

Abstract
PUMA (p53 upregulated modulator of apoptosis), a member of the B-cell lymphoma 2 (Bcl-2) protein family, is a pro-apoptotic protein. PUMA expression is modulated by the tumor suppressor p53. PUMA has a role in rapid cell death via p53-dependent and -independent mechanisms. To evaluate whether p53 is required for PUMA-mediated apoptosis in prostate cancer cells, p53 protein was silenced in human prostate cancer PC-3 cells by using p53 small interfering RNA (siRNA). The interference efficiency of p53 on RNA and protein levels was detected by reverse transcription-quantitative polymerase chain reaction and western blotting. Cell proliferation and p21 expression were subsequently examined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and western blot analysis, respectively. p53-silenced or control PC-3 cells were transfected with pCEP4-(hemagglutinin)-PUMA plasmid, or non-carrier plasmid. Enzyme-linked immunosorbent assay was used to determine cell apoptosis by measuring histone release and caspase-3 activation, and MTT assay was used to measure cell viability. In addition, the expression of pro-apoptosis protein Bax and anti-apoptosis protein Bcl-2 were evaluated. The results of the present study revealed that p53 siRNA significantly suppressed p53 RNA and protein expression in PC-3 cells. Deficiency of p53 increased the cell growth rate and decreased p21 expression. However, PUMA overexpression remained able to induce apoptosis in p53-silenced and control cells by increasing Bax expression and decreasing Bcl-2 expression, leading to the activation of caspase-3. These results suggest that PUMA may mediate apoptosis of prostate cancer PC-3 cells, potentially independently of p53. Furthermore, PUMA gene treatment to induce cancer cell apoptosis may be more efficient compared with p53-dependent apoptosis, where loss of p53 expression or function may lead to limited efficacy of PUMA expression. Therefore, the present study proposes the significant hypothesis that increasing PUMA expression may be an effective approach for the treatment of prostate cancer, regardless of p53 status.
Keywords: p53 upregulated modulator of apoptosis, p53, prostate cancer, apoptosis, caspase-3, B-cell lymphoma 2, Bcl-2-like protein 4
Introduction
Prostate cancer is one of the most common types of cancer in men, with >40,000 new cases diagnosed every year in the UK (1). Prostate cancer is typically slow-growing and the majority of men do not notice the symptoms until the cancer has become large enough to press against the urethra and interfere with urination. The standard treatment approach for prostate cancer is surgical removal of the gland or prostatectomy, coupled with chemotherapy and radiotherapy (1). These approaches may be followed by hormone therapy, which interrupts the cancer cells' supply of testosterone, and therefore inhibits their ability to grow. The high incidence of prostate cancer and the lack of treatment efficacy of traditional treatment seriously affects the normal life of men (1).
Tumors are caused by abnormal cell proliferation, differentiation and apoptosis due to the activation of certain proto-oncogenes, inactivation of tumor suppressors and changes to apoptosis-associated genes (2,3). p53 upregulated modulator of apoptosis (PUMA), also known as Bcl-2-binding component 3 (BBC3), is a pro-apoptotic protein and is a member of the B-cell lymphoma 2 (Bcl-2) protein family (4,5). The underlying mechanism of PUMA-mediated apoptosis has been extensively evaluated (6–9). The tumor suppressor protein p53 has a dual role associated with PUMA gene expression and function. The efficiency of PUMA as an apoptosis-inducing protein and its association with p53 depends on the cell lineage, the status of p53 (deficiency vs. mutation) and the type of stimulus. Thus, PUMA activates apoptosis through p53-dependent and -independent signaling pathways, induced by a range of signals (10–13). In the majority of cell types, p53 expression results in increased PUMA gene expression, and subsequent PUMA-mediated apoptosis requires functional p53. The majority of PUMA-induced apoptosis occurs via activation of p53 (10–12). p53 is activated by survival signals, including glucose deprivation (14), which leads to an increase in the expression levels of PUMA. PUMA expression leads to apoptosis by displacement of p53 from Bcl-extra large (xL), allowing p53 to increase mitochondrial permeability (15). The subsequent increase in PUMA levels is able to induce apoptosis via mitochondrial dysfunction. p53, as well as PUMA, is activated as a result of DNA damage caused by a range of genotoxic agents. Alternative agents that are able to induce p53-dependent apoptosis are neurotoxins (16,17), proteasome inhibitors (18), microtubule poisons (19) and transcription inhibitors (20). However, in certain cell types, PUMA apoptosis may additionally be induced independently of p53 activation by alternative stimuli, including oncogenic stress (21,22), growth factor and/or cytokine withdrawal and kinase inhibition, ER stress, altered redox status, ischemia, immune modulation, and infection (23,24). Regardless of the method of signaling pathway activation, PUMA interacts with the anti-apoptotic proteins Bcl-2, Bcl-xL, Bcl-W and myeloid cell leukemia 1, via its BH3 structural domain, to induce cell apoptosis (5,25) and obstructs the interaction of these proteins with the pro-apoptotic molecules Bcl-2-like protein 4 (Bax) and Bcl-2 homologous antagonist/killer (Bak), therefore freeing Bax and/or Bak, which are subsequently able to signal apoptosis to the mitochondria (5,23,25). Following mitochondrial dysfunction, the caspase cascade is activated, which leads to cell death (23).
