

Abstract
The aim of the present study was to investigate hypoxia-induced apoptosis and autophagy in vascular smooth muscle cells (VSMCs) and the underlying molecular mechanisms of microRNA (miR)-17-5p responses in an anaerobic environment. The results revealed that miR-17-5p expression was significantly upregulated in VSMCs subjected to hypoxic conditions (P<0.05) and lower miR-17-5p levels were observed in ethyl 3,4-dihydroxybenzoate-treated and hypoxia inducible factor-1 loss-of-function cells. Additionally, it was demonstrated that miR-17-5p is associated with hypoxia-induced autophagy, which was confirmed by upregulating the light chain 3-II/LC3-I ratio and downregulating nucleoporin p62. Cell apoptosis was inhibited in response to hypoxia, and levels of pro-apoptotic proteins B-cell lymphoma 2-associated X protein and p-caspase were markedly decreased when VSMCs were subjected to hypoxic conditions. Furthermore, expression of signal transducer and activator of transcription 3 (STAT3) decreased when cells were transfected with overexpressing miR-17-5p and subjected to hypoxic conditions, and the combination of miR-17-5p loss-of-function and hypoxia induced greater upregulation in the protein expression of STAT3 compared with a single treatment for hypoxia in VSMCs. In conclusion, miR-17-5p may be a novel hypoxia-responsive miR and hypoxia may induce protective autophagy and anti-apoptosis in VSMCs by targeting STAT3.
Keywords: microRNA, hypoxia, vascular smooth muscle cells, autophagy
Introduction
Vascular smooth muscle cells (VSMCs) comprise the medial layer of blood vessels, control vessel tone and blood flow, and thereby serve a fundamental role in blood pressure regulation and substance exchange (1,2). Under pathological conditions, VSMCs typically exhibit increased proliferative, migratory and extracellular matrix-synthesizing capacities, indicating that they convert to the synthetic phenotype (3). Autophagy is a cellular catabolic process responsible for the destruction of long-lived proteins and organelles via a lysosome-dependent pathway that maintains cellular homeostasis. Deregulated autophagy has been implicated in the pathogenesis of various diseases, including vascular disorders (4). A number of in vitro studies have demonstrated that various pro-atherogenic stimuli are able to induce autophagy in vascular cells (5,6). A recent study indicated that there may be a correlation between autophagy and hypoxia in the development and progression of vasculopathy (4). In pulmonary vascular cells, autophagy activation inhibits proliferation when cells are exposed to hypoxic conditions. Hypoxia has been demonstrated to activate autophagy via regulation of adenosine monophosphate-activated protein kinase (AMPK) in human pulmonary smooth muscle cells. Furthermore, suppressing AMPK expression prevents hypoxia-mediated autophagy and the induction of cell death (7). In human umbilical vein endothelial cells (HUVECs), autophagy is induced when cells are subjected to hypoxic conditions, which is enhanced by hypoxia-inducible factor 1 (HIF-1) gene overexpression and inhibited by HIF-1 loss-of-function (8). However, the extent to which molecular mechanisms of hypoxia contribute to the induction of autophagy in VSMCs remains to be determined.
MicroRNA (miR) are small non-coding RNA that serve as important post-transcriptional gene regulators, with a primary function of controlling cell proliferation and differentiation of various cell types. A number of previous studies have demonstrated that pathogenic changes in tissues, including cardiac hypertrophy, heart failure, cardiac fibrosis and vascular atherosclerosis, may be associated with miR (9,10). A recent study has indicated that miR-137 inhibits hypoxia-induced mitophagy by reducing expression of the mitophagy receptor, thereby leading to inadequate interaction between the receptor and light chain (LC) 3 (11), further indicating that miR has a role in modulating autophagy. Furthermore, it has been demonstrated that miR-20a-5p mediates hypoxia-induced autophagy by targeting the autophagy related 16-like 1 gene in ischemic kidney injury (12), and in cardiomyocytes, the inhibition of miR-497 is able to ameliorate anoxia/reoxygenation injury by suppressing cell apoptosis and enhancing autophagy (13). miR-17-5p is a member of the miR-17–92 cluster, which is located on human chromosome 13q31 (14). A previous study revealed the widespread overexpression of miR-17-5p in diverse tumor tissues, suggesting that miR-17-5p exhibits oncogenic activity (15). Notably, miR-17-5p promotes oxidative-stress-induced cardiomyocyte apoptosis in animal models of ischemia/reperfusion-induced cardiac injury and cellular models of cardiomyocyte injury (16). However, the roles of miR-17-5p in hypoxia-induced autophagy and apoptosis in VSMCs have not been investigated until recently.
