

Abstract
Cervical cancer is one of the most common types of cancer among women worldwide. In order to identify the microRNAs (miRNAs/miRs) and mRNAs associated with the carcinogenesis of cervical cancer, and to investigate the molecular mechanisms of cervical cancer, an miRNA microarray, GSE30656, and 3 mRNA microarrays, GSE63514, GSE39001 and GSE9750, for cervical cancer were retrieved from Gene Expression Omnibus. These datasets were analyzed in order to obtain differentially-expressed genes (DEGs) and miRNAs using the GEO2R tool. Gene Ontology (GO) and pathway enrichment analysis for DEGs were performed using the Database for Annotation, Visualization and Integrated Discovery. Protein-protein interaction (PPI) analysis for DEGs was conducted using The Search Tool for the Retrieval of Interacting Genes software and visualized using Cytoscape, followed by hub gene identification, and biological process and pathway enrichment analysis of the module selected from the PPI network using the Molecular Complex Detection plugin. In addition, miRecords was applied to predict the targets of differentially-expressed miRNAs. A total of 44 DEGs and 15 differentially-expressed miRNAs were identified. These DEGs were mainly enriched in GO terms associated with the cell cycle. In the PPI network, cyclin-dependent kinase 1, topoisomerase DNA IIα, aurora kinase A (AURKA) and minichromosome maintenance complex component 2 (MCM2) had higher degrees of connectivity. A significant module was detected from the PPI network. AURKA, MCM2 and kinesin family member 20A exhibited higher degrees in this module, while the genes in the module were mainly involved in the cell cycle and the DNA replication pathway. In addition, estrogen receptor 1 was predicted as the potential target of 13 miRNAs. A total of 10 DEGs were identified as potential targets of miR-203. In conclusion, the results indicated that microarray dataset analysis may provide a useful method for the identification of key genes and patterns to successfully identify determinants of the carcinogenesis of cervical cancer. The functional studies of candidate genes and miRNAs from these databases may lead to an increased understanding of the development of cervical cancer.
Keywords: cervical cancer, differentially-expressed genes, differentially-expressed microRNAs, protein-protein interaction, enrichment analysis
Introduction
Cervical cancer is one of the most common types of gynecological malignancy worldwide, with an annual incidence rate of ~454,000 cases in 2010 (1,2). Epidemiological and clinical studies have established a causal association between persistent infection of high-risk human papilloma virus (HR-HPV) types (including HPV 16 and 18) and cervical carcinogenesis (3), with the contribution of additional cofactors including smoking or the use of oral contraceptives (4). It is known that the integration of HR-HPV DNA into the host cell genome results in elevated expression of HPV E6 and E7 oncoproteins (5), which subsequently allows the virus to replicate through inhibitory effects on the tumor suppressor proteins p53 and retinoblastoma protein, respectively (6,7). The inhibition of these tumor suppressors results in uncontrolled cellular proliferation and the accumulation of specific epigenetic changes in the host cell genome, driving progression to a malignant phenotype (3). However, the mechanism of the progression from preneoplastic lesions and cervical intraepithelial neoplasia to carcinoma remains unknown.
HPV infection is necessary, but the E6-p53 and E7-Rb model is not sufficient to inevitably produce cervical carcinoma (8). Only a small number of women who are infected with HR-HPV will develop cervical cancer, highlighting the multistep nature of cervical carcinogenesis and the variety of cofactors required. A range of genetic and epigenetic events contribute to the initiation of cervical cancer. However, more insight is required into the genetic and epigenetic alterations that occur during cervical carcinogenesis in order for the identification of genes that are involved in the development and progression of cervical cancer (4,9).
MicroRNAs (miRNAs/miRs) are small non-coding single-stranded RNAs that regulate gene expression and perform an important role in the regulation of cellular differentiation, proliferation and apoptosis (10,11). Furthermore, certain miRNAs are considered to be oncogenes or tumor suppressor genes, and exhibit altered expression profiles in cervical cancer tumors (1).
A large amount of data on the transcriptome and the proteome states of cells has recently been created via the wide use of a number of high-throughput technologies. The large amount of publically available molecular data allows for the bioinformatic investigation of numerous factors, including biochip data extraction, sequence alignment, biological data clustering and pathway analysis. These analyses methods provide ways to study the molecular pathogenesis of various types of cancer.
The present study aimed to identify candidate biomarkers or therapeutic targets of cervical cancer, by investigating microarray data detailing the mRNA HR-HPV oncogenes and miRNA expression profiles in cervical cancer. These expression profiles data were retrieved from the Gene Expression Omnibus (GEO) database (12) and were analyzed to identify differentially-expressed genes (DEGs) and miRNAs. Combined bioinformatics methods, including functional annotation and pathway enrichment analysis, protein-protein interaction (PPI) network construction and mRNA-miRNA interaction analysis, were performed to obtain the key genes or miRNAs involved in cervical cancer.