A number of studies have demonstrated that PUMA functioning is altered or absent in cancer cells (26,27). In addition, the loss of p53-mediated apoptosis has been implicated as a significant occurrence during tumor progression (26,28). The majority of cancer types exhibit p53 gene deficiency or mutations, meaning that the application of gene therapies that target this gene is not possible (28). However, an alternative potential method may involve focusing on PUMA as a therapeutic target, leading to the induction of apoptosis in cancer cells. In the present study, the influence of exogenous PUMA on proliferation and apoptosis was investigated in prostate cancer PC-3 cells, and it was determined whether PUMA requires functional p53 in these cells.
Materials and methods
Cell culture and treatments
PC-3 human prostate adenocarcinoma cancer cells were purchased from American Type Culture Collection (Manassas, VA, USA). Cells were cultured with RPMI-1640 supplemented with 10% heat-inactivated fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA). Cells were maintained in culture at 37°C in a humidified atmosphere of 5% CO2 and 95% humidity.
Scrambled RNA and p53 small interfering RNA (siRNA) were obtained from Dharmacon (GE Healthcare Life Sciences, Chalfont, UK). pCEP4-[hemagglutinin (HA)] 2-PUMA recombinant plasmid and non-carrier plasmid pCEP4-(HA) 2-C1 were kindly provided by Dr B. Vogelstein (Johns Hopkins Oncology Center, Baltimore, MD, USA). A total of 2×106 cells were transfected by scrambled RNA or p53 siRNA using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) for 72 h according to the manufacturer's protocol. Subsequently, the cells were cultured in 100-mm dishes and transfected by PUMA or pCEP4 plasmid and left to grow for 24 h, prior to detection.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
RNA was isolated from PC-3 cells with the Total RNA Isolation kit (A&A Biotechnology, Gdynia, Poland) according to the manufacturer's protocol. First strand complementary DNA (cDNA) was obtained by reverse transcription of 1 µg of total RNA with the RevertAid First Strand cDNA synthesis kit (Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. Amplification of the cDNA was performed using the TaqMan® Gene Expression Assay (Applied Biosystems; Thermo Fisher Scientific, Inc.), with fluorogenic fluorescein amidite-labeled probes, sequence-specific primers of gene coding p53 and the internal control glyceraldehyde-3-phosphate dehydrogenase (GAPDH), according to the manufacturer's protocol. Samples with no cDNA or no reverse transcription were used as controls. Genes were amplified by a first step of 120 sec at 95°C, followed by 45 cycles of 30 sec at 95°C, 30 sec at 60°C and 30 sec at 72°C. The real-time fluorescence detection was performed with the ABI PRISM 7700 Sequence Detector (Perkin-Elmer Applied Biosystem; Thermo Fisher Scientific, Inc.). Fold differences in p53 expression, normalized to the level of GAPDH, were calculated with the formula 2ΔΔCq (29). Relative quantities of messenger RNA (mRNA) in siRNA-treated cells were indicated as a percentage of the amount of mRNA in the untreated cells.
Western blotting
Cells were lysed in cell lysis buffer (50 mM HEPES, pH 8.0; 150 mM NaCl; 1% Triton X-100; 10% glycerol; 1 mM MgCl2; 1.5 mM ethylenediaminetetraacetic acid; 20 mM β-glycerophosphate; 50 mM NaF; 1 mM Na3VO4; 10 µg/ml aprotonin; 1 µM pepstatin A and 1 mM phenylmethylsulfonyl fluoride; catalogue no. P0013; Beyotime Institute of Biotechnology, Haimen, China) containing a protease inhibitor cocktail (Roche Applied Science, Madison, WI, USA). Proteins were isolated from the total cells using 14,000 × g centrifugation for 10 min at 4°C and the supernatant was collected. Protein samples (50 µg) were separated by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to Immobilon-P polyvinyl difluoride membranes (EMD Millipore, Billerica, MA, USA). The membrane was blocked with 5% non-fat dried milk in Tris-buffered saline with Tween 20 for 1 h at room temperature and then incubated overnight at 4°C with mouse monoclonal anti-p53 (1:1,000 dilution; catalog no. sc-126), anti-HA tag (1:2,000 dilution; catalog no. sc-7392), rabbit polyclonal anti-p21 (1:2,000 dilution; catalog no. sc-397), anti-Bcl-2 (1:2,000 dilution; catalog no. sc-783), anti-Bax (1:2,000 dilution; catalog no. sc-526) and anti-GAPDH (1:2,000 dilution; catalog no. sc-25778) antibodies (Santa Cruz Biotechnology Inc., Dallas, TX, USA). Goat anti-mouse (1:2,000 dilution; catalog no. sc-2005) and goat anti-rabbit immunoglobulin G (1:2,000 dilution; catalog no. sc-2004) were used for incubation for 2 h at room temporature. Enhanced chemiluminescence-detecting reagent (GE Healthcare Life Sciences) was used to visualize the membranes. The protein blots were quantified by densitometry using QuantityOne software (Bio-Rad Laboratories, Inc., Hercules, CA, USA), and the amounts were expressed relative to the internal reference GAPDH.
