

Abstract
Diabetes mellitus (DM) is a chronic disease in which the body either does not use or produce the glucose metabolising hormone insulin efficiently. Calcification of elastin in the arteries of diabetics is a major predictor of cardiovascular diseases. It has been previously shown that elastin degradation products work synergistically with transforming growth factor-beta 1 (TGF-β1) to induce osteogenesis in vascular smooth muscle cells. In this study, we tested the hypothesis that high concentration of glucose coupled with elastin degradation products and TGF-β1 (a cytokine commonly associated with diabetes) will cause a greater degree of osteogenesis compared to normal vascular cells. Thus, the goal of this study was to analyse the effects of high concentration of glucose, elastin peptides and TGF-β1 on bone-specific markers like alkaline phosphatase (ALP), osteocalcin (OCN) and runt-related transcription factor 2 (RUNX2). We demonstrated using relative gene expression and specific protein assays that elastin degradation products in the presence of high glucose cause the increase in expression of the specific elastin–laminin receptor-1 (ELR-1) and activin receptor-like kinase-5 (ALK-5) present on the surface of the vascular cells, in turn leading to overexpression of typical osteogenic markers like ALP, OCN and RUNX2. Conversely, blocking of ELR-1 and ALK-5 strongly suppressed the expression of the osteogenic proteins. In conclusion, our results indicate that glucose plays an important role in amplifying the osteogenesis induced by elastin peptides and TGF-β1, possibly by activating the ELR-1 and ALK-5 signalling pathways.
Keywords: High glucose, vascular calcification, elastin peptides, transforming growth factor-β1, vascular smooth muscle cells
Introduction
Cardiovascular calcification has been shown to be an independent marker for mortality in patients with advanced cardiovascular diseases.1,2 Two pathological patterns of vascular calcification include intimal calcification and medial calcification. Intimal calcification occurs mostly in association with atherosclerosis subsequent to lipid deposition, macrophage infiltration and smooth muscle cell proliferation.3 On the contrary, medial arterial calcification (MAC) may exist with or without atherosclerosis and is characterised by the presence of calcific deposits on the elastic lamellae.4 As MAC progresses, it forms dense circumferential sheets in the medial layer of the artery, which depicts features very similar to that of physiological calcification in bone. MAC is most commonly observed in the distal arteries of patients with diabetes and end-stage renal failure or with advanced age. It can lead to stroke and lower limb amputations.
In recent years, it has been established that vascular calcification is an active and tightly regulated biological process that involves crosstalk between cells and extracellular matrix of the arteries and resembles physiological bone formation.1 In vitro cell culture studies with high concentration of glucose exposure have been shown to accelerate vascular smooth muscle cell (VSMC) calcification. For example, Chen et al. showed that bovine VSMCs when incubated with high concentrations of glucose (25 mM), coupled with inorganic calcifying agents like β-glycerophosphate and ascorbic acid, underwent a more pronounced osteogenic differentiation compared to cells with normal glucose (5 mM) concentrations, indicating the role of hyperglycaemia in the increased vascular calcification.5
The role of elastin degradation on medial calcification as observed in MAC is not understood well, specifically in diabetic high-glucose conditions. Matrix metalloproteinases (MMPs) are known to degrade elastic fibre, and it has been shown that MMP activity is increased in diabetic arteries.6,7 This may lead to an accelerated degradation of elastic fibres. Elastin-derived peptides (EDPs), a product of elastin degradation, can be detected in the serum of diabetic patients.8,9 In fact, a direct positive correlation has been established between the concentrations of serum EDPs and the development of microvascular complications in diabetic patients.8,9 Elastic fibre degradation in arteries, along with degradation of transforming growth factor-beta (TGF-β) binding protein associated with elastic fibres, releases sequestered TGF-β.10,11 Thus, elastic fibre degradation can increase the concentration of free TGF-β. In our previous study, to test the effect of EDPs and TGF-β1 on VSMCs, we incubated rat VSMCs in vitro with increased concentrations of TGF-β1 and EDPs. We showed that EDP and TGF-β1 have a synergistic effect in the process of osteogenesis of VSMCs, which was characterised by the overexpression of runt-related transcription factor 2 (RUNX2), alkaline phosphatase (ALP) and osteocalcin (OCN).12 We found increased osteogenesis in the absence of any external calcifying agents added to the cell culture such as β-glycerophosphate. However, previous studies were performed in high-glucose culture medium, and the effects of normal glucose levels were not clearly examined. To elucidate the effects of glucose concentrations, we compared the degree of osteogenesis of VSMCs in the presence of EDPs and TGF-β1 in high and low concentrations of glucose.
Materials and methods
Cell isolation and cell culture
Primary rat aortic smooth muscle cells (VSMCs) were purchased from Cell Applications, Inc. (San Diego, CA, USA). Passage numbers 4–8 were used for all the experiments. Cells were cultured in six-well plates (1.5 × 105/well) in either low-glucose Dulbecco’s modified Eagle’s medium (DMEM; HyClone Laboratories, Inc., Novato, CA, USA) or high-glucose DMEM (Cellgro-Mediatech, Herndon, VA, USA), containing 10% foetal bovine serum (HyClone Laboratories, Inc.), 100 units/mL penicillin and 100 units/mL streptomycin (Cellgro-Mediatech) in a humidifier incubator at 37°C, with 5% CO2. Media were replenished every 3 days.