The present study aimed to demonstrate that miR-17-5p is a regulator of signal transducer and activator of transcription 3 (STAT3) by using miRanda, TargetScan and picTar databases, and to elucidate if miR-17-5p is able to mediate hypoxia-induced autophagy and inhibit apoptosis by targeting STAT3 in VSMCs.
Materials and methods
Cell culture
VSMCs were obtained from the Cell Resource Center, Shanghai Institutes for Biological Sciences (Shanghai, China) and maintained in RPMI-1640 supplemented with 10% fetal bovine serum (FBS; both Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37°C in a humidified incubator (Thermo Fisher Scientific, Inc.), in an atmosphere containing 5% CO2. Ethyl 3,4-dihydroxybenzoate (EDHB) was purchased from Sigma-Aldrich; Merck Millipore (Darmstadt, Germany) and dissolved in ethanol. VSMCs were exposed to EDHB (50 µg/ml) for 24 h.
Caspase-3 activity and cell apoptosis assay
VSMC lysates were prepared and measured using a caspase-3 ELISA Kit (no. KGA203, Nanjing KeyGEN BioTECH, Co., Ltd., Nanjing China). In brief, 50 µl supernatant was mixed with 2X reaction buffer (50 µl) and dithiothreitol (0.5 µl). Immunocomplexes were incubated with 5 µl peptide substrate in assay buffer for 2 h at 37°C. Release of p-nitroaniline was measured at 405 nm using an ELISA reader (SpectraMax M5; Molecular Devices, LLC., Sunnyvale, CA, USA) according to the manufacturer's instructions.
Quantitative assessment of apoptotic cells was performed using the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) method, examining DNA-strand breaks during apoptosis with the ApoAlert DNA Fragmentation Assay kit (BD Biosciences, Franklin Lakes, NJ, USA). Briefly, cells were incubated at 37°C in hypoxic conditions for 48 h. Cells were trypsinized, fixed with 4% paraformaldehyde at room temperature for 24 h and permeabilized with 0.1% Triton-X-100 in 0.1% sodium citrate. Cells were washed and incubated with the reaction mixture for 60 min at 37°C, and immediately analyzed using FACScan and the Cellquest program ver. 5.1 (BD Biosciences).
Overexpression and small interfering (si) RNA
Lentiviral vectors containing miR-17-5p were constructed to transfect VSMCs. Briefly, VSMCs were cultured in McCoy's 5α medium containing 10% FBS (MP, Biomedicals, Santa Ana, CA, USA) and when the exponential growth phase was reached, 1.0×105 cells/well were seeded in 96-well plates. A total of 300 µl complete culture medium, containing recombinant lentiviruses, control lentiviruses or McCoy's 5α medium (all of which contained 6 µg/ml polybrene; Sigma-Aldrich; Merck Millipore) was added to the plates when the cells reached 50–60% confluence. The virus-containing medium was replaced with fresh complete medium two days later.
The siRNAs or antagomirs for miR-17-5p, were obtained from GE Healthcare Dharmacon, Inc. (Pittsburgh, PA, USA), and were designed with the following sequences: miR-17-5p, 5′-CUGAGGUCCAGGACACACA-3′; scramble, 5′-AGAGAUGACUCACUGUCAC-3′. VSMCs were transfected with siRNA oligonucleotides using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol.