Cell proliferation assay
Cell proliferation was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, cells were seeded (1×104 cells per well) into a 96-well flat-bottom plate following transfection. Cells were cultured at 37°C in an atmosphere of 5% CO2 for 48 h, followed by an additional 4 h of incubation subsequent to the addition of 20 µl 5 mg/ml MTT (Sigma-Aldrich; Merck Millipore, Darmstadt, Germany) to each well. Cells were lysed by addition of 200 µl dimethylsulfoxide (Sigma-Aldrich; Merck Millipore). Absorbance was measured at 490 nm with an enzyme-linked immunosorbent assay (ELISA) reader (Tecan Austria GmbH, Grödig, Austria).
Cell apoptosis assay
Apoptosis was assessed using a Cell Death Detection ELISAPLUS kit (Sigma-Aldrich; Merck Millipore). PC-3 cells (4×103) were seeded into each well of a 96-well plate following transfection. A total of 9 h later, samples were collected and ELISA was performed in accordance with the manufacturer's protocol. The results are presented as the fold induction compared with the control. To confirm the role of apoptosis, caspase-3 activation was additionally determined in transfected cells using the Caspase-3 (Active) Human ELISA kit (Thermo Fisher Scientific, Inc.). PUMA or non-carrier plasmids were transfected into p53 siRNA-transfected or scrambled siRNA-transfected cells. The cells were subsequently cultured at 4.5×105 cells per well in 6-well plates. A total of 8 h later, the cells were lysed and assayed for western blotting.
Statistical analysis
SPSS (version 11.0; SPSS, Inc., Chicago, IL, USA) was used to analyze the experimental data. Data are presented as the mean ± standard error. Statistical significance was analyzed by Student's t-test using SPSS 11.0 software (SPSS, Inc., Chicago, IL, USA). All experiments were repeated at least 3 times. P<0.05 was considered to indicate a statistically significant difference.
Results
Silencing of p53 protein in prostate cancer PC-3 cells
To determine whether siRNA was able to knockdown p53 expression in cultured prostate cancer PC-3 cells, p53 RNA expression was examined by RT-qPCR (Fig. 1A) and protein expression was detected by western blotting (Fig. 1B). A representative time course and dose response is presented in Fig. 1B. The results demonstrated that the relative p53 mRNA amount was significantly decreased in p53 siRNA-treated cells relative to non-silencing scrambled siRNA-transfected cells (P=0.002; Fig. 1A), and the residual expression of p53 protein in the cells with 2 µg of siRNA transfected for 72 h was markedly suppressed. For the subsequent experiments, 2 µg of siRNA transfected for 72 h was selected as the optimum condition.
Figure 1.
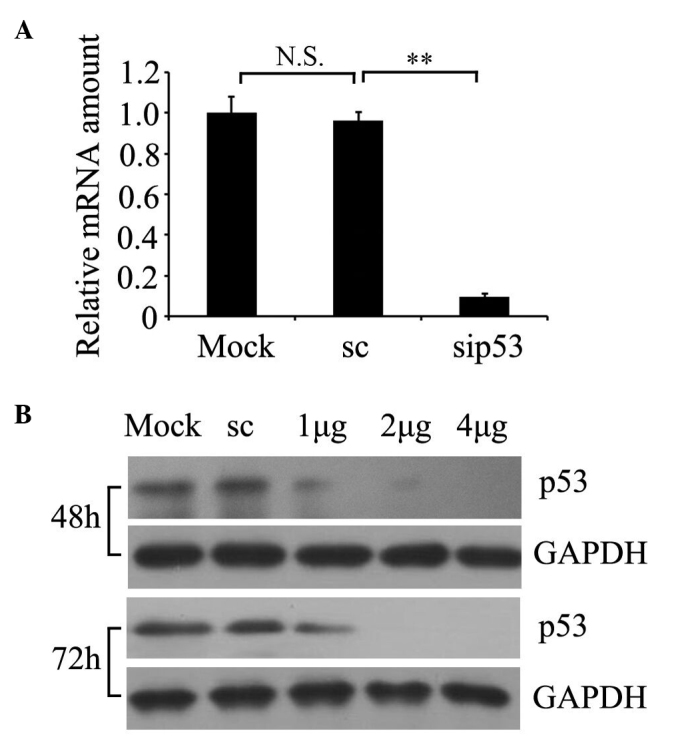
siRNA-mediated downregulation of p53. PC-3 cells were treated with siRNA duplexes or non-silencing scrambled siRNA. Mock-transfected cells were treated in the same manner except that no siRNA was added. (A) Relative amounts of p53 mRNA 48 h subsequent to cell transfection with 2 µg siRNA were assessed by reverse transcription-quantitative polymerase chain reaction. **P<0.01 vs. non-silencing scrambled siRNA (Student's t-test). (B) Western blot analysis of changes in p53 protein expression level in lysates of PC-3 cells treated with 1 µg, 2 µg and 4 µg siRNA for 48 h or 72 h. Western blot analysis was performed with p53 and GAPDH antibodies. siRNA, small interfering RNA; sip53, siRNA duplexes; sc, non-silencing scrambled siRNA; mRNA, messenger RNA; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; N.S., no significance.
Effect of p53 deficiency on cellular function
The functional effects of p53 knockdown were assessed by detection of cell proliferation and p21 protein expression, which is normally induced by p53 (30). The results revealed that the growth of prostate cancer PC-3 cells was increased following p53 siRNA transfection compared with the mock-transfected cells (P=0.007; Fig. 2A) and the expression of p21 protein was blocked in p53-silenced cells (Fig. 2B). These data confirmed that p53 deficiency with siRNA contributed to relevant functional alterations, which were consistent with p53 interference.