Elastin peptides were purchased from Elastin Products Company (Owensville, MO, USA), which were a mixture of elastin fragments, purified from bovine neck ligament, ranging between a molecular weight of 1000–60,000 kDa and were highly soluble in water. Recombinant TGF-β1 was purchased from Peprotech (Rocky Hill, NJ, USA). Cells were also treated with SB-431542 and lactose (Sigma, St. Louis, MO, USA) to block the activin receptor-like kinase-5 (ALK-5) and elastin–laminin receptor (ELR-1), respectively. In addition, cells were treated with GF109203X (Enzo Life Sciences, Farmingdale, NY, USA) to block intracellular protein kinase C beta II (PKCβII).
Experimental design and time points
VSMCs were grown in low-glucose baseline control DMEM (LGC), low-glucose DMEM with elastin peptides and TGF-β1 (LGET), high-glucose control DMEM (HGC) and high-glucose DMEM with elastin peptides and TGF-β1 (HGET). Test additives were administered to sub-confluent cells (n = 6 per group) as follows: 100 μg/mL elastin peptides and 10 ng/mL TGF-β1.
Additionally, to study the response of inhibition of the respective receptors of elastin peptides (ELR-1) and TGF-β1 (ALK-5), 5 mM lactose or 10 ng/mL SB-431542 was added along with elastin peptides and TGF-β1. For PKCβII inhibition studies, 10 μM GF109203X was added to cell cultures, and relative gene expression of representative osteogenic genes was investigated.
Cells were grown for 1, 3 and 7 days at the end of which total cellular protein and total cellular RNA were isolated. The spent media were collected for protein analyses at the end of each time point.
Gene expression
At each time point, cell monolayers were scraped and homogenised using a PowerGen 125 homogeniser (Fisher Scientific, Pittsburgh, PA, USA). The total RNA from the cells was isolated using the RNeasy mini kit (Qiagen, Valencia, CA, USA). The quality and quantity of the RNA were analysed by Agilent 2100 Bioanalyzer using 6000 Nano lab-on-a chip kit (Agilent Technologies, Foster City, CA, USA). A quantity of 1μg of RNA was reverse transcribed using Qiagen RT kit. The complementary DNA (cDNA) samples were then amplified using Qiagen SYBR green kit in Rotor-Gene reverse transcription polymerase chain reaction (RT-PCR) machine (Corbett Research, Mortlake, NSW, Australia). The primer sets used (forward and reverse primers) are tabulated in Table 1 along with their accession numbers and PCR product sizes. All primers were procured from Integrated DNA Technologies (IDT; Coralville, IA, USA). Each sample was normalised to the expression of beta 2-microglobulin (β2-MG) as a housekeeping gene and compared to LGC group cells (cells cultured in low glucose DMEM alone), using the 2−ΔΔCT method.13
Table 1.
PCR primers used in the study.
| Gene | Name | Primer (forward) | Primer (reverse) | Product size (bp) | Accession number |
|---|---|---|---|---|---|
| β2-MG | Beta 2-microglobulin | CGTGATCTTTCTGGTGCTTGTC | ACGTAGCAGTTGAGGAAGTTGG | 123 | NM_012512 |
| RUNX2 | Runt-related transcription factor 2 | CAACCACAGAACCACAAGTGC | CACTGACTCGGTTGGTCTCG | 120 | AF053950 |
| ALP | Alkaline phosphatase | TCCCAAAGGCTTCTTCTTGC | ATGGCCTCATCCATCTCCAC | 108 | J03572 |
| OCN | Osteocalcin | TATGGCACCACCGTTTAGGG | CTGTGCCGTCCATACTTTCG | 123 | NM_013414 |
| ELR-1 | Elastin–laminin receptor-1 | GTCAGCGTCATCTCCTCCAG | GAAGGTCCCAGGTGTGAAGC | 105 | NM_017138 |
Protein isolation
Cell monolayers were washed twice in phosphate-buffered saline (PBS), and cells were isolated in a mammalian extraction buffer. To prepare the buffer, one tablet of protease inhibitor cocktail (Sigma) was added to 10 mL of SoluLyze-M mammalian extraction buffer (Genlantis, San Diego, CA, USA). Cell layers were homogenised using PowerGen 125 homogeniser and centrifuged at 10,000g for 15 min. The supernatant was collected and assayed for different proteins of interest.
Immunofluorescence for ELR-1 and ALK-5
VSMCs were grown on glass chamber slides (Lab-Tek II Chamber Slide System; Nalge Nunc, Thermo Fisher Scientific, Rochester, NY, USA) and then incubated with elastin peptides and TGF-β1 for 3 days. The cells were then washed twice with PBS and fixed in 4% formaldehyde for 10 min in room temperature followed by incubation with a 5% bovine serum albumin blocking serum. The primary antibody, a rabbit polyclonal anti-67 kDa laminin receptor (Abcam, Cambridge, MA, USA) or a rabbit polyclonal anti-TGF-β1 antibody (Abcam) at 1:100 dilution was applied overnight at 4°C. Alexa Fluor 488 chicken anti-rabbit immunoglobulin G (IgG) secondary antibody (Molecular Probes, Eugene, OR, USA) was applied at a dilution of 8 μg/mL for 2 h at room temperature. Coverslips were mounted in glass slides using aqueous mounting medium with anti-fading agents (Biomedia Corp., Foster City, CA, USA). The slides were examined by fluorescent microscopy.