HIF-1α-siRNA transfection
The HIF-1α-siRNA oligonucleotide fragment was designed and synthesized according to the human HIF-1α gene sequence (GenBank no. NM001530). The sequence was as follows: Forward, 5′-GATCCCGAGGAAGAACTATGAACATAATTCAAGAGATTATGTTCATAGTTCTTCCTCTTTTTGGAT-3′ and reverse, 5′-AGCTATCCAAAAAGAGGAAGAACTATGAACATAATCTCTTGAATTATGTTCATAGTTCTTCCTCGG-3′. Scramble and HIF-1α-siRNA oligonucleotide were cloned into the pSIREN-RetroQ plasmid (Addgene, Inc., Cambridge, MA, USA) for retrovirus production. After 48 h, infected cells were selected with puromycin (2 mg/ml) and the clones were selected and cultured for further experiment.
Luciferase reporter gene activity assay
The three prime untranslated region (3′UTR) of the STAT3 gene containing the predicated target sites for miR-17-5p was obtained by polymerase chain reaction (PCR) amplification. The fragment was inserted into the multiple cloning sites of the pMIR-REPORT luciferase miR expression reporter vector (Ambion; Thermo Fisher Scientific, Inc.). Cells from the human embryonic kidney cell line HEK-293 (Cell Resource Center, Shanghai Institutes for Biological Sciences) were co-transfected with 0.1 µg luciferase reporters containing STAT3 3′UTR and microR-17-5p mimics using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.). Cell lysates were harvested 48 h post-transfection and luciferase activity was measured with a dual luciferase reporter assay kit (no. RG028. Beyotime Institute of Biotechnology, Haimen, China) according to the manufacturer's protocol.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
RNA extraction was performed using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. Synthesis of cDNA was performed by RT reactions with 2 µg total RNA using Moloney murine leukemia virus reverse transcriptase (Invitrogen; Thermo Fisher Scientific, Inc.) with oligo dT (15) primers (Fermentas; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. miR-17-5p level was measured using qPCR with the mirVana RT-qPCR miR detection kit (Ambion; Thermo Fisher Scientific, Inc.) in conjunction with SYBR Green (Invitrogen; Thermo Fisher Scientific, Inc.). Following the circle reaction, the threshold cycle (Cq) was determined and the relative miR-17-5p level was calculated based on the Cq values and normalized to U6 level in each sample. PCR was performed with the following primers: miR-17-5p, forward 5′-TCTAGATCCCGAGGACTG-3′ and reverse, 5′-ATCGTGACCTGAACC-3′; U6, forward 5′-CTCGCTTTGGCAGCACA-3′ and reverse 5′-AACGCTTCACGAATTTGCGT-3′.
Autophagy detection in VSMCs
Autophagy in VSMCs was detected via western blotting and fluorescence microscopy, and cultured VSMCs were prepared for observation under a transmission electron microscope (TEM) to investigate the formation of autophagosomes. Cells were washed with phosphate-buffered saline, fixed with 4% formaldehyde/1% glutaraldehyde at room temperature overnight, and then rinsed three times with 0.1 M sodium cacodylate/0.2 M sucrose buffer. Following fixation, cells were incubated in 1% osmium tetroxide for 1 h, dehydrated in a series of ethanol washes, and then embedded in epoxy resin. When the epoxy resin had polymerized at 65°C for 24 h, sections 100-nm thick were then cut onto a carbon-coated copper grid. Cells were stained with uranyl acetate and lead citrate (SPI Supplies, West Chester, PA, USA) and examined under a TEM (H-800; Hitachi, Ltd., Tokyo, Japan).
Western blotting
VSMCs were homogenized and extracted in NP-40 buffer (Thermo Fisher Scientific, Inc.), followed by 5–10 min boiling and centrifugation at 7,500 × g, for 15 min at 4°C to obtain the supernatant. Protein samples were quantified using the bicinchoninic Acid kit for Protein Determination, (no. BCA1-1KT; Sigma-Aldrich; Merck Millipore).
