

Abstract
Neutropenia in the setting of acute hematological malignancies may impact disease prognosis, thus affecting therapy dose intensity. This is often due to chemotherapy-induced aplasia as well as to the disease itself. However, chronic neutropenia deserves further investigation, as the management of reversible concomitant causes may avoid treatment delay. The present study describes a case of an acute promyelocytic leukemia patient with chronic severe neutropenia of multifactorial origin, including acute leukemia itself, chemotherapy, autoimmune activation with anti-platelets and anti-neutrophil antibodies positivity, and the rare association of large granular lymphocyte (LGL) expansion. As neutropenia may challenge the diagnosis and treatment of acute malignancies, clinicians and hematopathologists must discuss the differential diagnosis in order to avoid misdiagnosing and undertreating concomitant diseases. In particular, LGL chronic expansion and autoimmunity should be considered.
Keywords: acute myeloid leukemia, chronic neutropenia, natural killer chronic expansion, autoimmunity, chemotherapy-induced neutropenia
Introduction
In patients undergoing chemotherapy for hematological malignancies, neutropenia severity and duration are important tools to monitor bone marrow recovery, and may significantly affect the treatment dose intensity, with a possible impact on disease prognosis (1). In this setting, other causes of neutropenia may be difficult to diagnose, possibly leading to therapy delay and undertreatment. Chronic neutropenia, lasting >3 months, may be primary (idiopathic or autoimmune) or secondary to other conditions, including malignancies, chemicals or radiation exposure (2). The present report describes a case of an acute promyelocytic leukemia (APL) patient who developed a long-lasting multifactorial neutropenia [associated with chemotherapy, autoimmunity and large granular lymphocyte (LGL) expansion], which was difficult to diagnose due to unusual disease association, and interfered with the leukemia treatment.
Case report
A 53-year-old male patient was referred to the outpatient service of Fondazione IRCCS Cà Granda, Ospedale Maggiore Policlinico (Milan, Italy) in March 2013 due to neutropenia [absolute neutrophil count (ANC), 1.1×103 cells/mm3; normal range, 1.5–6.5×103/mm3) and severe thrombocytopenia (18×103 cells/mm3; normal range, 130–400×103/mm3). The medical history was positive for rheumatic fever in his childhood, prostatic hypertrophy and chronic autoimmune thyroiditis. In 2004, the patient had been diagnosed with oligoastrocytoma and had been treated with chemotherapy (vincristine, procarbazine and lomustine), obtaining persistent remission. During this period, the patient had an oxcarbazepine drug-induced neutropenia (1.2×103 cells/mm3), driven by epilepsy prophylaxis, which recovered upon substitution for levetiracetam. The patient's platelets and neutrophil levels during the follow-up are shown in Fig. 1. Physical examination at presentation revealed diffuse petechiae and an 8-cm diameter hematoma on the left gluteus. The liver (aspartate transaminase/alanine transaminase) and kidney (creatinine) values were normal, as well as the lactate dehydrogenase levels, coagulation times and fibrinogen; C reactive protein was increased (7.75 mg/dl; normal range, <0.5 mg/dl), while hepatitis B and C virus serology was negative. Anti-platelets autoantibodies were positive at low titer, suggesting primary immune thrombocytopenia. Thus, first-line intravenous steroid therapy with methylprednisolone at 1 mg/kg/day was started, but after 7 days, the blood counts values further declined [hemoglobin (Hb), 10.7 g/dl; normal range, 13.5–17.5 g/dl; neutrophils, 0.9×103 cells/mm3; and platelets, 19×103 cells/mm3], and the fibrinogen values started to decrease (99 mg/dl; normal range, 165–350 mg/dl). Bone marrow aspirate revealed 