ALP assay
After quantifying the total cellular protein, cell lysates were analysed for ALP using para-nitrophenol phosphate (pNPP) (Thermo Fisher Scientific) as a substrate in diethanolamine buffer (Pierce, Rockford, IL, USA). ALP level was calculated using a para-nitrophenol standard curve and was normalised to the total protein content. Additionally, cells in culture were stained with 0.2 mg/mL 5-bromo-4-chloro-3-indolyl-phosphate (BCIP), 0.4 mg/mL 4-nitroblue tetrazolium (NBT) and 5 mM MgCl2 in 50 mM Tris buffer, pH 9.5 (Sigma) to localise the activity of ALP in cell cultures.
OCN assay
Culture medium samples from each group were analysed in triplicates for secreted soluble OCN, using a rat OCN enzyme-linked immunoassay kit (Biomedical Technologies, Stoughton, MA, USA), and values were normalised to the total protein.
Inhibition of ELR-1 and ALK-5 receptors
Lactose (5 mM) was used to block the ELR-1, the concentrations of which were optimised in previous studies.12 SB-431542 (10 μM) was used to block ALK-5 receptor.14
Live–dead assay
A live–dead assay (Molecular Probes) was performed as per the manufacturer’s protocol. Cells were observed under a fluorescent microscope. Red fluorescence indicated dead cells while green fluorescence indicated live cells.
Statistical data analysis
Results are expressed as means ± standard error of the mean (SEM). Statistical analyses of the data were performed using single-factor analysis of variance (ANOVA). Subsequently, differences between means were determined using the least significant difference (LSD) with an alpha value of 0.05.
Results
To examine the cytotoxicity of the concentrations of elastin peptides and TGF-β1 used in this study, we performed a live–dead assay. None of the groups were significantly more cytotoxic compared to the low glucose control group at the end of 3 days (Figure 1). However, there was a distinct morphological change in the VSMCs from a typical spindle shaped one to that of an atypical rounded morphology (Figure 1). This effect was evident only in the groups treated with EDP and TGF-β1, and high concentrations of glucose did not seem to contribute to the effect.
Figure 1.
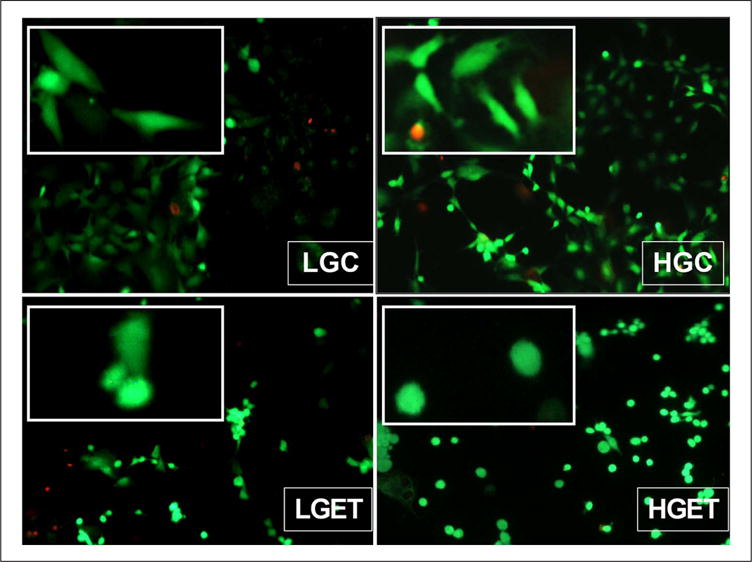
Live–dead assay of VSMCs cultured in the presence of low-glucose baseline control (LGC), high-glucose control (HGC), low glucose + elastin peptides + TGF-β1 (LGET) and high glucose + elastin peptides + TGF-β1 (HGET). Live and dead cells are indicated by green and red fluorescence, respectively. Original magnification, 100×. Inset demonstrates cellular morphological differences among the groups. (For color figure, refer to the online version of the paper.)
VSMC: vascular smooth muscle cell; TGF-β1: transforming growth factor-beta 1.