Samples containing 30 µg protein were separated by 10% SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Following blocking for 2 h at room temperature with 5% (w/v) non-fat dry milk in tris-buffered saline and 0.1% (w/v) Tween 20 (TBST), membranes were incubated with primary antibodies for LC3 (sc-292,354, dilution, 1:1,000), P62 (sc-48389, dilution, 1:1,000), β-actin (sc-81178, dilution, 1:2,000), Bax (sc-6236, dilution, 1:1,000), p-caspase3 (sc-22171-R, dilution, 1,000), caspase3 (sc-7272, dilution, 1:1,000), STAT2 (sc-483, dilution, 1:1,000) at 4°C overnight. All primary antibodies were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). The membranes were washed three times with TBST and incubated with secondary antibodies donkey anti-mouse immunoglobulin (Ig) G (sc-2096, dilution, 1:10,000) and goat anti-rabbit IgG (sc-2004, dilution, 1:10,000; both Santa Cruz Biotechnology, Inc.) for 2 h at room temperature and visualized with an Amersham ECL Western blotting Detection reagent (GE Healthcare Life Sciences, Chalfont, UK). Signals were densitometrically assessed using Quantity One® software ver. 4.5 (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Each experiment was performed four times.
Statistical analysis
The data from these experiments were presented as the mean ± standard deviation for each group. Statistical analyses were performed using PRISM version 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). Inter-group differences were analyzed via one-way analysis of variance. P<0.05 was considered to indicate a statistically significant difference.
Results
miR-17-5p expression in response to hypoxia and inhibitors
Significant upregulation of miR-17-5p expression was observed in VSMCs subjected to hypoxic conditions (P<0.05; Fig. 1A). HIF-1 is stabilized under hypoxic conditions as a cellular response to low oxygen concentration. Furthermore, EDHB has been shown to have a cytoprotective effect against oxidative stress in various types of cells, including VSMCs (17). In the present study, lower miR-17-5p levels were observed in EDHB-treated and HIF-1α loss-of-function cells (Fig. 1B). These results indicate that miR-17-5p may be a novel hypoxia-responsive miR that has not been reported in previous miR profiling data.
Figure 1.
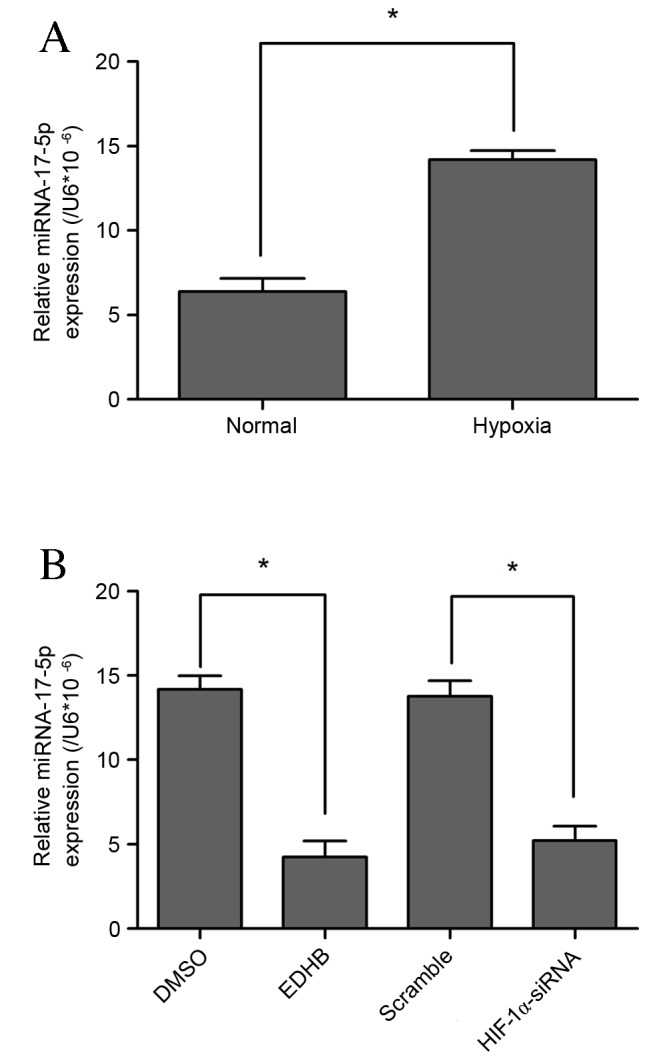
(A) The expression of miR-17-5p in VSMCs was measured using reverse transcription-quantitative polymerase chain reaction in the presence of hypoxia. (B) miR-17-5p levels were observed in EDHB-treated and HIF-1 loss-of-function VSMCs. Data are presented as the mean ± standard deviation, n=3 in each group. *P<0.05. miR, microRNA; VSMC, vascular smooth muscle cell; DMSO, dimethyl sulfoxide; EDHB, ethyl 3,4-dihydroxybenzoate; HIF-1, hypoxia inducible factor 1; siRNA, small interfering RNA.