40% infiltrate of myeloid blasts with immune phenotype and cytogenetics consistent with APL. Fresh frozen plasma and platelets transfusion were administered, as well as pathogenic therapy with all-trans retinoic acid (ATRA) and steroids. Polymerase chain reaction evaluation was positive for the promyelocytic leukemia protein/retinoic acid receptor α transcript, and induction chemotherapy with idarubicin (12 mg/m2) in addition to ATRA was therefore started. Induction was complicated by neutropenic fever of unknown origin, requiring antimicrobial broad-spectrum therapy and granulocyte-colony stimulating factor (G-CSF). At dischargement (April 15th, 2013), the blood counts had almost recovered (Hb, 10.2 g/dl; ANC, 1.17×103 cells/mm3; and platelets, 107×103 cells/mm3), and bone marrow evaluation revealed complete hematologic, morphologic, cytogenetic and molecular response. The first cycle of consolidation chemotherapy with idarubicin alongside ATRA was then administered. Due to prolonged moderate neutropenia (ANCs, 0.74×103 cells/mm3 after 2 months), levetiracetam was temporary stopped, but no ANCs recovery was observed. In July 2013, as a spontaneous increase in ANCs values occurred, a second chemotherapy consolidation course with ATRA and mitoxantrone was administered, which was complicated by severe febrile neutropenia (ANCs, 0.05×103 cells/mm3) and controlled by intravenous broad-spectrum antibiotics and G-CSF. However, in November 2013, severe neutropenia persisted, thus forcing consolidation chemotherapy interruption and maintenance with ATRA only. In January 2014, a mild lymphocytosis (absolute lymphocyte count, 6.5×103 cells/mm3) was observed. Peripheral blood flow cytometry demonstrated a cluster of differentiation (CD)3+ CD8+ CD57+ phenotype in 99% of lymphocytes, consistent with T-LGL cells. However, no lymphocyte infiltrate was observed by bone marrow aspirate and histology on April 2014, and computed tomography scan was negative for splenomegaly and lymphadenopathies. In addition, anti-neutrophils antibodies were positive by direct granulocyte immunofluorescence test and negative in serum. A year later, bone marrow histology revealed for the first time a 25% T-LGL infiltrate, consistent with T-LGL chronic lymphocytic disorder, according to the World Health Organization criteria (Fig. 2A) (3). From September 2013 to the time of writing this study, the clinical conditions of the patient remained stable, with moderate neutropenia and mild LGL lymphocytosis, without specific therapy (Fig. 1). A re-evaluation of the bone marrow histology sample obtained at the time of APL diagnosis was positive for ~10% T-LGL infiltrate, which was missed at the first pathological evaluation (Fig. 2B).
Figure 1.

Patient absolute neutrophil counts (black line) and platelets (grey line) during the follow-up. Dotted arrow indicates all-trans retinoic acid treatment, including induction, consolidation and maintenance. Cross symbol represents grade 2 infections. ANC, absolute neutrophil counts; BM, bone marrow; APL, acute promyelocytic leukemia; LGL, large granular lymphocyte; PB, peripheral blood; ATRA, all-trans retinoic acid; PLT, platelet; I, induction chemotherapy; C, consolidation chemotherapy.
Figure 2.
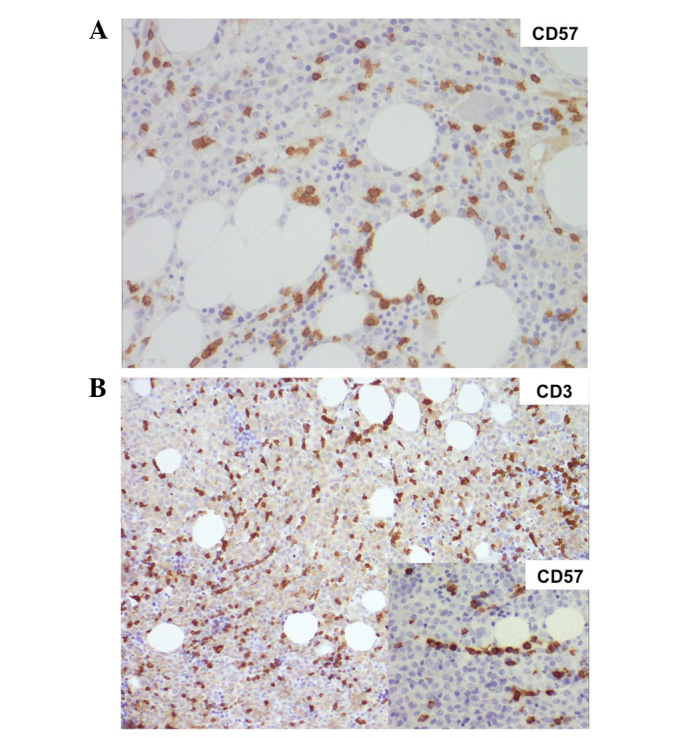
(A) Bone marrow histology at large granular lymphocyte diagnosis: Increased CD3+ CD57+ lymphoid infiltrate, both interstitial and intra-sinusoidal, in the context of normally proliferating bone marrow, with cells at all differentiating stages. Magnification, ×200. (B) Bone marrow histology at acute promyelocytic leukemia diagnosis: CD3+ CD57+ lymphoid infiltrate, both interstitial and intra-sinusoidal, in the context of CD13+ CD33+ CD38+ CD64+ CD117+ CD14− CD34− human leukocyte antigen-antigen D related− CD11a-b− promyelo-blast marrow invasion. Magnification, ×100 (insert, ×200). CD, cluster of differentiation.