Figure 2.
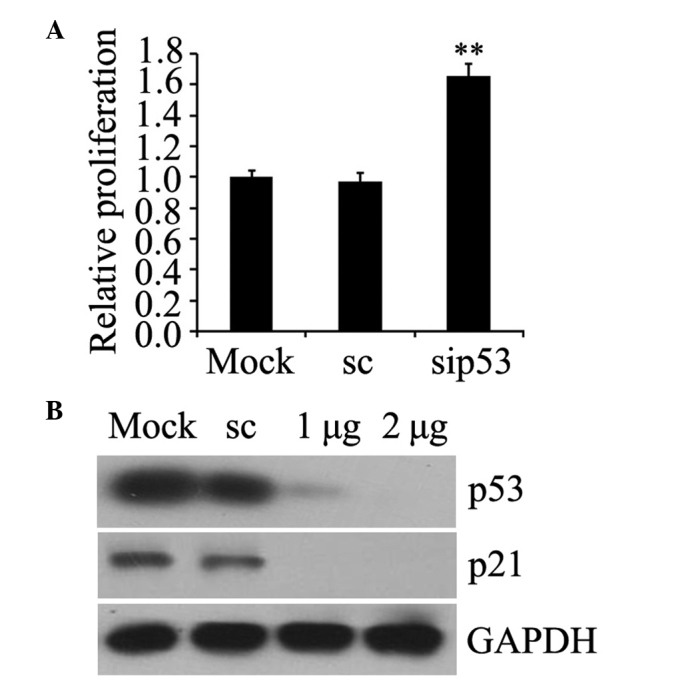
Effect of p53 deficiency on the functioning of PC-3 cells. (A) p53 was knocked down by siRNA and cell growth was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. The fold induction in p53 siRNA-transfected PC-3 cells is shown relative to mock-transfected cells. **P<0.01 (Student's t-test) for p53 siRNA-transfected cells compared with scrambled siRNA. Data are presented as the mean ± standard error. (B) p53 was knocked down by using siRNA, and p21 protein expression was determined by western blot analysis. p21 expression decreased in p53-silenced cells. siRNA, small interfering RNA; sc, scrambled siRNA; sip53, siRNA duplexes; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
PUMA induces apoptosis independently of p53
To confirm whether p53 was required for PUMA-induced apoptosis, prostate cancer PC-3 cells were transfected with p53 siRNA. Once p53 reached its minimum level of expression 72 h subsequent to transfection, cells were transfected with pCEP4-(HA)-PUMA, or empty pCEP4 plasmids. As demonstrated in Fig. 3A and B, PUMA-induced apoptosis was assessed by measuring cell viability or histone release. The results indicated that p53-silenced cells did not show significant differences in PUMA-induced apoptosis levels compared with scrambled siRNA-transfected cells (P=0.095 and P=0.126; Fig. 3A and B, respectively). PUMA-induced apoptosis in scrambled siRNA-transfected cells was similar to that in p53 siRNA-transfected cells. As additional evidence of PUMA-mediated apoptosis, caspase-3 activation was evaluated in p53-deficient PC-3 cells. The data demonstrated that PUMA induced higher caspase-3 activation in p53-deficient PC-3 cells compared with scrambled siRNA-transfected cells (P=0.045 and P=0.021; Fig. 3C), suggesting that PUMA did not require the participation of p53 to induce this apoptosis signaling pathway.
Figure 3.
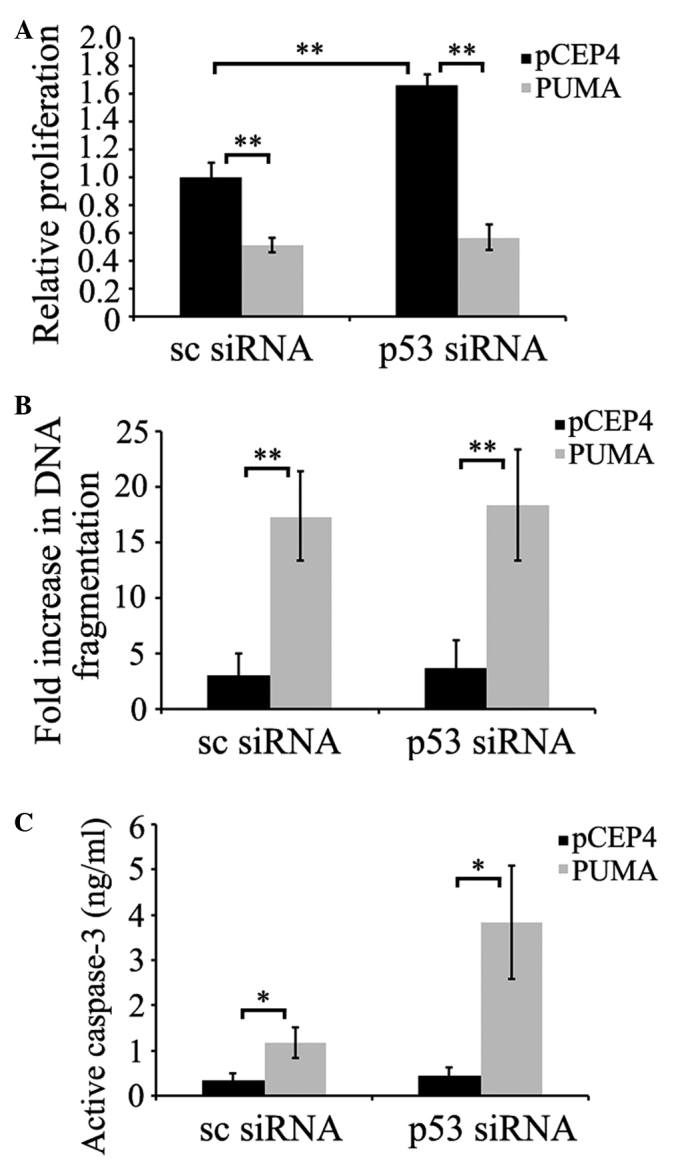
PUMA induces apoptosis in p53-deficient PC-3 cells. PC-3 cells were treated with 2 µg siRNA for 72 h, and subsequently were transfected with 8 µg PUMA or pCEP4 plasmid. (A) PUMA-induced apoptosis was initially evaluated by inhibition of cell survival. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay was performed 24 h subsequent to complementary DNA transfection in siRNA-treated PC-3 cells. The fold inductions are shown relative to mock-transfected cells. (B) DNA fragmentation was determined by histone release. Histone release was measured by enzyme-linked immunosorbent assay in samples collected 9 h subsequent to the second transfection. The fold induction of DNA fragmentation in PC-3 cells transfected with PUMA plasmids is shown relative to the control value of pCEP4-transfected cells. (C) Caspase-3 activation was determined in control and p53-deficient PC-3 cells 8 h subsequent to transfection with PUMA. *P<0.05, **P<0.01. PUMA, p53 upregulated modulator of apoptosis; siRNA, small interfering RNA; sc, control.
Influence of exogenous PUMA on Bcl-2 and Bax protein expression in human prostate cancer PC-3 cells
The western blot analysis results revealed HA-PUMA protein expression in scrambled siRNA-transfected and p53 siRNA-transfected cells, indicating successful transfection (Fig. 4A). Compared with the control transfected with empty pCEP4 plasmid, the PUMA group demonstrated significantly increased expression of the pro-apoptotic protein Bax and significantly reduced expression of the anti-apoptotic protein Bcl-2, regardless of p53 deficiency (Fig. 4A). In addition, the Bax/Bcl-2 ratios were significantly increased compared with the control with empty plasmid pCEP4 (P=0.003 and P=0.002; Fig. 4B). These results verified that PUMA was able to engage the apoptotic signaling pathway by modulating Bcl-2 and Bax protein expression independently of p53.
Figure 4.
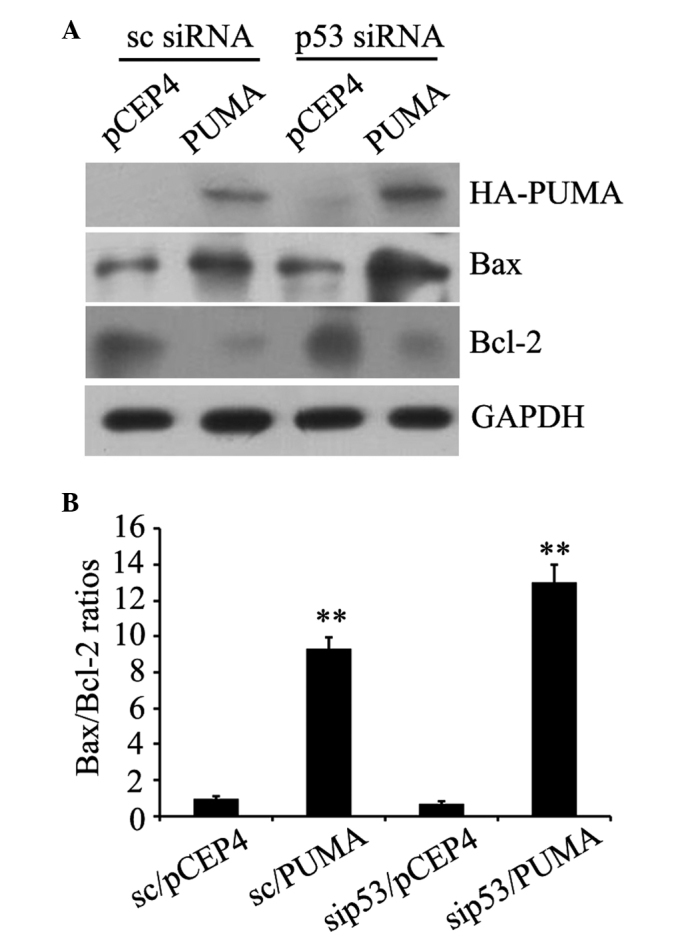
Influence of exogenous PUMA on Bcl-2 and Bax protein expression in human prostate cancer PC-3 cells. (A) Exogenous PUMA expression, and alterations in Bax or Bcl-2 protein expression, in control and p53-deficient PC-3 cells were assessed by western blot analysis, respectively, with anti-HA, anti-Bax or anti-Bcl-2 antibodies. (B) Bax/Bcl-2 ratios were counted by the quantitation of Bax protein blots divided by the quantitation of Bcl-2 protein blots. **P<0.01 vs. pCET4 empty vector-transfected control cells. Data are presented as the mean ± standard error. PUMA, p53 upregulated modulator of apoptosis; Bcl-2, B-cell lymphoma 2; Bax, bcl-2-like protein 4; HA, hemagglutinin; sc, control; siRNA, small interfering RNA; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; sip53; siRNA duplexes.