Hypoxia induces autophagy and inhibits apoptosis in VSMCs
The ultrastructure of VSMCs was observed under a TEM to assess levels of autophagy in response to hypoxia and miR-17-5p loss-of-function. An increase in typical autophagic vacuoles containing extensively degraded organelles, including mitochondria and endoplasmic reticulum, was observed in the cytoplasm of VSMCs following treatment with hypoxia as compared with the control group (Fig. 2). However, this effect was reversed by miR-17-5p antagomir in hypoxic conditions (Fig. 2). To assess autophagosome formation, LC3-II expression was measured via immunofluorescent staining and western blotting was used to measure the LC3-II/LC3-I ratio and p62 levels. The results demonstrated that the LC3-II intensity of green fluorescence in response to hypoxia was greater than that of the control group following incubation for 24 h in RPMI-1640 (Fig. 3A). Consistent with the fluorescence imaging results, the LC3-II/LC3-I ratio increased in response to hypoxia (Fig. 3B) and the expression of p62 was markedly decreased following exposure of cells to hypoxic conditions. However, the results of LC3-II degeneration and p62 accumulation demonstrated that miR-17-5p loss-of-function inhibited hypoxia-induced autophagy (Fig. 3A and B). Furthermore, it was investigated whether hypoxia induced apoptosis in VSMCs via an apoptotic mechanism using a caspase-3 activity assay and TUNEL staining following the exposure of VSMCs for 24 h. The results indicate that caspase-3 activity in the hypoxia-treated group was significantly lower than that of the control group (P<0.05; Fig. 3C), and cell apoptosis was significantly inhibited in response to hypoxia (P<0.05; Fig. 3D); however, these reductions were significantly reversed by miR-17-5p antagomir transfection (both P<0.05). The apoptotic response was further investigated by measuring the expression of apoptosis-related proteins. Western blot analysis demonstrated that treatment of VSMCs with hypoxia markedly decreased pro-apoptotic proteins BAX and p-caspase levels (Fig. 3E). These results suggest that hypoxia induced protective autophagy in VSMCs via inhibition of cell apoptosis as a response to an anaerobic environment, which is consistent with the literature (18).
Figure 2.
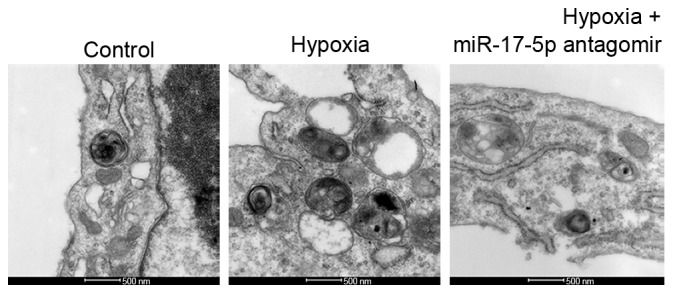
Presence of autophagic vacuoles in response to different treatments observed using transmission electron microscopy (scale bar, 500 nm). miR, micro RNA.
Figure 3.

(A) Immunofluorescence staining of LC3-II (scale bar, 50 µm). (B) Western blot analysis of LC3I, LC3-II and p62. (C) VSMCs were incubated with hypoxia for 48 h and caspase 3 activity was measured using an ELISA assay. (D) TUNEL staining was measured by flow cytometry. (E) The protein expression levels of BAX, p-caspase 3 and caspase 3 were determined via western blotting. Data are presented as the mean ± standard deviation, n=3 in each group. *P<0.05. LC3, light chain 3; VSMC, vascular smooth muscle cell; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; miR, microRNA.