Materials and methods
Acquisition of microarray datasets
Microarray data detailing cervical cancer-associated the miRNA microarray dataset GSE30656 and mRNA microarray datasets GSE63514, GSE39001 and GSE9750 were retrieved and downloaded from the National Center for Biotechnology Information GEO database (http://www.ncbi.nlm.nih.gov/geo). The dataset GSE30656, based on the platform of GPL 6955 Agilent-016436 Human miRNA Microarray 1.0 (Agilent Technologies, Inc., Santa Clara, CA, USA), included 10 patients with HR-HPV-positive cervical cancer and 10 normal cervical (NC) tissues samples. The dataset GSE63514, using the GPL570 Affymetrix Human Genome U133 Plus 2.0 Array (Affymetrix, Inc., Santa Clara, CA, USA), included 28 patients with cervical cancer and 24 NC samples. GSE39001 included 43 patients with HPV16-positive cervical cancer and 12 NC samples, and GSE9750 included 33 patients with cervical cancer that were mainly marked by HPV16 or HPV18 and 24 NC samples, based on the GPL201 Affymetrix Human HG-Focus Target Array and the GPL96 Affymetrix Human Genome U133A Array (both from Affymetrix, Inc.), respectively.
Data processing
In the present study, GEO2R (http://www .ncbi.nlm.nih.gov/geo/geo2r/) was applied to screen differentially-expressed miRNAs and DEGs between cervical cancer and NC samples. GEO2R is an R programming language-based dataset analysis tool based on a t-test or analysis of variance. GEO2R is an interactive tool that allows users to compare two groups of samples in a GEO series in order to identify DEGs or miRNAs under the same experimental conditions (13). In total, >90% of GEO data can be accessed and analyzed in this way, and results are presented as a table of genes in sequence of significance and may be viewed as profile graphs. GEO2R handles a large quantity of experimental designs and data types, and applies the adjusted P-value (adj. P) to assist in correcting for the occurrence of false-positives. In the present study, differentially-expressed miRNAs and DEGs between cervical cancer and NC tissues were screened using an adj. P<0.05 and a fold-change of >2 as the threshold values.
Gene ontology and pathway enrichment analysis
Significantly changed DEGs were submitted to the Database for Annotation, Visualization and Integrated Discovery (DAVID; http://david .abcc.ncifcrf.gov/), an online tool for functional annotation analysis (14). The significant enrichment analysis of DEGs was assessed based on the gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/kegg2.html) with P<0.05 as the cutoff. GO terms consisted of 3 aspects: Biological process, cellular component and molecular function.
PPI network construction
High-quality protein interaction networks can provide key insights into the functional and biological properties of cellular systems. The Search Tool for the Retrieval of Interacting Genes (STRING; http://string.embl.de/) database (15) is an online tool of known and predicted protein interactions, including physical and indirect functional associations. The PPI network of DEGs was constructed using STRING with a combined score of >0.4 and visualized using Cytoscape (16), an open source software platform for visualizing molecular interaction networks and integrating data.
Modules selection from PPI network
As a plugin to Cytoscape, Molecular Complex Detection (MCODE) (17) was used to detect densely connected regions from the PPI network with the following cutoff values: Degree cutoff, 2; node score cutoff, 0.2; k-core, 2; and maximum depth, 100. Subsequently, based on modules selected from the PPI network, functional enrichment analysis was performed with the criterion of P<0.05.
mRNA-miRNA interaction analysis
Targets of differentially-expressed miRNAs were predicted using miRecords (http://c1.accurascience.com/miRecords/) (18), an integrated resource for miRNA-target interactions, which stores miRNA targets predicted by 11 established target prediction tools. The overlap predicted by at least 3 of these target prediction programs were selected as targets of miRNAs.
Results
Identification of DEGs
In the present study, the mRNA expression profiling datasets GSE63514, GSE39001 and GSE9750 were screened using the GEO2R tool to identify genes that were differentially expressed between normal and cancerous tissues. A total of 939, 113 and 2,117 genes were identified as the DEGs from these datasets, respectively (Fig. 1). There were 44 DEGs that exhibited the same expression trends in all 3 datasets; 20 of these were downregulated and 24 were upregulated in cervical cancer (Table I).
Figure 1.
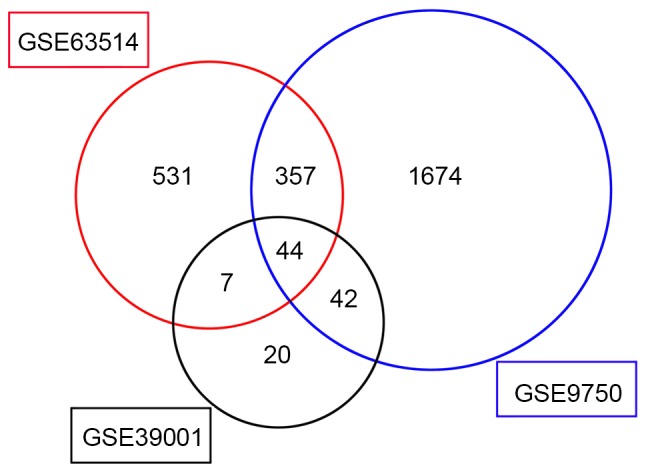
Identification of differentially-expressed genes in mRNA expression profiling datasets GSE63514, GSE39001 and GSE9750 using the GEO2R tool. Thresholds used in the GEO2R analysis were adjusted P<0.05, fold-change >2.