High glucose is required for expression of osteogenic markers in VSMCs
To investigate the effect of high glucose concentration coupled with growth factor TGF-β1 (10 ng/mL) and elastin peptides (EDP) (100 μg/mL), VSMCs were incubated with both these additives either under the presence of high concentrations of glucose (4.5 g/L) or under the presence of normal concentrations (1.0 g/L) of glucose. Cells were cultured for 1, 3 and 7 days to study the relative gene expression of RUNX2, OCN and ALP, all three of which are known to be commonly associated with osteogenesis.15,16 At the end of 1 day, there was ~ 1.5 times (p < 0.05) overexpression of the RUNX2 relative gene expression in HGC, LGET and HGET groups with additives when compared to cells grown in low glucose control DMEM alone (Figure 2(a)). This early overexpression was independent of glucose concentration, and by the end of 3 days, its expression was significantly suppressed. RUNX2 is a transcription factor and it is the earliest marker for osteogenesis; therefore, its overexpression at 1 day was expected. In contrast to RUNX2, the relative gene expression for ALP, a key enzyme in the physiological and pathological calcification, and OCN, bone-specific marker, were significantly greater in the presence of HGET groups compared to the LGET groups at 3 and 7 days, clearly showing that high glucose levels are essential for the overexpression of these important bone markers (Figure 2(b) and (c)). We also measured the protein activity of ALP by a colorimetric quantitative method using a BCIP/NBT that is a specific for ALP.12 At 7 days, there was almost a fourfold (p < 0.05) increase in the activity of ALP enzyme seen in the HGET groups (Figure 3(a)). Staining for ALP in cell cultures showed notably higher purple colouration in the HGET samples around the cells indicating a higher activity of ALP (Figure 3(b)). In addition, we observed a clustering of VSMCs under the influence of EDP and TGF-β1, which indicates the probable initiation of calcification nodules (Figure 3(b)). OCN protein activity was also found to be twofold (p < 0.05) higher in the cell culture media of HGET group when compared to any other group at 7 days (Figure 3(c)). The protein expression was higher in HGET group when compared to HGC, clearly showing that high glucose levels were not sufficient to increase the osteogenic protein activity in SMCs, and exposure to high glucose in combination with high EDP and TGF-β1 was necessary.
Figure 2.
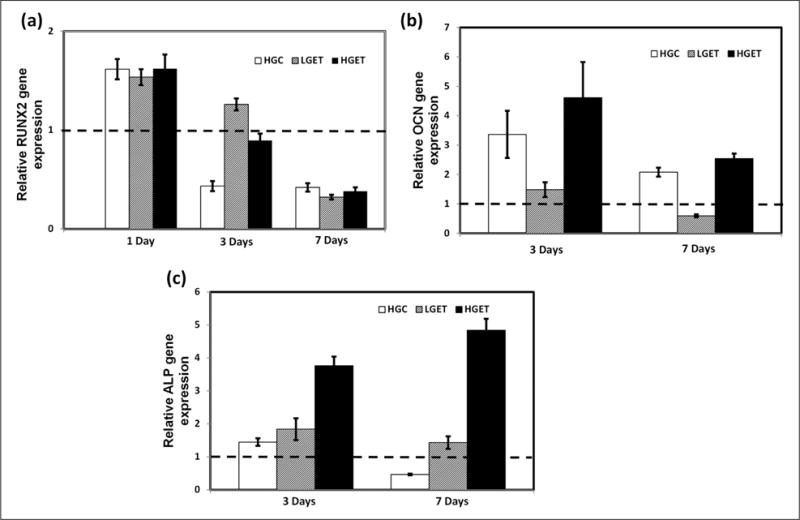
Relative gene expression of VSMCs cultured in the presence of low-glucose baseline control (LGC), high-glucose control (HGC), low glucose + elastin peptides + TGF-β1 (LGET) and high glucose + elastin peptides + TGF-β1 (HGET): (a) relative RUNX2 gene expression after 1, 3 and 7 days of exposure to glucose, elastin peptides and TGF-β1, (b) relative ALP gene expression after 3 and 7 days of exposure to glucose, elastin peptides and TGF-β1 and (c) relative OCN gene expression after 3 and 7 days of exposure to glucose, elastin peptides and TGF-β1. Bars represent gene expression relative to cells exposed to low glucose baseline control.
VSMC: vascular smooth muscle cell; TGF-β1: transforming growth factor-beta 1; RUNX2: runt-related transcription factor 2; ALP: alkaline phosphatase; OCN: osteocalcin.
Figure 3.
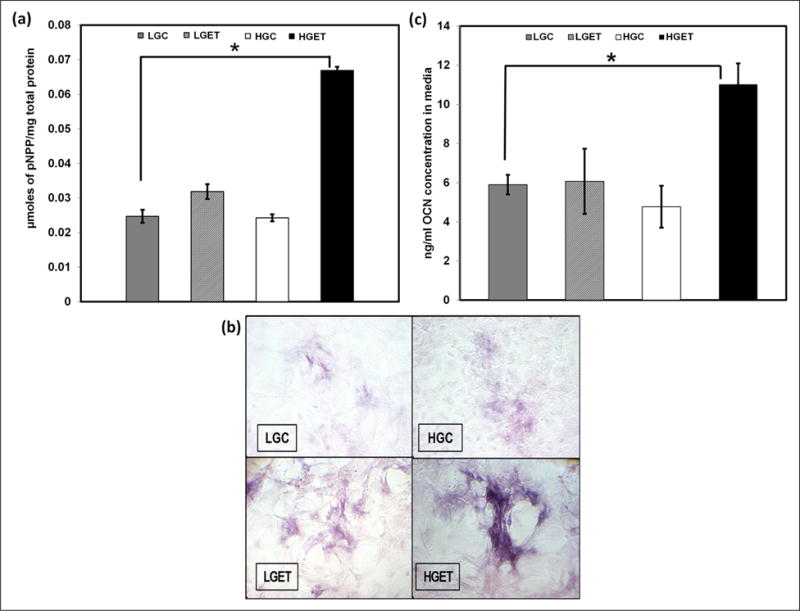
Regulation of osteogenic protein activity by VSMCs cultured in the presence of low-glucose baseline control (LGC), high-glucose control (HGC), low glucose + elastin peptides + TGF-β1 (LGET) and high glucose + elastin peptides + TGF-β1 (HGET): (a) alkaline phosphatase activity in cell lysates, (b) histochemical staining of enzyme activity in VSMCs and (c) levels of OCN protein secreted by VSMCs in the culture media. (For color figure, refer to the online version of the paper.)