miR-17-5p directly targets the 3′-UTR of STAT3
To determine whether miR-17-5p regulated STAT3 through the predicted binding sites in its 3′-UTR (Fig. 4A), a luciferase construct was designed by incorporating wild-type or mutant 3′-UTR of STAT3, which expressed luciferase unless repressed by the incorporated 3′-UTR. No significant difference was observed between the control group and the group that underwent cotransfection of VSMCs with the pMIR-REPORT construct containing mutant STAT3 3′-UTR and PLemiR-17-5p. Cotransfection with the luciferase construct containing wild-type STAT3 3′-UTR and PLemiR-17-5p resulted in significantly lower luciferase activity than the control group, leading to a ~70% decline in luciferase activity compared with control group (P<0.05; Fig. 4B). Western blotting results indicated that the expression of STAT3 was significantly decreased following cotransfection with the luciferase construct containing wild-type STAT3 3′-UTR and overexpressing miR-17-5p compared with that of the wild-type control group (P<0.05; Fig. 4C). Furthermore, the combination of miR-17-5p loss-of-function and hypoxia was demonstrated to significantly upregulate the protein expression of STAT3 compared with hypoxia single treatment in VSMCs (P<0.05), which was confirmed by immunofluorescent staining (Fig. 5A) and western blotting (Fig. 5B).
Figure 4.

(A) Schematic representation of the putative miR-17-5p binding site in the STAT3 3′UTR. (B) Luciferase activity assay. (C) The protein expression of STAT3 was measured using western blotting in the presence of miR-17-5p overexpression. *P<0.05. miR, microRNA; STAT3, signal transducer and activator of transcription 3; UTR, untranslated region; wild, wild type; mut, mutant.
Figure 5.

The protein expression of STAT3 in VSMCs was measured using (A) immunofluorescent staining (scale bar, 50 µm) and (B) western blotting in the presence of hypoxia or miR-17-5p antagomir. Data are presented as the mean ± standard deviation, n=3 in each group. *P<0.05. miR, microRNA; STAT3, signal transducer and activator of transcription 3; VSMC, vascular smooth muscle cell.
Discussion
The present study identified miR-17-5p as a novel hypoxia-responsive miR of VSMC autophagy and apoptosis via targeting STAT3. MiR-17-5p was significantly upregulated in response to hypoxia, which also significantly increased autophagic vacuoles and decreased apoptosis. However, miR-17-5p loss-of-function was able to reverse hypoxia-induced increased autophagic vacuoles and decreased apoptosis, indicating that miR-17-5p may be associated with hypoxia-induced protective autophagy and anti-apoptosis in VSMCs.
Autophagy has dual roles in cardiovascular disease: Physiological autophagy serves as a protective mechanism to maintain normal cardiovascular function, whereas impaired autophagy contributes to the development and progression of various diseases (19). A previous study indicated that in cardiac HL-1 cells, ischemia/reperfusion-induced apoptosis is decreased when autophagy is increased, suggesting that it serves a cardioprotective role in response to cardiac injury (20). Conversely, pro-apoptosis may synergistically increase autophagy in oxidated low-density lipoprotein-treated VSMCs via the suppression of miR hsa-let-7g (5). In HUVECs, HIF-1 is able to reduce cell viability by inducing autophagy (8). The present study demonstrated that increasing autophagy may inhibit apoptosis in hypoxia-treated VSMCs, suggesting that autophagy may serve as a protective mechanism for hypoxia-treated VSMCs via the inhibition of cell apoptosis.
miR-17-5p is a member of the microR-17–92 cluster and is associated with the regulation of tumor proliferation and progression (21,22). Notably, miR-17-5p is associated with ischemia/reperfusion and cardiomyocyte injury under oxidative stress in mice, and overexpression of miR-17-5p aggravates cardiomyocyte injury leading to reduced cell viability and enhanced H2O2-induced apoptotic cell death, whereas inhibition of miR-17-5p by its anti-miR oligonucleotide, AMO-17-5p, abrogates the deleterious changes (16). The present study demonstrated that miR-17-5p is upregulated in VSMCs when cells are exposed to hypoxia and, furthermore, that miR-17-5p antagomir was able to restore the LC3-II/LC3-I ratio, which increases in response to hypoxia in VSMCs. The conversion of LC3-I into LC3-II is an essential step in autophagosome formation and the abundance of LC3-II is correlated with the number of autophagosomes (5,18). These results indicated that LC3-II degenerates in response to miR-17-5p loss-of-function inhibited hypoxia-induced autophagy.