Table I.
All 44 differentially-expressed genes that exhibited the same expression trends in all three datasets.
| Log FC | |||
|---|---|---|---|
| Gene name | GSE39001 | GSE9750 | GSE63514 |
| CFD | 3.99 | 2.83 | 2.91 |
| APOD | 3.76 | 2.55 | 3.53 |
| CRNN | 3.36 | 8.07 | 9.51 |
| CXCL14 | 3.22 | 2.27 | 3.77 |
| MAL | 3.18 | 6.35 | 6.35 |
| CRISP3 | 3.18 | 7.21 | 8.91 |
| ESR1 | 3.10 | 3.15 | 4.12 |
| CRCT1 | 3.02 | 5.54 | 5.34 |
| DEFB1 | 2.79 | 1.70 | 2.68 |
| SPINK5 | 2.78 | 4.98 | 5.85 |
| NDN | 2.69 | 1.53 | 2.38 |
| ID4 | 2.63 | 2.31 | 2.07 |
| KRT13 | 2.61 | 4.50 | 4.67 |
| ALOX12 | 2.50 | 5.56 | 5.27 |
| SPRR3 | 2.50 | 4.64 | 5.56 |
| PPP1R3C | 2.47 | 4.54 | 5.12 |
| EDN3 | 2.36 | 5.13 | 5.40 |
| MPPED2 | 2.31 | 1.51 | 3.19 |
| FCGBP | 2.22 | 1.60 | 4.72 |
| CYP2B6 | 2.08 | 1.73 | 2.63 |
| NEK2 | −2.05 | −3.40 | −2.64 |
| PLOD2 | −2.08 | −1.92 | −3.23 |
| KIF20A | −2.10 | −1.78 | −2.09 |
| CXCL9 | −2.12 | −1.50 | −3.71 |
| CENPA | −2.13 | −1.77 | −2.70 |
| AURKA | −2.18 | −2.41 | −2.52 |
| CXCL10 | −2.20 | −1.34 | −2.97 |
| APOBEC3B | −2.29 | −1.50 | −2.58 |
| MCM2 | −2.39 | −2.33 | −2.61 |
| SMC4 | −2.45 | −1.50 | −3.02 |
| MMP1 | −2.46 | −5.49 | −2.82 |
| CDK1 | −2.49 | −2.41 | −2.88 |
| SYCP2 | −2.51 | −3.34 | −5.15 |
| CDKN3 | −2.58 | −1.06 | −2.53 |
| TOP2A | −2.60 | −2.24 | −2.53 |
| CDKN2A | −2.61 | −3.78 | −6.41 |
| RFC4 | −2.75 | −1.76 | −3.15 |
| KRT17 | −2.80 | −1.95 | −2.63 |
| UBD | −2.81 | −3.46 | −2.00 |
| PRC1 | −2.86 | −1.65 | −2.25 |
| TYMS | −3.22 | −2.03 | −2.09 |
| MMP12 | −3.23 | −2.68 | −5.05 |
| NUSAP1 | −3.35 | −1.73 | −2.36 |
| RRM2 | −3.48 | −1.94 | −2.16 |
A positive log FC value indicates that the gene expression level was downregulated in the cervical cancer samples, whereas a negative log FC value indicates that the gene expression level was upregulated in the cervical cancer samples. Thresholds used in the GEO2R analysis were adjusted P<0.05, fold >2. Gene names were listed in line with the gene database at the national center for biological information. FC, fold-change.
Significant functions and pathway enrichment analysis
To reveal the biological significance of the common DEGs in the carcinogenesis of cervical cancer, GO functional and pathway enrichment analysis were performed on the 44 aforementioned DEGs using DAVID. As demonstrated in Table II, DEGs are involved in a number of biological processes, including the cell cycle, cellular division, DNA replication and chemotaxis. In terms of cellular components, the DEGs were mainly enriched in the microtubular cytoskeleton, spindle and cytoskeleton. The molecular function terms in the DEGs were chemokine activity and chemokine receptor binding. Based on the KEGG pathway enrichment analysis, the DEGs were identified to be significantly enriched in the p53 signaling pathway.
Table II.
Gene ontology and pathway enrichment analysis for DEGs.
| Category | GO term | Description | Count | P-value |
|---|---|---|---|---|
| BP | 0000278 | Mitotic cell cycle | 10 | 7.97×10−7 |
| BP | 0022403 | Cell cycle phase | 10 | 2.02×10−6 |
| BP | 0022402 | Cell cycle process | 11 | 3.17×10−6 |
| BP | 0007049 | Cell cycle | 12 | 7.86×10−6 |
| BP | 0048015 | Phosphoinositide-mediated signaling | 5 | 1.16×10−4 |
| CC | 0015630 | Microtubular cytoskeleton | 8 | 7.31×1044 |
| CC | 0005819 | Spindle | 5 | 7.43×10−5 |
| CC | 0044430 | Cytoskeletal part | 10 | 1.02×10−3 |
| CC | 0005694 | Chromosome | 7 | 1.64×10−3 |
| CC | 0043228 | Non-membrane-bound organelle | 16 | 2.83×10−3 |
| MF | 0008009 | Chemokine activity | 3 | 7.17×10−3 |
| MF | 0042379 | Chemokine receptor binding | 3 | 8.10×10−3 |
| KEGG | hsa04115 | p53 signaling pathway | 3 | 1.87×10−2 |
If there were >5 terms enriched by DEGs in this category, the top 5 terms were selected according to the P-value. Count refers to the number of genes significantly enriched in this term. DEGs, differentially-expressed genes; GO, gene ontology; BP, biological process; CC, cellular component; MF, molecular function; KEGG, Kyoto Encyclopedia of Genes and Genomes.