VSMC: vascular smooth muscle cell; TGF-β1: transforming growth factor-beta 1; OCN: osteocalcin; pNPP: p-nitrophenol phosphate.
Role of ELR-1and ALK-5 in osteogenesis
It has been shown that TGF-β1 binds to the ALK-5 receptor on cells to cause a series of cellular cascade that are responsible for a variety of cellular phenomenon.17 Similarly, EDPs bind to elastin–laminin receptor (ELR) on VSMCs. Activation of ELR-1 has been shown to regulate a variety of biological and pathological phenomena including cell proliferation18 and chemotaxis.19 Thus, we wanted to test whether these receptors are involved in rendering osteogenic response in VSMCs. When cells were exposed to high concentration of glucose, along with EDP and TGF-β1, both ELR-1 (Figure 4(a)) and ALK-5 (Figure 4(b)) receptors on VSMCs were overexpressed, showing that these receptors may be involved in signalling towards osteogenic pathway. Glucose concentration in the culture medium did not affect the expression of ELR-1 and ALK-5 as negligible signal was detected in the LGC and HGC groups and no detectable difference was observed between the LGET and HGET treatments. We further blocked the respective receptors by incubating the cells with agents known to block their activities: ELR-1 with 5 mM lactose and TGF-β1 receptor ALK-5 by 10 μM SB-431542. Relative gene expression was studied at the end of 1 and 3 days. Figure 5(a) shows that inhibition of ELR-1 and ALK-5 caused significant down-regulation in the RUNX2, ALP and OCN relative gene expression in these cells, even though they were exposed to high concentration of glucose, EDPs and TGF-β1. These data clearly suggest that ELR-1 and ALK-5-mediated signalling is required for osteogenesis of VSMCs. Interestingly, blocking only one of the two receptors had similar effect in inhibition of osteogenic markers (Figure 5(a)). These data are suggestive of crosstalk between the two receptors: suppression of one leading to the down-regulation of the other.
Figure 4.
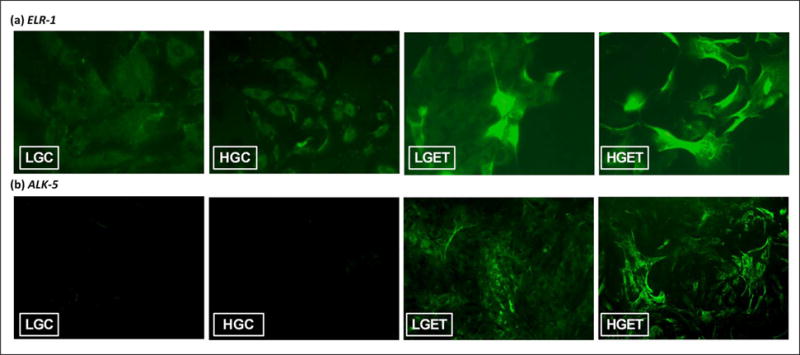
Immunocytochemical detection of (a) ELR-1 and (b) ALK-5 on VSMCs when exposed low-glucose baseline control (LGC), high-glucose control (HGC), low glucose + elastin peptides + TGF-β1 (LGET) and high glucose + elastin peptides + TGF-β1 (HGET). (For color figure, refer to the online version of the paper.)
ELR-1: elastin–laminin receptor-1; ALK-5: activin receptor-like kinase-5; VSMC: vascular smooth muscle cell; TGF-β1: transforming growth factor-beta 1. Original magnification, 200×.
Figure 5.
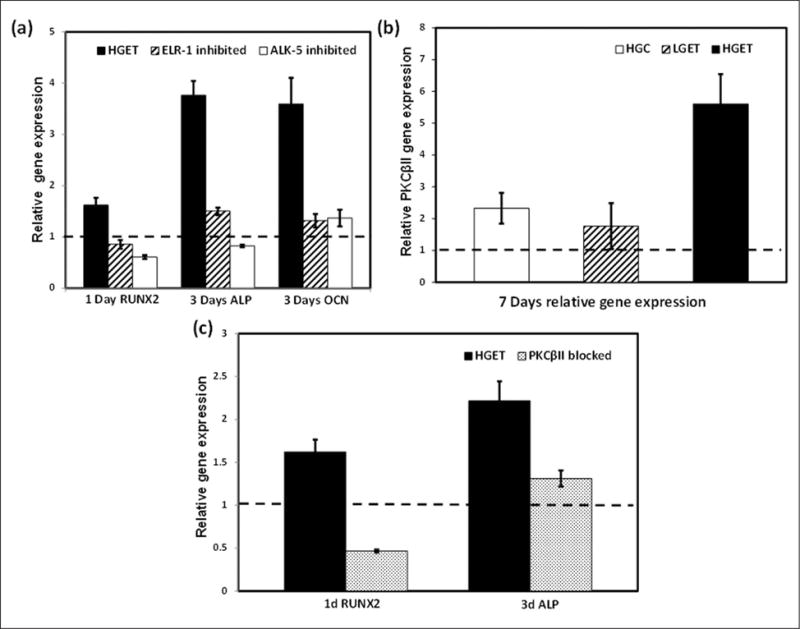
(a) Relative gene expression of RUNX2, ALP and OCN by VSMCs when exposed to high glucose + elastin peptides + TGF-β1 (HGET), 5 mM lactose (ELR-1 inhibited) and 10 μM SB-431542 (ALK-5 inhibited), (b) PKCβII expression showing highest levels in HGET group when compared to others relative to low-glucose baseline control (LGC) and (c) blocking of PKCβII reduces gene expression of RUNX2 and ALP.
