab 2014; 20:840-55; PMID:25440059; http://dx.doi.org/ 10.1016/j.cmet.2014.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mortusewicz O, Ame JC, Schreiber V, Leonhardt H. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res 2007; 35:7665-75; PMID:17982172; http://dx.doi.org/ 10.1093/nar/gkm933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].McCord RA, Michishita E, Hong T, Berber E, Boxer LD, Kusumoto R, Guan S, Shi X, Gozani O, Burlingame AL, et al.. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging (Albany NY) 2009; 1:109-21; PMID:20157594; http://dx.doi.org/ 10.18632/aging.100011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kaidi A, Weinert BT, Choudhary C, Jackson SP. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science 2010; 329:1348-53; PMID:20829486; http://dx.doi.org/ 10.1126/science.1192049 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [41].Jeong J, Juhn K, Lee H, Kim SH, Min BH, Lee KM, Cho MH, Park GH, Lee KH. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp Mol Med 2007; 39:8-13; PMID:17334224; http://dx.doi.org/ 10.1038/emm.2007.2 [DOI] [PubMed] [Google Scholar]
- [42].Li K, Casta A, Wang R, Lozada E, Fan W, Kane S, Ge Q, Gu W, Orren D, Luo J. Regulation of WRN protein cellular localization and enzymatic activities by SIRT1-mediated deacetylation. J Biol Chem 2008; 283:7590-8; PMID:18203716; http://dx.doi.org/ 10.1074/jbc.M709707200 [DOI] [PubMed] [Google Scholar]
- [43].Uhl M, Csernok A, Aydin S, Kreienberg R, Wiesmuller L, Gatz SA. Role of SIRT1 in homologous recombination. DNA Repair (Amst) 2010; 9:383-93; PMID:20097625; http://dx.doi.org/ 10.1016/j.dnarep.2009.12.020 [DOI] [PubMed] [Google Scholar]
- [44].Gerhold CB, Hauer MH, Gasser SM. INO80-C and SWR-C: guardians of the genome. J Mol Biol 2015; 427:637-51; PMID:25451604; http://dx.doi.org/ 10.1016/j.jmb.2014.10.015 [DOI] [PubMed] [Google Scholar]
- [45].Ramanathan B, Smerdon MJ. Changes in nuclear protein acetylation in u.v.-damaged human cells. Carcinogenesis 1986; 7:1087-94; PMID:3087643; http://dx.doi.org/ 10.1093/carcin/7.7.1087 [DOI] [PubMed] [Google Scholar]
- [46].Tjeertes JV, Miller KM, Jackson SP. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J 2009; 28:1878-89; PMID:19407812; http://dx.doi.org/ 10.1038/emboj.2009.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].O'Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet 2008; 4:e1000155; PMID:18704159; http://dx.doi.org/ 10.1371/journal.pgen.1000155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhang PY, Li G, Deng ZJ, Liu LY, Chen L, Tang JZ, Wang YQ, Cao ST, Fang YX, Wen F, et al.. Dicer interacts with SIRT7 and regulates H3K18 deacetylation in response to DNA damaging agents. Nucleic Acids Res 2016; 44:3629-42; PMID:26704979; http://dx.doi.org/ 10.1093/nar/gkv1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kim J, Sturgill D, Tran AD, Sinclair DA, Oberdoerffer P. Controlled DNA double-strand break induction in mice reveals post-damage transcriptome stability. Nucleic Acids Res 2016; 44:e64; PMID:26687720; http://dx.doi.org/ 10.1093/nar/gkv1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bosch-Presegue L, Vaquero A. Sirtuins in stress response: guardians of the genome. Oncogene 2014; 33:3764-75; PMID:23995787; http://dx.doi.org/ 10.1038/onc.2013.344 [DOI] [PubMed] [Google Scholar]
- [51].Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, et al.. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 2008; 40:897-903; PMID:18552846; http://dx.doi.org/ 10.1038/ng.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hsiao KY, Mizzen CA. Histone H4 deacetylation facilitates 53BP1 DNA damage signaling and double-strand break repair. J Mol Cell Biol 2013; 5:157-65; PMID:23329852; http://dx.doi.org/ 10.1093/jmcb/mjs066 [DOI] [PubMed] [Google Scholar]
- [53].Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV, Mer G, Greenberg RA. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol 2013; 20:317-25; PMID:23377543; http://dx.doi.org/ 10.1038/nsmb.2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jacquet K, Fradet-Turcotte A, Avvakumov N, Lambert JP, Roques C, Pandita RK, Paquet E, Herst P, Gingras AC, Pandita TK, et al.. The TIP60 complex regulates bivalent chromatin recognition by 53BP1 through direct H4K20me binding and H2AK15 acetylation. Mol Cell 2016; 62:409-21; PMID:27153538; http://dx.doi.org/ 10.1016/j.molcel.2016.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lottersberger F, Bothmer A, Robbiani DF, Nussenzweig MC, de Lange T. Role of 53BP1 oligomerization in regulating double-strand break repair. Proc Natl Acad Sci U S A 2013; 110:2146-51; PMID:23345425; http://dx.doi.org/ 10.1073/pnas.1222617110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, Zhang Y, Bedford MT. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep 2006; 7:397-403; PMID:16415788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tsai YC, Greco TM, Boonmee A, Miteva Y, Cristea IM. Functional proteomics establishes the interaction of SIRT7 with chromatin remodeling complexes and expands its role in regulation of RNA polymerase I transcription. Mol Cell Proteomics 2012; 11:60-76; PMID:22586326; http://dx.doi.org/ 10.1074/mcp.A111.015156 [DOI] [PMC free article] [PubMed] [Google Scholar]