PPI network of DEGs
The PPI network was constructed to dissect the interactions between DEGs. There were 24 nodes and 75 edges in the network (Fig. 2). A number of upregulated genes had higher node degrees: Cyclin-dependent kinase 1 (CDK1; degree, 15), topoisomerase DNA II alpha (TOP2A; degree, 14), aurora kinase A (AURKA; degree, 13) and kinesin family member 20A (KIF20A; degree, 9). Notably, CDK1 exhibited interactions with TOP2A, AURKA and KIF20A, as well as minichromosome maintenance complex component 2 (MCM2) and estrogen receptor 1 (ESR1), respectively. AURKA was also observed to be associated with KIF20A.
Figure 2.
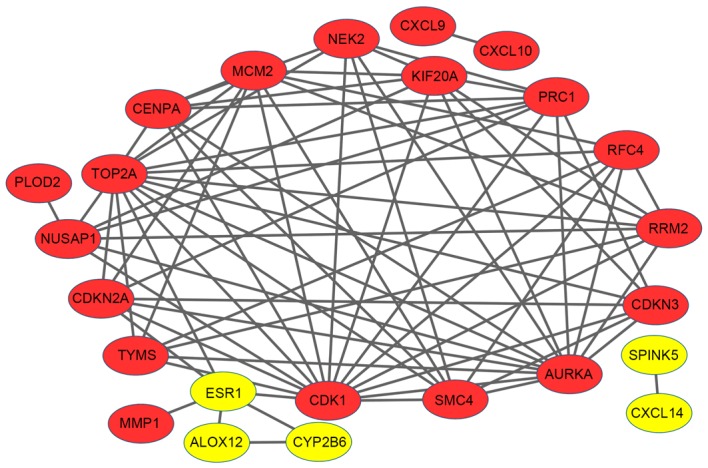
Protein-protein interaction network of differentially-expressed genes. Red nodes represent upregulated genes, whilst yellow nodes represent downregulated genes. The lines (edges) represent interactions between genes.
Module analysis
A significant module that included 11 nodes and 26 edges was identified from the PPI network with an MCODE score ≥4 (Fig. 3). AURKA (degree, 9), MCM2 (degree, 6) and KIF20A (degree, 6) possessed higher degrees of connectivity. MCM2 had interactions with AURKA and KIF20A, respectively. Based on DAVID software, overrepresented biological functions were identified for these genes of the module. A total of 37 GO functions were enriched. As demonstrated in Table III, these genes were mainly enriched in the cell cycle, which is similar to the aforementioned result. The KEGG pathway enrichment analysis revealed that these genes were associated with the DNA replication pathway.
Figure 3.
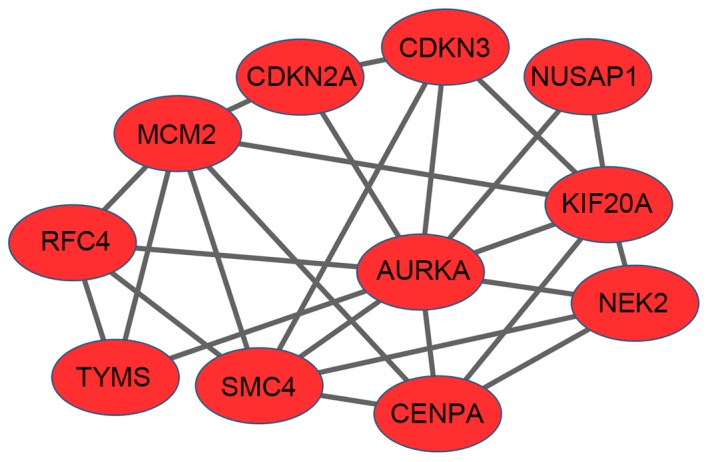
Significant module in protein-protein interaction network with a molecular complex detection score ≥4. The nodes represent the genes. All genes were upregulated in cervical cancer. The lines (edges) represent interactions between genes.
Table III.
Biological process terms of DEGs from the module.
| GO term | Description | Count | P-value |
|---|---|---|---|
| 0000278 | Mitotic cell cycle | 7 | 7.70×10−8 |
| 0007049 | Cell cycle | 8 | 2.05×10−7 |
| 0022402 | Cell cycle process | 7 | 9.41×10−7 |
| 0022403 | Cell cycle phase | 6 | 5.82×10−6 |
| 0007017 | Microtubule-based process | 5 | 2.30×10−5 |
If there were >5 terms enriched by DEGs in this category, the top 5 terms were selected according to the P-value. Count refers to the number of genes significantly enriched in this term. DEGs, differentially-expressed genes; GO, gene ontology.