It is known that STAT3 is required to regulate cell proliferation, differentiation, and apoptosis, and activation of STAT3 by phosphorylation increases cell survival in response to various cytokines and stress stimuli (23). In VSMCs, STAT3 is associated with cell senescence and calcification, and STAT3 expression is significantly decreased in response to miR-135a overexpression (24). Recent in vitro and in vivo research has demonstrated that miR-17-5p overexpression promotes cardiomyocyte apoptosis, which is induced by oxidative stress via the targeting of STAT3 (16). In the present study, hypoxia-induced miR-17-5p upregulation was able to inhibit STAT3 expression in vitro; therefore STAT3, as a target for miR-17-5p, may be associated with hypoxia-induced protective autophagy.
In conclusion, the present study demonstrated that miR-17-5p is associated with hypoxia-induced protective autophagy and apoptosis and that the underlying mechanism was mediated, at least partially, by targeting STAT3.
Acknowledgements
This study was supported by the National Natural Science Foundation for the Youth of China (grant no.81500198)
References
- 1.Salabei JK, Hill BG. Autophagic regulation of smooth muscle cell biology. Redox biology. 2015;4:97–103. doi: 10.1016/j.redox.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Meyer GR, Grootaert MO, Michiels CF, Kurdi A, Schrijvers DM, Martinet W. Autophagy in vascular disease. Circ Res. 2015;116:468–479. doi: 10.1161/CIRCRESAHA.116.303804. [DOI] [PubMed] [Google Scholar]
- 3.Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15:713–720. doi: 10.1038/ncb2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vindis C. Autophagy: An emerging therapeutic target in vascular diseases. Br J Pharmacol. 2015;172:2167–2178. doi: 10.1111/bph.13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding Z, Wang X, Schnackenberg L, Khaidakov M, Liu S, Singla S, Dai Y, Mehta JL. Regulation of autophagy and apoptosis in response to ox-LDL in vascular smooth muscle cells, and the modulatory effects of the microRNA hsa-let-7g. Int J Cardiol. 2013;168:1378–1385. doi: 10.1016/j.ijcard.2012.12.045. [DOI] [PubMed] [Google Scholar]
- 6.Peng N, Meng N, Wang S, Zhao F, Zhao J, Su L, Zhang S, Zhang Y, Zhao B, Miao J. An activator of mTOR inhibits oxLDL-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E-/− mice. Sci Rep. 2014;4:5519. doi: 10.1038/srep05519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ibe JC, Zhou Q, Chen T, Tang H, Yuan JX, Raj JU, Zhou G. Adenosine monophosphate-activated protein kinase is required for pulmonary artery smooth muscle cell survival and the development of hypoxic pulmonary hypertension. Am J Respir Cell Mol Biol. 2013;49:609–618. doi: 10.1165/rcmb.2012-0446OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu J, Lei Z, Yu J. Hypoxia induces autophagy in human vascular endothelial cells in a hypoxia-inducible factor 1dependent manner. Mol Med Rep. 2015;11:2677–2682. doi: 10.3892/mmr.2014.3093. [DOI] [PubMed] [Google Scholar]
- 9.Pan ZW, Lu YJ, Yang BF. MicroRNAs: A novel class of potential therapeutic targets for cardiovascular diseases. Acta Pharmacol Sin. 2010;31:1–9. doi: 10.1038/aps.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Urbich C, Kuehbacher A, Dimmeler S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc Res. 2008;79:581–588. doi: 10.1093/cvr/cvn156. [DOI] [PubMed] [Google Scholar]