Discussion
Prostate cancer is one of the most common malignant tumors in men (1). Chemotherapy kills tumor cells by activating apoptosis, while apoptotic signaling pathway defects are associated with tumor resistance to chemotherapy. Due to side effects and tumor cell insensitivities, the clinical application of chemotherapeutic drugs may be greatly restricted (31). A number of studies concerning gene therapy have attracted widespread attention (32–34). Therefore, searching for ideal target genes has become a significant issue to address.
The introduction of apoptotic genes has the potential to increase sensitivity to chemotherapy drugs. The loss of p53-mediated apoptosis has been implicated as a significant event in tumor progression. p53 is able to induce or potentiate apoptosis via a number of mechanisms, including by regulating the expression of genes that are able to participate in the apoptotic response and via transcriptionally independent means. p53 is a notable potential therapeutic gene as it is able to induce apoptosis in a range of cell types. Defects in p53 structure and function in specific unhealthy cells have been described, suggesting that forced expression of this tumor suppressor protein may be beneficial (35–38). However, enhancing p53 gene expression has been observed to have only modest efficacy (39). PUMA has a significant role in apoptotic signaling pathways, is rapidly induced by p53 and has powerful apoptosis-promoting effects. Therefore, a possible explanation for the limited efficacy of p53 gene enhancement may be that p53 did not readily induce apoptosis, potentially because PUMA expression was not increased (40). Thus, directly targeting PUMA may be an effective treatment approach to cause rapid cell death. In vitro studies have demonstrated that PUMA affects tumor cell proliferation and induces cellular apoptosis independently of p53, which has led to hopes of using PUMA for tumor therapy (41,42). Yu et al (43) reported that PUMA induced lung cancer cell apoptosis and inhibited cell proliferation via caspase activation and cytochrome c release in non-chemoradiotherapy sensitive lung cancer cells, providing evidence that PUMA is able to increase sensitivity to radiotherapy and chemotherapy drugs.
In the present study, using siRNA to decrease p53 expression demonstrated that prostate cancer PC-3 cells are sensitive to PUMA-induced death. Western blot analysis revealed that, following PUMA gene transfection, pro-apoptotic protein Bax significantly increased and anti-apoptotic protein Bcl-2 significantly decreased. In the Bcl-2 family, the ratio of pro-apoptotic to anti-apoptotic proteins is a significant factor in determining the occurrence and level of apoptosis (44). The proportion of pro-apoptotic proteins in cells additionally determines the cellular response to death signals and cell fate. It has been reported that decreased Bax protein expression is associated with the sensitivity of tumor cells to chemotherapy and the length of patient survival times (45,46). Therefore, the results of the present study suggested that PUMA was an effective mediator of apoptosis, regardless of the p53 status in PC-3 cells.
In conclusion, the results of the present study imply that PUMA is able to efficiently decrease the growth of prostate cancer PC-3 cells by promoting apoptosis independently of p53. This supports the potential use of PUMA as a novel and promising target for prostate cancer therapy. A number of cancer types exhibit deletions or mutations in p53, so PUMA shows great significance in cancer treatment. In future studies, we will verify whether the phenomenon exists in other cell lines of prostate cancer. Furthermore, the potential functions of PUMA in the chemosensitivity and treatment of protate cancer remain to be investigated.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (grant no. 81302234), the Shandong Provincial Natural Science Foundation, China (Grant no. ZR2016HP38), Projects of Medical and Health Technology Development program in Shandong province (Grant no. 2016WS0714), Science and Technology Project of Yantai (grant nos. 2016ZH084 and 2016WS018) and the Scientific research startup Project of Binzhou Medical University (grant no. BY2014KYQD24).