Identification of differentially-expressed miRNAs and potential target genes
In the present study, the miRNA profiling dataset GSE30656 was analyzed to screen the differentially-expressed miRNAs in cervical cancer using the GEO2R tool. As presented in Table IV, a total of 15 differentially-expressed miRNAs were identified from this microarray dataset. A total of 11 miRNAs were significantly downregulated in cervical cancer, whereas the expression levels of the remaining 4 miRNAs were upregulated. The miRNAs with the most significant difference in downregulated and upregulated miRNAs were miR-203 and miR-21, respectively.
Table IV.
Differentially-expressed miRNAs in cervical cancer and their targets.
| miRNAs | P-value | Log FC | Target genes |
|---|---|---|---|
| miR-203 | 4.76×10−5 | 2.73 | TOP2A, PRC1, CXCL14, CRISP3, ID4, RRM2, TYMS, NDN, ESR1, SMC4 |
| miR-370 | 7.32×10−4 | 2.62 | AURKA, ESR1, CRCT1 |
| miR-638 | 8.63×10−5 | 2.16 | CXCL9, ESR1 |
| miR-494 | 6.72×10−4 | 1.80 | ESR1, PLOD2, CXCL14, MAL |
| miR-575 | 5.54×10−4 | 1.69 | ESR1, SMC4, EDN3, CXCL9, CYP2B6, CDKN2A |
| miR-125b | 1.42×10−5 | 1.67 | ESR1, RRM2, SPRR3, CDKN2A |
| miR-99a | 8.47×10−4 | 1.41 | ESR1 |
| miR-572 | 1.6×10−3 | 1.31 | CRCT1 |
| miR-375 | 7.39×10−6 | 1.19 | TYMS |
| miR-193b | 1.09×10−3 | 1.06 | CXCL14, ESR1 |
| miR-188 | 5.91×10−3 | 1.01 | ESR1, MMP1, ID4 |
| miR-106b | 6.91×10−8 | −1.07 | ESR1, CXCL14, CENPA, RRM2, CDKN2A, NDN, CRCT1 |
| miR-15b | 2.87×10−4 | −1.16 | ESR1, CXCL10, MAL, EDN3, CENPA |
| miR-16 | 1.39×10−4 | −1.22 | ESR1, CXCL10, EDN3, CENPA, ID4, PPP1R3C |
| miR-21 | 6.34×10−6 | −2.51 | CXCL14, EDN3, ESR1, NUSAP1 |
A positive log FC value represents the downregulation of that miRNA in cervical cancer, whilst a negative log FC value represents the upregulation of that miRNA in cervical cancer. Target gene names were listed according to the gene database at national center for biological information. miRNA/miR, microRNA; FC, fold-change.
The intersections of 44 DEGs in 3 microarray datasets and potential target genes identified by miRecords were determined. As demonstrated in Table IV, by comparing predicted targets of differentially-expressed miRNAs with DEGs, ESR1 was predicted as the potential target of 13 miRNAs and CXCL14 was the potential target of 5 miRNAs, including miR-203, miR-494, miR-193b, miR-106b and miR-21. CXCL9 was identified as a potential target of miR-638 and miR-575, for which their expression levels were negatively associated in cervical cancer tissues. In addition, 10 genes (including TOP2A, SMC4 and RRM2) of 44 DEGs were potentially targeted by miR-203.
Discussion
The present study identified 44 DEGs and 15 differentially-expressed miRNAs in cervical cancer compared with control samples. These genes were mainly enriched in the cell cycle, DNA replication and other relevant pathways that were closely associated with cancer, indicating that the present method was effective in identifying key genes.
Through PPI network construction, it was revealed that CDK1, TOP2A, AURKA, MCM2 and KIF20A had higher degrees of connectivity. As a protein kinase of the CDK family, CDK1 performed an important role in cell cycle progression (19). The cell cycle is a strictly regulated process. The main executor protein of the G2/M phase transition is CDK1, the functional activity of which is dependent on the expression of cyclin B proteins. A knockout experiment in mice revealed that CDK1 was required to drive mammalian cellular proliferation (20). Cellular growth of cells was also observed to be successfully inhibited via the functional loss of CDK1 in cervical cancer (21). The TOP2A gene is located on locus q21 of chromosome 17 and encodes a nuclear enzyme involved in DNA replication, transcription and chromosome condensation by altering the topological structure of DNA. Increased expression of TOP2A was identified in cervical cancer using different high-throughput expression profiling technologies (22). The TOP2A and MCM2 genes have been identified as molecular markers for the diagnosis of cervical cancer (23,24). Similar studies have also demonstrated that TOP2A was upregulated in cervical cells and tissues with elevated expression of the HPV E6/E7 proteins (25,26). Notably, the present study revealed that CDK1 interacted with TOP2A and MCM2, suggesting the joint function in cervical cancer.