The patient is presently in a good general condition. Complete remission has been maintained for acute promyelocytic leukemia for 21 months since the end of therapy, and the patient undergoes regular clinical check-ups every 6 months. The patient provided written informed consent.
Discussion
The present study describes a case of three different causes of cytopenia in the same patient: Acute myeloid leukemia (AML), autoimmunity and LGL chronic expansion. In addition, a history of previous chemotherapy and the use of drugs possibly interfering with ANCs complicated the clinical setting.
LGL expansion presents with neutropenia in 70–80% of cases, and it has been reported in ≤35% of patients with idiopathic cytopenia of uncertain significance (4).
An association between LGL leukemia and solid or hematological malignancies has been described in various series; in particular, chronic B-cell dyscrasia is often observed in LGL patients, including monoclonal gammopathy of undetermined significance (MGUS), chronic lymphocytic leukemia, monoclonal B lymphocytosis, hairy cell leukemia and multiple myeloma (5). Additionally, LGL may be associated with myeloid malignancies such as myelodysplastic syndrome and myeloproliferative disease (6,7). However, to the best of our knowledge, the present study is the first report of an LGL/APL association in the same patient.
Considering the therapeutic approach, no killing was observed in LGL cells following APL chemotherapy; on the contrary, LGL clones continued to increase once the marrow leukemic blasts had been cleared. The prolonged neutropenia observed led to consolidation and maintenance therapy delay and modification. In particular, no methotrexate maintenance for APL was administered. Notably, methotrexate is also used in LGL, and it may have cleared LGL cells (5). In a recent report of six LGL/multiple myeloma cases, bortezomib-based therapy was able to inhibit LGL clones, whereas lenalidomide, which is known to activate T-natural killer cells, was ineffective (5,6).
In the present case, no LGL specific treatment was performed. Instead, a patient monitoring approach was preferred due to the potential side effects of immunosuppression both on infectious risk and cerebral tumors. Treatment is usually required in 30–70% of LGL patients due to cytopenia-related manifestations and/or organomegaly, B symptoms and LGL raising counts (7,8). Notably, all the infectious episodes observed in the present patient occurred during induction/consolidation, and were of grade ≤2.
Regarding possible pathogenic mechanisms, it has been hypothesized that clonal cytotoxic T-cell population could play a role in antitumor immunosurveillance by controlling concomitant neoplasia development, as observed in previous MGUS/LGL cases (5). Otherwise, LGL could derive from a ‘proliferation escape’ following a chronic immune response against the concomitant tumor (9). It is difficult to assess whether LGL expansion is a primary or secondary event, and both conditions have been described (6). In the present patient, a posteriori evaluation of the bone marrow histology from the diagnosis of APL demonstrated that LGL expansion was already present, suggesting that it may have been an attempt to control APL tumor growth. Anti-APL chemotherapy and consequent promyelocytic lysis may have conferred a growing advantage, and may have ultimately led to a clone expansion of LGL cells. Of note, there was no peripheral blood lymphocytosis at that time, and a limited LGL clone is not easy to observe unless specific immunohistochemistry is performed. Consistently, bone marrow re-evaluation demonstrated a previously misdiagnosed LGL clone in patients with chronic idiopathic neutropenia, which subsequently evolved to LGL chronic expansion (2).