References
- 1.Mandal A: Prostate Cancer, corp-author. NEWS MEDICAL. http://www.news-medical.net/health/Prostate-Cancer.aspx. The Latest Developments in Life Sciences & Medicine. 2015 Feb 27; Accessed. [Google Scholar]
- 2.Tsai SC, Lu CC, Lee CY, Lin YC, Chung JG, Kuo SC, Amagaya S, Chen FN, Chen MY, Chan SF, Yang JS. AKT serine/threonine protein kinase modulates bufalin-triggered intrinsic pathway of apoptosis in CAL 27 human oral cancer cells. Int J Oncol. 2012;41:1683–1692. doi: 10.3892/ijo.2012.1605. [DOI] [PubMed] [Google Scholar]
- 3.Umit UM, Berna T, Handan K, Ipek E, Berrak Y, Can E, Bahadir GM. Role of melatonin and luzindole in rat mammary cancer. J Invest Surg. 2012;25:345–353. doi: 10.3109/08941939.2012.665570. [DOI] [PubMed] [Google Scholar]
- 4.Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/S1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 5.Han J, Flemington C, Houghton AB, Gu Z, Zambetti GP, Lutz RJ, Zhu L, Chittenden T. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc Natl Acad Sci USA. 2001;98:11318–11323. doi: 10.1073/pnas.201208798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu H, Li W, Yu X, Gao F, Duan Z, Ma X, Tan S, Yuan Y, Liu L, Wang J, et al. EZH2-mediated Puma gene repression regulates non-small cell lung cancer cell proliferation and cisplatin-induced apoptosis. Oncotarget. 2016 Jul 26; doi: 10.18632/oncotarget.10841. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jang Y, Kim J, Ko JW, Kwon YH. Homocysteine induces PUMA-mediated mitochondrial apoptosis in SH-SY5Y cells. Amino Acids. 2016;48:2559–2569. doi: 10.1007/s00726-016-2280-6. [DOI] [PubMed] [Google Scholar]
- 8.Ma J, Feng Y, Liu Y, Li X. PUMA and survivin are involved in the apoptosis of HepG2 cells induced by microcystin-LR via mitochondria-mediated pathway. Chemosphere. 2016;157:241–249. doi: 10.1016/j.chemosphere.2016.05.051. [DOI] [PubMed] [Google Scholar]
- 9.Sun Y, Xia P, Zhang H, Liu B, Shi Y. P53 is required for Doxorubicin-induced apoptosis via the TGF-beta signaling pathway in osteosarcoma-derived cells. Am J Cancer Res. 2015;6:114–125. [PMC free article] [PubMed] [Google Scholar]
- 10.Wang P, Yu J, Zhang L. The nuclear function of p53 is required for PUMA-mediated apoptosis induced by DNA damage. Proc Natl Acad Sci USA. 2007;104:4054–4059. doi: 10.1073/pnas.0700020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/S1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 12.Avila JL, Grundmann O, Burd R, Limesand KH. Radiation-induced salivary gland dysfunction results from p53-dependent apoptosis. Int J Radiat Oncol Biol Phys. 2009;73:523–529. doi: 10.1016/j.ijrobp.2008.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niizuma K, Endo H, Nito C, Myer DJ, Chan PH. Potential role of PUMA in delayed death of hippocampal CA1 neurons after transient global cerebral ischemia. Stroke. 2009;40:618–625. doi: 10.1161/STROKEAHA.108.524447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Y, Coloff JL, Ferguson EC, Jacobs SR, Cui K, Rathmell JC. Glucose metabolism attenuates p53 and Puma-dependent cell death upon growth factor deprivation. J Biol Che. 2008;283:36344–36353. doi: 10.1074/jbc.M803580200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309:1732–1735. doi: 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- 16.Gomez-Lazaro M, Galindo MF, Fernandez-Gomez FJ, Prehn JH, Jordán J. Activation of p53 and the pro-apoptotic p53 target gene PUMA during depolarization-induced apoptosis of chromaffin cells. Exp. Neurol. 2005;196:96–103. doi: 10.1016/j.expneurol.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 17.Wong HK, Fricker M, Wyttenbach A, Villunger A, Michalak EM, Strasser A, Tolkovsky AM. Mutually exclusive subsets of BH3-only proteins are activated by the p53 and c-Jun N-terminal kinase/c-Jun signaling pathways during cortical neuron apoptosis induced by arsenite. Mol Cell Biol. 2005;25:8732–8747. doi: 10.1128/MCB.25.19.8732-8747.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu J, Wang P, Ming L, Wood MA, Zhang L. SMAC/Diablo mediates the proapoptotic function of PUMA by regulating PUMA-induced mitochondrial events. Oncogene. 2007;26:4189–4198. doi: 10.1038/sj.onc.1210196. [DOI] [PubMed] [Google Scholar]
- 19.Giannakakou P, Nakano M, Nicolaou KC, O'Brate A, Yu J, Blagosklonny MV, Greber UF, Fojo T. Enhanced microtubule-dependent trafficking and p53 nuclear accumulation by suppression of microtubule dynamics. Proc Natl Acad Sci USA. 2002;99:10855–10860. doi: 10.1073/pnas.132275599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kalousek I, Brodska B, Otevrelova P, Röselova P. Actinomycin D upregulates proapoptotic protein Puma and downregulates Bcl-2 mRNA in normal peripheral blood lymphocytes. Anticancer Drugs. 2007;18:763–772. doi: 10.1097/CAD.0b013e3280adc905. [DOI] [PubMed] [Google Scholar]
- 21.Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B. Genomic targets of the human c-Myc protein. Genes Dev. 2003;17:1115–1129. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maclean KH, Keller UB, Rodriguez-Galindo C, Nilsson JA, Cleveland JL. c-Myc augments gamma irradiation-induced apoptosis by suppressing Bcl-XL. Mol Cell Biol. 2003;23:7256–7270. doi: 10.1128/MCB.23.20.7256-7270.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu J, Zhang L. PUMA, a potent killer with or without p53. Oncogene. 2008;27:S71–S83. doi: 10.1038/onc.2009.45. (Suppl 1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castedo M, Perfettini JL, Piacentini M, Kroemer G. p53 - A pro-apoptotic signal transducer involved in AIDS. Biochem Biophys Res Commun. 2005;331:701–706. doi: 10.1016/j.bbrc.2005.03.188. [DOI] [PubMed] [Google Scholar]