RUNX2: runt-related transcription factor 2; ALP: alkaline phosphatase; OCN: osteocalcin; VSMC: vascular smooth muscle cell; TGF-β1: transforming growth factor-beta 1; ELR-1: elastin–laminin receptor-1; ALK-5: activin receptor-like kinase-5; PKCβII: protein kinase C beta II.
Bars represent gene expression relative to cells exposed to LGC baseline control.
Involvement of PKCβII in osteogenesis
Involvement of the protein kinase C (PKC) signalling pathway in the up-regulation of RUNX2, OCN and ALP has also been studied in the literature.5 We wanted to investigate the involvement of PKCβII in our cell culture model of osteogenesis. At the end of 7 days, cells exposed to high glucose, elastin peptides and TGF-β1 showed a five times greater PKCβII relative gene expression (Figure 5(b)), which was confirmed by higher immunofluorescence staining for PKCβII in HGET (data not shown). In addition to exposing cells to GF109203X (a potent inhibitor of PKCβ) in the presence of high glucose, elastin peptides and TGF-β1, a strong down-regulation of two representative osteogenic markers (RUNX2 and ALP) was noted (Figure 5(c)).
Discussion
Our results indicate that the presence of high concentration of glucose amplifies the osteogenesis of VSMCs in the presence of elastin peptides and TGF-β1. Our data also indicate that a crosstalk in the ELR-1 and ALK-5-mediated transduction pathways is involved in osteogenic response.
MAC is a commonly observed pathology in diabetes, end-stage renal disease (ESRD) and ageing. MMP-mediated elastin degradation releases soluble elastin peptides, which have been shown to induce a wide range of chemotactic effects, angiogenesis,20 cell proliferation21 and proteolytic activity.22 In previous publications, we have demonstrated that TGF-β1, when coupled with elastin peptides in a dose-dependent manner, induces osteogenesis in VSMCs12 and skin fibroblasts23 by activating the ELR-1 and overexpressing three representative osteogenic markers namely, RUNX2, ALP and OCN. In addition, these additives also increase the cellular production of MMP-2, which can further exacerbate elastin degradation and release of soluble elastin peptides. However, in that study, normal DMEM was used that contains a very high level of glucose. It was unclear whether the osteogenic transformation effects were due to exposure of cells to high glucose.
Our results here indicate that under high glucose conditions, relative gene expression of RUNX2 is up-regulated by 1.5 (p < 0.05) times compared to baseline low glucose control group at 1 day. RUNX2 expression decreases at 3 days onwards; however, two other osteogenic genes namely ALP and OCN show a 4.7-fold and 2.5-fold (p < 0.05) overexpression, respectively, at 3 days and continually overexpressed at 7 days. Interestingly, although RUNX2 is equally up-regulated in both LGET and HGET groups after 24 h, the sustenance of ALP and OCN relative gene expression is visible only in the presence of high glucose. In addition, the ALP enzymatic activity is significantly greater in the HGET groups at the end of 7 days. Our data suggest that exposure to elastin peptides and TGF-β1 alone may initiate the process of osteogenic transformation by overexpression of RUNX2, earliest transcription factor marker for osteogenesis; however, progression of osteogenic pathway needs high-glucose conditions. ALP and OCN genes and proteins were only overexpressed when cells were exposed to EDPs and TGF-β1 in the presence of high glucose.
It should be noted that high glucose alone did not affect osteogenic gene expression. Hyperglycaemia has been implicated in vascular calcification in several in vitro studies. Chen et al.5 have shown the increase in the relative gene expression of ALP and OCN and formation of calcifying nodules by VSMCs in a hyperglycaemic culture medium. Liu et al.24 have also corroborated the importance of RUNX2 in diabetic vascular calcification. They have shown an increase in intracellular calcium content, greater relative expression of RUNX2, ALP and bone morphogenetic protein (BMP2) in VSMCs in the presence of high glucose concentrations.24 Subsequent BMP2 inhibition via noggin partially blocked RUNX2 expression and osteoblastic differentiation of smooth muscle cells. BMPs, which belong to the TGF-β superfamily, have also shown to strongly mediate arterial calcification in diabetic rats as well as in a cell culture model of hyperglycaemia.25 Our results are in agreement with current understanding of hyperglycaemic induction of calcification of smooth muscle cells; however, it is of notable importance that our experimental set-up is devoid of any external calcifying/phosphate-elevating agents, such as β-glycerophosphate, that are used in aforementioned in vitro cell culture studies. Addition of these agents itself can cause cells to show osteogenic activity.26,27 We show that exposure of elastin peptides (due to degradation of elastic fibres8) and TGF-β1 (due to its release from TGF-β1-binding protein associated with elastic fibre11), and high local concentration of glucose (diabetic), conditions known to occur in diabetic vascular pathology, can cause smooth muscle cellular transformation to osteoblast-like cellular behaviour.