Additionally, AURKA and KIF20A also had higher degrees in modules of the PPI network. AURKA belongs to serine/threonine protein kinase family. The protein encoded by AURKA is a cell cycle-regulated kinase that is involved in microtubule formation and/or stabilization at the spindle pole during chromosome segregation. Previous studies demonstrated that the overexpression of AURKA promoted tumorigenesis in multiple types of cancer, including neuroblastoma, and ovarian and cervical cancer (27,28). Inhibition of AURKA could improve the sensitivity of cervical cancer cells to taxol by inducing cell cycle arrest and apoptosis (29). In the module of the PPI network, AURKA was observed to interact with KIF20A, a member of the kinesin superfamily of motor proteins. KIF20A attracted attention for its important role in the cell cycle and in cell motility (30,31). KIF20A silencing with small interfering RNA (siRNA) could reduce the proliferation, migration and invasion of pancreatic cancer cells (32). Therefore, these studies support the present finding that AURKA and KIF20A are overexpressed, and are therefore closely associated with cervical cancer.
In the present study, ESR1 was predicted as a potential target of 13 differentially-expressed miRNAs and was downregulated in cervical cancer. ESR1 encodes a ligand-active transcription factor composed of several domains important for hormone binding, DNA binding and the activation of transcription. ESR1 is necessary for sexual development and reproductive function, and performs an important role in cellular development and differentiation. A previous study indicated that the loss of ESR1 enhanced cervical cancer progression and invasion (33). The epigenetic alteration by DNA methylation of ESR1 promoters is associated with the response of patients with advanced invasive cervical carcinoma who are treated with chemoradiation (34). Furthermore, the present study identified that 3 members of the chemokine family (CXCL9, CXCL10 and CXCL14) were potentially targeted by various differentially-expressed miRNAs. Multiplex Luminex immunoassays for cervical cancer cells of patients at different stages demonstrated that CXCL9 levels progressively increased with the advancement of cervical cancer (35).
Emerging evidence suggests that miRNAs, which regulate gene expression by targeting the 3′-untranslated region of mRNAs to cause translational repression and/or degradation, may be involved in the pathogenesis of several types of human cancer, including cervical carcinoma. Increasing evidence indicates that the dysregulation of miRNAs has been frequently observed in the carcinogenesis of cervical cancer, including metastasis and the drug resistance of tumor cells. Several miRNAs, including miR-9, miR-127, miR-145, miR-146a, miR-199a, miR-200a and miR-424, have been observed to be dysregulated in cervical carcinoma (36). In the present study, 11 downregulated miRNAs and 4 upregulated miRNAs in cervical cancer were identified, as demonstrated in Table IV. miR-203 was observed to be significantly downregulated in cervical cancer, which is consistent with a previous study by Zhao et al (37), which demonstrated the lower expression of miR-203 in cervical cancer tissues by quantitative-polymerase chain reaction. miR-203 has been identified as a skin-specific miRNA and it exhibits differential expression between cervical cancer and matched-nontumor cervical tissues (37). miR-203 inhibited cellular proliferation by directly targeting E2F1 in esophageal cancer cells (38). miR-203 has also been reported as being a tumor suppressor by inhibiting tumor growth and angiogenesis in cervical cancer (39).
Among the miRNAs identified in the present study, other miRNAs also have a close association with cervical cancer; for example, miR-375 was revealed to be downregulated >2 fold in cervical cancer samples. Consistent with this finding, Wang et al (40) observed that in 170 cervical cancer tissues, miR-375 expression was significantly decreased compared with that in 68 normal tissues. This finding suggested that the miR-375 downregulation should be involved in cervical cancer progression. Additionally, the present study predicted that the TOP2A and ESR1 genes were potential targets of miR-203, as determined by miRecords software, suggesting that miR-203 may affect the progression of cervical cancer by regulating its potential targets of TOP2A, ESR1 or others genes. Therefore, the present results suggested that the downregulation of miRNAs may perform an important role in the progression of cervical cancer, and that novel candidate markers may be presented by the evaluation of specific miRNAs for cancer screening and prognostic purposes in patients with cervical cancer. Furthermore, certain miRNAs may possess the potential to be used as predictors and promising therapeutic targets in the treatment of cervical cancer.
In summary, high-throughput technologies such as microarray analysis aid the identification of the molecular determinants of tumorigenesis. The present study identified 44 DEGs and 15 differentially-expressed miRNAs that may be useful for future studies focused on the assessment of the molecular mechanisms of cervical cancer via comprehensive bioinformatics analysis. In particular, CDK1, TOP2A, MCM2, AURKA, KIF20A, ESR1 and several miRNAs may be involved in the carcinogenesis of cervical cancer. The results of the present study also demonstrate the value of data mining in multi-dimensional omics data. The present findings provide novel insights into the development and progression of cervical cancer. However, the network of interactions of miRNAs and mRNAs is extremely complex and expression profiling analysis is a relatively new tool. Therefore, additional experimental studies are essential to confirm the present findings.
Acknowledgements
The present study was supported by the Hubei Province health and family planning scientific research project (China; grant no. WJ2017M001) and the Natural Science Foundation of Hubei Province of China (grant no. 2016CFB457).