- 25.Yee KS, Vousden KH. Contribution of membrane localization to the apoptotic activity of PUMA. Apoptosis. 2008;13:87–95. doi: 10.1007/s10495-007-0140-2. [DOI] [PubMed] [Google Scholar]
- 26.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 27.Yu J, Zhang L. The transcriptional targets of p53 in apoptosis control. Biochem Biophys Res Commun. 2005;331:851–858. doi: 10.1016/j.bbrc.2005.03.189. [DOI] [PubMed] [Google Scholar]
- 28.Hao, Cho WC. Battle against cancer: An everlasting saga of p53. Int J Mol Sci. 2014;15:22109–22127. doi: 10.3390/ijms151222109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Livak and Schmittgen, corp-author. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCt method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 30.Pap T, Aupperle KR, Gay S, Firestein GS, Gay RE. Invasiveness of synovial fibroblasts is regulated by p53 in the SCID mouse in vivo model of cartilage invasion. Arthritis Rheum. 2001;44:676–681. doi: 10.1002/1529-0131(200103)44:3<676::AID-ANR117>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 31.Pommier Y, Sordet O, Antony S, Hayward RL, Kohn KW. Apoptosis defects and chemotherapy resistance: Molecular interaction maps and networks. Oncogene. 2004;23:2934–2949. doi: 10.1038/sj.onc.1207515. [DOI] [PubMed] [Google Scholar]
- 32.Hirscheler B. http://www.arabtimesonline.com/wp-content/uploads/pdf/2016/apr/03/29.pdf. Accessed. Europe gives green light to first gene therapy for children. 2016 Apr 13; [Google Scholar]
- 33.Coghlan A. Gene Therapy Approved. New Scientist. 2016;230:8–9. doi: 10.1016/S0262-4079(16)30593-0. [DOI] [Google Scholar]
- 34.Cyranoski D. Chinese scientists to pioneer first human CRISPR trial. Nature. 2016;535:476–477. doi: 10.1038/nature.2016.20302. [DOI] [PubMed] [Google Scholar]
- 35.Yamanishi Y, Boyle DL, Rosengren S, Green DR, Zvaifler NJ, Firestein GS. Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc Natl Acad Sci USA. 2002;99:10025–10030. doi: 10.1073/pnas.152333199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Firestein GS, Echeverri F, Yeo M, Zvaifler NJ, Green DR. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc Natl Acad Sci USA. 1997;94:10895–10900. doi: 10.1073/pnas.94.20.10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rème T, Travaglio A, Gueydon E, Adla L, Jorgensen C, Sany J. Mutations of the p53 tumour suppressor gene in erosive rheumatoid synovial tissue. Clin Exp Immunol. 1998;111:353–358. doi: 10.1046/j.1365-2249.1998.00508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kullmann F, Judex M, Neudecker I, Lechner S, Jüsten HP, Green DR, Wessinghage D, Firestein GS, Gay S, Schölmerich J, Müller-Ladner U. Analysis of the p53 tumor suppressor gene in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 1999;42:1594–1600. doi: 10.1002/1529-0131(199908)42:8<1594::AID-ANR5>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 39.Yao Q, Wang S, Glorioso JC, Evans CH, Robbins PD, Ghivizzani SC, Oligino TJ. Gene transfer of p53 to arthritic joints stimulates synovial apoptosis and inhibits inflammation. Mol Ther. 2001;3:901–910. doi: 10.1006/mthe.2001.0343. [DOI] [PubMed] [Google Scholar]
- 40.Cha HS, Rosengren S, Boyle DL, Firestein GS. PUMA regulation and proapoptotic effects in fibroblast-like synoviocytes. Arthritis Rheum. 2006;54:587–592. doi: 10.1002/art.21631. [DOI] [PubMed] [Google Scholar]
- 41.Thin TH, Li L, Chung TK, Sun H, Taneja R. Stra13 is induced by genotoxic stress and regulates ionizing-radiation-induced apoptosis. EMBO Rep. 2007;8:401–407. doi: 10.1038/sj.embor.7400912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dudgeon C, Peng R, Wang P, Sebastiani A, Yu J, Zhang L. Inhibiting oncogenic signaling by sorafenib activates PUMA via GSK3β and NF-κB to suppress tumor cell growth. Oncogene. 2012;31:4848–4858. doi: 10.1038/onc.2011.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu J, Yue W, Wu B, Zhang L. PUMA sensitizes lung cancer cells to chemotherapeutic agents and irradiation. Clin Cancer Res. 2006;12:2928–2936. doi: 10.1158/1078-0432.CCR-05-2429. [DOI] [PubMed] [Google Scholar]
- 44.Gupta S, Afaqa F, Mukhtar H. Involvement of nuclear factor-kappa B, Bax and Bcl-2 in induction of cell cycle arrest and apoptosis by apigenin in human prostate carcinoma cells. Oncogene. 2002;21:3727–3738. doi: 10.1038/sj.onc.1205474. [DOI] [PubMed] [Google Scholar]
- 45.Xiao D, Vogel V, Singh SV. Benzyl isothiocyanate-induced apoptosis in human breast cancer cells is initiated by reactive oxygen species and regulated by Bax and Bak. Mol Cancer Ther. 2006;5:2931–2945. doi: 10.1158/1535-7163.MCT-06-0396. [DOI] [PubMed] [Google Scholar]
- 46.Xiao D, Lew KL, Kim YA, Zeng Y, Hahm ER, Dhir R, Singh SV. Diallyl trisulfide suppresses growth of PC-3 human prostate cancer xenograft in vivo in association with Bax and Bak induction. Clin Cancer Res. 2006;12:6836–6843. doi: 10.1158/1078-0432.CCR-06-1273. [DOI] [PubMed] [Google Scholar]