TGF-β1 is a cytokine with a wide range of biological functions regulating cell–matrix interactions, cellular proliferation and inflammation. In particular, TGF-β1 has been observed in association with vascular calcification and smooth muscle cell transdifferentiation.28,29 TGF-β1 has also been shown to mediate apoptosis-induced calcification of sheep aortic valve interstitial cells.30 These cells develop characteristic calcifying nodules under the effect of TGF-β1 and express ALP and apoptosis markers. Our results indicate that TGF-β1 and elastin peptides cause a visible morphological shift in the VSMCs, which is independent of glucose levels in the culture medium. This is in agreement with previous publications where VSMCs have undergone a spindle to cuboidal morphological shift under the effect of the cytokine tumour necrosis factor-alpha (TNFα).31 It is unknown how high glucose levels alter cellular interactions of growth factors and EDPs. Some previous studies shed light on these events. For example, TGF-β1 has been shown to increase the cellular uptake of glucose by activating the glucose transporter 1 (GLUT1) receptors in mesangial cells.32 This TGF-β1–GLUT1 axis can influence the cellular glucose metabolism resulting in vascular pathology. Induction of PKC pathway in diabetic tissues has been co-related with several vascular complications. Up-regulated activation of the diacylglycerol (DAG)–PKC pathway in the aorta and heart has been demonstrated in diabetic animal models,33,34 and subsequent blocking of PKC has shown mitigation of vascular35 and glomerular dysfunction.36 In vitro cellular PKC activation by vascular endothelial and smooth muscle cells under hyperglycaemic conditions has also been established.37–39 Blocking of PKCβ has also shown a significant down-regulation in the TGF-β1 activity in diabetic rat models, suggesting positive correlation between them.36,40
ELR-1 is also suspected to increase cellular DAG concentrations, thereby facilitating PKC translocation in the cells.41 Thus, EDPs in the presence of high glucose may enhance ELR-1 activity and PKC translocation and thus increase ALK-5 activity. Consistent with the current understanding, our data indicate an increased PKCβII expression by VSMCs under the influence of high glucose, EDP and TGF-β1. Finally, involvement of the PKC signalling pathway in the up-regulation of RUNX2, OCN and ALP has also been identified.5 We show that blocking PKCβII does decrease osteogenic gene expression in SMCs. Taken together, these findings suggest the possible role of PKCβII in the osteogenic responses of VSMCs. Based on current research, we hypothesise that EDPs and TGF-β1 released by elastic fibre degradation under high glycaemic condition in vessels can lead to increased ELR-1, ALK-5 and PKCβII expression in SMCs and lead to osteogenic transformation of SMCs. Overexpression of bone proteins can lead to mineral deposition on degraded elastin. Elastin is known to have calcium-binding sites.42 This in turn can lead to elastin-specific medial calcification, as seen in diabetic patients. Further in vivo research with diabetic animal models is needed to confirm the findings of this in vitro cell culture results.
In conclusion, we have demonstrated that glucose plays a fundamental role in the osteogenesis of smooth muscle cells when coupled with EDP and TGF-β1. Blocking of ELR-1, ALK-5 or PKCβII suppresses the osteogenic response in the cells, thereby suggesting a possible crosstalk among various pathways. This glucose-mediated accelerated osteogenesis of VSMCs can broaden our understanding of MAC in diabetic patients.
Acknowledgments
The authors would like to thank Dr Agneta Simionescu for immunocytochemistry and Dr Ken Webb for PCR primers.
Funding
This study was partially supported by the grant from the National Institutes of Health (P20GM103444) and Hunter Endowment at Clemson University.
Footnotes
Conflict of interest
The authors declare that there is no conflict of interest.