- 11.Li W, Zhang X, Zhuang H, Chen HG, Chen Y, Tian W, Wu W, Li Y, Wang S, Zhang L, et al. MicroRNA-137 is a novel hypoxia-responsive microRNA that inhibits mitophagy via regulation of two mitophagy receptors FUNDC1 and NIX. J Biol Chem. 2014;289:10691–10701. doi: 10.1074/jbc.M113.537050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang IK, Sun KT, Tsai TH, Chen CW, Chang SS, Yu TM, Yen TH, Lin FY, Huang CC, Li CY. MiR-20a-5p mediates hypoxia-induced autophagy by targeting ATG16L1 in ischemic kidney injury. Life Sci. 2015;136:133–141. doi: 10.1016/j.lfs.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Li X, Zeng Z, Li Q, Xu Q, Xie J, Hao H, Luo G, Liao W, Bin J, Huang X, Liao Y. Inhibition of microRNA-497 ameliorates anoxia/reoxygenation injury in cardiomyocytes by suppressing cell apoptosis and enhancing autophagy. Oncotarget. 2015;6:18829–18844. doi: 10.18632/oncotarget.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mogilyansky E, Rigoutsos I. The miR-17/92 cluster: A comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013;20:1603–1614. doi: 10.1038/cdd.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu J, Ohuchida K, Mizumoto K, Fujita H, Nakata K, Tanaka M. MicroRNA miR-17-5p is overexpressed in pancreatic cancer, associated with a poor prognosis, and involved in cancer cell proliferation and invasion. Cancer Biol Ther. 2010;10:748–757. doi: 10.4161/cbt.10.8.13083. [DOI] [PubMed] [Google Scholar]
- 16.Du W, Pan Z, Chen X, Wang L, Zhang Y, Li S, Liang H, Xu C, Zhang Y, Wu Y, et al. By targeting Stat3 microRNA-17-5p promotes cardiomyocyte apoptosis in response to ischemia followed by reperfusion. Cell Physiol Biochem. 2014;34:955–965. doi: 10.1159/000366312. [DOI] [PubMed] [Google Scholar]
- 17.Nimker C, Kaur G, Revo A, Chaudhary P, Bansal A. Ethyl 3,4-dihydroxy benzoate, a unique preconditioning agent for alleviating hypoxia-mediated oxidative damage in L6 myoblasts cells. J Physiol Sci. 2015;65:77–87. doi: 10.1007/s12576-014-0348-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem J. 2012;441:523–540. doi: 10.1042/BJ20111451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mei Y, Thompson MD, Cohen RA, Tong X. Autophagy and oxidative stress in cardiovascular diseases. Biochim Biophys Acta. 2015;1852:243–251. doi: 10.1016/j.bbadis.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–29787. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 21.Spaccarotella E, Pellegrino E, Ferracin M, Ferreri C, Cuccuru G, Liu C, Iqbal J, Cantarella D, Taulli R, Provero P, et al. STAT3-mediated activation of microRNA cluster 17~92 promotes proliferation and survival of ALK-positive anaplastic large cell lymphoma. Haematologica. 2014;99:116–124. doi: 10.3324/haematol.2013.088286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Novotny GW, Sonne SB, Nielsen JE, Jonstrup SP, Hansen MA, Skakkebaek NE, Rajpert-De Meyts E, Kjems J, Leffers H. Translational repression of E2F1 mRNA in carcinoma in situ and normal testis correlates with expression of the miR-17–92 cluster. Cell Death Differ. 2007;14:879–882. doi: 10.1038/sj.cdd.4402090. [DOI] [PubMed] [Google Scholar]
- 23.Liao XH, Wang N, Zhao DW, Zheng DL, Zheng L, Xing WJ, Ma WJ, Bao LY, Dong J, Zhang TC. STAT3 Protein Regulates Vascular Smooth Muscle Cell Phenotypic Switch by Interaction with Myocardin. J Biol Chem. 2015;290:19641–19652. doi: 10.1074/jbc.M114.630111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin L, He Y, Xi BL, Zheng HC, Chen Q, Li J, Hu Y, Ye MH, Chen P, Qu Y. miR-135a suppresses calcification in senescent VSMCs by regulating KLF4/STAT3 pathway. Curr Vasc Pharmacol. 2016;14:211–218. doi: 10.2174/1570161113666150722151817. [DOI] [PMC free article] [PubMed] [Google Scholar]