References
- 1.Forouzanfar MH, Foreman KJ, Delossantos AM, Lozano R, Lopez AD, Murray CJ, Naghavi M. Breast and cervical cancer in 187 countries between 1980 and 2010 A systematic analysis. Lancet. 2011;378:1461–1484. doi: 10.1016/S0140-6736(11)61351-2. [DOI] [PubMed] [Google Scholar]
- 2.Cesur Ö, Nicol C, Groves H, Mankouri J, Blair GE, Stonehouse NJ. The subcellular localisation of the human papillomavirus (HPV) 16 E7 protein in cervical cancer cells and its perturbation by RNA aptamers. Viruses. 2015;7:3443–3461. doi: 10.3390/v7072780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.zur Hausen H. Papillomaviruses in the causation of human cancers-a brief historical account. Virology. 2009;384:260–265. doi: 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]
- 4.Jensen KE, Schmiedel S, Frederiksen K, Norrild B, Iftner T, Kjaer SK. Risk for cervical intraepithelial neoplasia grade 3 or worse in relation to smoking among women with persistent human papillomavirus infection. Cancer Epidemiol Biomarkers Prev. 2012;21:1949–1955. doi: 10.1158/1055-9965.EPI-12-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johansson H, Bjelkenkrantz K, Darlin L, Dilllner J, Forslund O. Presence of high-risk HPV mRNA in relation to future high-grade lesions among high-risk HPV DNA positive women with minor cytological abnormalities. PloS One. 2015;10:e0124460. doi: 10.1371/journal.pone.0124460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naucler P, Ryd W, Törnberg S, Strand A, Wadell G, Elfgren K, Rådberg T, Strander B, Johansson B, Forslund O, et al. Human papillomavirus and Papanicolaou tests to screen for cervical cancer. N Engl J Med. 2007;357:1589–1597. doi: 10.1056/NEJMoa073204. [DOI] [PubMed] [Google Scholar]
- 7.Galloway DA. Human papillomaviruses: A growing field. Genes Dev. 2009;23:138–142. doi: 10.1101/gad.1765809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woodman CB, Collins SI, Young LS. The natural history of cervical HPV infection: Unresolved issues. Nat Rev Cancer. 2007;7:11–22. doi: 10.1038/nrc2050. [DOI] [PubMed] [Google Scholar]
- 9.Lee HS, Yun JH, Jung J, Yang Y, Kim BJ, Lee SJ, Yoon JH, Moon Y, Kim JM, Kwon YI. Identification of differentially-expressed genes by DNA methylation in cervical cancer. Oncol Lett. 2015;9:1691–1698. doi: 10.3892/ol.2015.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ivey KN, Srivastava D. MicroRNAs as regulators of differentiation and cell fate decisions. Cell Stem Cell. 2010;7:36–41. doi: 10.1016/j.stem.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 11.Sirotkin AV, Lauková M, Ovcharenko D, Brenaut P, Mlyncek M. Identification of microRNAs controlling human ovarian cell proliferation and apoptosis. J Cell Physiol. 2010;223:49–56. doi: 10.1002/jcp.21999. [DOI] [PubMed] [Google Scholar]
- 12.Davis S, Meltzer PS. GEOquery: A bridge between the gene expression omnibus (GEO) and BioConductor. Bioinformatics. 2007;23:1846–1847. doi: 10.1093/bioinformatics/btm254. [DOI] [PubMed] [Google Scholar]
- 13.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. NCBI GEO Archive for functional genomics data sets-update. Nucleic Acids Res. 2013;41(Database issue):D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiao X, Sherman BT, da W Huang, Stephens R, Baseler MW, Lane HC, Lempicki RA. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics. 2012;28:1805–1806. doi: 10.1093/bioinformatics/bts251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiao F, Zuo Z, Cai G, Kang S, Gao X, Li T. miRecords: An integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37(Database issue):D105–D110. doi: 10.1093/nar/gkn851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130–146. doi: 10.1038/nrd4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santamaria D, Barrière C, Cerqueira A, Hunt S, Tardy C, Newton K, Cáceres JF, Dubus P, Malumbres M, Barbacid M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448:811–815. doi: 10.1038/nature06046. [DOI] [PubMed] [Google Scholar]
- 21.Zhang W, Chen H, Chen Y, Liu J, Wang X, Yu X, Chen JJ, Zhao W. Cancerous inhibitor of protein phosphatase 2A contributes to human papillomavirus oncoprotein E7-induced cell proliferation via E2F1. Oncotarget. 2015;6:5253–5262. doi: 10.18632/oncotarget.2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santin AD, Zhan F, Bignotti E, Siegel ER, Cané S, Bellone S, Palmieri M, Anfossi S, Thomas M, Burnett A, et al. Gene expression profiles of primary HPV16- and HPV18-infected early stage cervical cancers and normal cervical epithelium: Identification of novel candidate molecular markers for cervical cancer diagnosis and therapy. Virology. 2005;331:269–291. doi: 10.1016/j.virol.2004.09.045. [DOI] [PubMed] [Google Scholar]