In the patient described in the present case report, anti-platelets and low titer anti-neutrophils antibodies were identified at different steps of the disease. Despite the low sensibility and specificity of the available tests, this may reflect an immune activation/dysregulation in the course of a hematological malignancy. Autoantibodies were previously observed to be positive in several patients, including those with AML. In particular, antinuclear antibody and anti-DNA positivity were detected in ≤90% in a study of 39 AML cases (10), and anti-cardiolipin antibodies were identified in 25 out of 37 AML cases (68%) (11).
In conclusion, in the setting of acute malignancies, it is important to consider concomitant causes of unexplained persistent neutropenia. In particular, LGL chronic expansion should be considered, and ad hoc immunohistochemistry should be performed. Furthermore, autoimmunity in the context of immune activation/dysregulation may contribute to the severity of neutropenia.
References
- 1.Morrison VA. Infections in patients with leukemia and lymphoma. Cancer Treat Res. 2014;161:319–349. doi: 10.1007/978-3-319-04220-6_11. [DOI] [PubMed] [Google Scholar]
- 2.Fattizzo B, Zaninoni A, Consonni D, Zanella A, Gianelli U, Cortelezzi A, Barcellini W. Is chronic neutropenia always a benign disease? Evidences from a 5-year prospective study. Eur J Intern Med. 2015;26:611–655. doi: 10.1016/j.ejim.2015.05.019. [DOI] [PubMed] [Google Scholar]
- 3.Jaffe ES. The 2008 WHO classification of lymphomas: Implications for clinical practice and translational research. Hematology Am Soc Hematol Educ Program. 2009;2009:523–531. doi: 10.1182/asheducation-2009.1.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bektas O, Uner A, Aydin SM, Eliacik E, Uz B, Işık A, Haznedaroğlu IC, Goker H, Sayinalp N, Aksu S, et al. High frequency of autonomous T-cell proliferation compatible with T-cell large granular lymphocytic leukemia in patients with cytopenia of unknown etiology. Int J Hematol. 2015;102:211–217. doi: 10.1007/s12185-015-1816-y. [DOI] [PubMed] [Google Scholar]
- 5.Zhang D, Loughran TP., Jr Large granular lymphocytic leukemia: Molecular pathogenesis, clinical manifestations, and treatment. Hematology Am Soc Hematol Educ Program. 2012;2012:652–659. doi: 10.1182/asheducation-2012.1.652. [DOI] [PubMed] [Google Scholar]
- 6.Cheng J, Talamo G, Malysz J, Ochmann M, Lamy T, Loughran TP., Jr Report of 6 cases of large granular lymphocytic leukemia and plasma cell dyscrasia. Clin Lymphoma Myeloma Leuk. 2014;14:e169–e172. doi: 10.1016/j.clml.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 7.Lamy T, Loughran TP., Jr How I treat LGL leukemia. Blood. 2011;117:2764–2774. doi: 10.1182/blood-2010-07-296962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dhodapkar MV, Li CY, Lust JA, Tefferi A, Phyliky RL. Clinical spectrum of clonal proliferations of T-large granular lymphocytes: A T-cell clonopathy undetermined significance? Blood. 1994;84:1620–1627. [PubMed] [Google Scholar]
- 9.Zambello R, Semenzato G. Large granular lymphocyte disorders: New etiopathogenetic clues as a rationale for innovative therapeutic approaches. Haematologica. 2009;94:1341–1345. doi: 10.3324/haematol.2009.012161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kostiala AA, Gripenberg M, Elonen E, Gripenberg G, Kostiala I. Follow-up of antibodies against single-stranded DNA in patients with haematological malignancies. Clin Exp Immunol. 1985;61:15–23. [PMC free article] [PubMed] [Google Scholar]
- 11.Lossos IS, BogomolskiYahalom V, Matzner Y. Anticardiolipin antibodies in acute myeloid leukemia: Prevalence and clinical significance. Am J Hematol. 1998;57:139–143. doi: 10.1002/(SICI)1096-8652(199802)57:2<139::AID-AJH8>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]