- 23.Tornesello ML, Buonaguro L, Giorgi-Rossi P, Buonaguro FM. Viral and cellular biomarkers in the diagnosis of cervical intraepithelial neoplasia and cancer. Biomed Res Int. 2013;2013:519619. doi: 10.1155/2013/519619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Del Pino M, Svanholm-Barrie C, Torné A, Marimon L, Gaber J, Sagasta A, Persing DH, Ordi J. mRNA biomarker detection in liquid-based cytology: A new approach in the prevention of cervical cancer. Mod Pathol. 2015;28:312–320. doi: 10.1038/modpathol.2014.106. [DOI] [PubMed] [Google Scholar]
- 25.Gibbons D, Fogt F, Kasznica J, Holden J, Nikulasson S. Comparison of topoisomerase II alpha and MIB-1 expression in uterine cervical squamous lesions. Mod Pathol. 1997;10:409–413. [PubMed] [Google Scholar]
- 26.Sahasrabuddhe VV, Luhn P, Wentzensen N. Human papillomavirus and cervical cancer: Biomarkers for improved prevention efforts. Future Microbiol. 2011;6:1083–1098. doi: 10.2217/fmb.11.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dahlhaus M, Burkovski A, Hertwig F, Mussel C, Volland R, Fischer M, Debatin KM, Kestler HA, Beltinger C. Boolean modeling identifies Greatwall/MASTL as an important regulator in the AURKA network of neuroblastoma. Cancer Lett. 2016;371:79–89. doi: 10.1016/j.canlet.2015.11.025. [DOI] [PubMed] [Google Scholar]
- 28.Umene K, Banno K, Kisu I, Yanokura M, Nogami Y, Tsuji K, Masuda K, Ueki A, Kobayashi Y, Yamagami W, et al. Aurora kinase inhibitors: Potential molecular-targeted drugs for gynecologic malignant tumors. Biomed Rep. 2013;1:335–340. doi: 10.3892/br.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun JM, Yang LN, Xu H, Chang B, Wang HY, Yang G. Inhibition of Aurora A promotes chemosensitivity via inducing cell cycle arrest and apoptosis in cervical cancer cells. Am J Cancer Res. 2015;5:1133–1145. [PMC free article] [PubMed] [Google Scholar]
- 30.Odrowaz Z, Sharrocks AD. The ETS transcription factors ELK1 and GABPA regulate different gene networks to control MCF10A breast epithelial cell migration. PloS One. 2012;7:e49892. doi: 10.1371/journal.pone.0049892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009;10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- 32.Stangel D, Erkan M, Buchholz M, Gress T, Michalski C, Raulefs S, Friess H, Kleeff J. Kif20a inhibition reduces migration and invasion of pancreatic cancer cells. J Surg Res. 2015;197:91–100. doi: 10.1016/j.jss.2015.03.070. [DOI] [PubMed] [Google Scholar]
- 33.Zhai Y, Bommer GT, Feng Y, Wiese AB, Fearon ER, Cho KR. Loss of estrogen receptor 1 enhances cervical cancer invasion. Am J Pathol. 2010;177:884–895. doi: 10.2353/ajpath.2010.091166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sood S, Patel FD, Ghosh S, Arora A, Dhaliwal LK, Srinivasan R. Epigenetic alteration by DNA methylation of ESR1, MYOD1 and hTERT gene promoters is useful for prediction of response in patients of locally advanced invasive cervical carcinoma treated by chemoradiation. Clin Oncol (R Coll Radiol) 2015;27:720–727. doi: 10.1016/j.clon.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 35.Zhi W, Ferris D, Sharma A, Purohit S, Santos C, He M, Ghamande S, She JX. Twelve serum proteins progressively increase with disease stage in squamous cell cervical cancer patients. Int J Gynecol Cancer. 2014;24:1085–1092. doi: 10.1097/IGC.0000000000000153. [DOI] [PubMed] [Google Scholar]
- 36.Gadducci A, Guerrieri ME, Greco C. Tissue biomarkers as prognostic variables of cervical cancer. Crit Rev Oncol Hematol. 2013;86:104–129. doi: 10.1016/j.critrevonc.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 37.Zhao S, Yao DS, Chen JY, Ding N. Aberrant expression of miR-20a and miR-203 in cervical cancer. Asian Pac J Cancer Prev. 2013;14:2289–2293. doi: 10.7314/APJCP.2013.14.4.2289. [DOI] [PubMed] [Google Scholar]
- 38.Zhang K, Dai L, Zhang B, Xu X, Shi J, Fu L, Chen X, Li J, Bai Y. miR-203 is a direct transcriptional target of E2F1 and causes G1 arrest in esophageal cancer cells. J Cell Physiol. 2015;230:903–910. doi: 10.1002/jcp.24821. [DOI] [PubMed] [Google Scholar]
- 39.Zhu X, Er K, Mao C, Yan Q, Xu H, Zhang Y, Zhu J, Cui F, Zhao W, Shi H. miR-203 suppresses tumor growth and angiogenesis by targeting VEGFA in cervical cancer. Cell Physiol Biochem. 2013;32:64–73. doi: 10.1159/000350125. [DOI] [PubMed] [Google Scholar]
- 40.Wang F, Li Y, Zhou J, Xu J, Peng C, Ye F, Shen Y, Lu W, Wan X, Xie X. miR-375 is down-regulated in squamous cervical cancer and inhibits cell migration and invasion via targeting transcription factor SP1. Am J Pathol. 2011;179:2580–2588. doi: 10.1016/j.ajpath.2011.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]