References
- 1.Johnson RC, Leopold JA, Loscalzo J. Vascular calcification. Circ Res. 2006;99:1044–1059. doi: 10.1161/01.RES.0000249379.55535.21. [DOI] [PubMed] [Google Scholar]
- 2.National Diabetes Fact Sheet. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention; 2011. [Google Scholar]
- 3.Bobryshev YV, Lord RSA, Warren BA. Calcified deposit formation in intimal thickenings of the human aorta. Atherosclerosis. 1995;118:9–21. doi: 10.1016/0021-9150(95)05588-n. [DOI] [PubMed] [Google Scholar]
- 4.Janzen J, Vuong PN. Arterial calcifications: morphological aspects and their pathological implications. Z Kardiol. 2001;90(Suppl 3):6–11. doi: 10.1007/s003920170044. [DOI] [PubMed] [Google Scholar]
- 5.Chen NX, Duan D, O’Neill KD, et al. High glucose increases the expression of Cbfa1 and BMP-2 and enhances the calcification of vascular smooth muscle cells. Nephrol Dial Transplant. 2006;21:3435–3442. doi: 10.1093/ndt/gfl429. [DOI] [PubMed] [Google Scholar]
- 6.Chung AWY, Yang HHC, Sigrist MK, et al. Matrix metalloproteinase-2 and -9 exacerbate arterial stiffening and angiogenesis in diabetes and chronic kidney disease. Cardiovasc Res. 2009;84:494–504. doi: 10.1093/cvr/cvp242. [DOI] [PubMed] [Google Scholar]
- 7.Signorelli SS, Malaponte G, Libra M, et al. Plasma levels and zymographic activities of matrix metalloproteinases 2 and 9 in type II diabetics with peripheral arterial disease. Vasc Med. 2005;10:1–6. doi: 10.1191/1358863x05vm582oa. [DOI] [PubMed] [Google Scholar]
- 8.Nicoloff G, Baydanoff S, Stanimorova N, et al. Relationship between elastin-derived peptides and the development of microvascular complications: a longitudinal study in children with Type 1 (insulin-dependent) diabetes mellitus. Gen Pharmacol. 2000;35:59–64. doi: 10.1016/s0306-3623(01)00088-x. [DOI] [PubMed] [Google Scholar]
- 9.Baydanoff S, Nicoloff G, Alexiev C. Age-related changes in the level of circulating elastin-derived peptides in serum from normal and atherosclerotic subjects. Atherosclerosis. 1987;66:163–168. doi: 10.1016/0021-9150(87)90192-4. [DOI] [PubMed] [Google Scholar]
- 10.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 11.Sinha S, Heagerty AM, Shuttleworth CA, et al. Expression of latent TGF-beta binding proteins and association with TGF-beta 1 and fibrillin-1 following arterial injury. Cardiovasc Res. 2002;53:971–983. doi: 10.1016/s0008-6363(01)00512-0. [DOI] [PubMed] [Google Scholar]
- 12.Simionescu A, Philips K, Vyavahare N. Elastin-derived peptides and TGF-beta1 induce osteogenic responses in smooth muscle cells. Biochem Biophys Res Commun. 2005;334:524–532. doi: 10.1016/j.bbrc.2005.06.119. [DOI] [PubMed] [Google Scholar]
- 13.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 14.Fei T, Zhu S, Xia K, et al. Smad2 mediates activin/nodal signaling in mesendoderm differentiation of mouse embryonic stem cells. Cell Res. 2010;20:1306–1318. doi: 10.1038/cr.2010.158. [DOI] [PubMed] [Google Scholar]
- 15.Khatiwala CB, Kim PD, Peyton SR, et al. ECM compliance regulates osteogenesis by influencing MAPK signaling downstream of RhoA and ROCK. J Bone Miner Res. 2009;24:886–898. doi: 10.1359/JBMR.081240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mimori K, Komaki M, Iwasaki K, et al. Extracellular signal-regulated kinase 1/2 is involved in ascorbic acid-induced osteoblastic differentiation in periodontal ligament cells. J Periodontol. 2007;78:328–334. doi: 10.1902/jop.2007.060223. [DOI] [PubMed] [Google Scholar]
- 17.Miyazono K. TGF-beta receptors and signal transduction. Int J Hematol. 1997;65:97–104. doi: 10.1016/s0925-5710(96)00542-7. [DOI] [PubMed] [Google Scholar]
- 18.Ghuysen-Itard AF, Robert L, Jacob MP. Effect of elastin peptides on cell proliferation. C R Acad Sci III. 1992;315:473–478. [PubMed] [Google Scholar]
- 19.Mecham RP, Griffin GL, Madaras JG, et al. Appearance of chemotactic responsiveness to elastin peptides by developing fetal bovine ligament fibroblasts parallels the onset of elastin production. J Cell Biol. 1984;98:1813–1816. doi: 10.1083/jcb.98.5.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinet A, Fahem A, Cauchard J-H, et al. Elastin-derived peptides enhance angiogenesis by promoting endothelial cell migration and tubulogenesis through upregulation of MT1-MMP. J Cell Sci. 2005;118:343–356. doi: 10.1242/jcs.01613. [DOI] [PubMed] [Google Scholar]
- 21.Jung S, Rutka JT, Hinek A. Tropoelastin and elastin degradation products promote proliferation of human astrocytoma cell lines. J Neuropathol Exp Neurol. 1998;57:439–448. doi: 10.1097/00005072-199805000-00007. [DOI] [PubMed] [Google Scholar]
- 22.Duca L, Floquet N, Alix AJ, et al. Elastin as a matrikine. Crit Rev Oncol Hematol. 2004;49:235–244. doi: 10.1016/j.critrevonc.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 23.Simionescu A, Simionescu DT, Vyavahare NR. Osteogenic responses in fibroblasts activated by elastin degradation products and transforming growth factor-beta1: role of myofibroblasts in vascular calcification. Am J Pathol. 2007;171:116–123. doi: 10.2353/ajpath.2007.060930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu F, Zhong H, Liang JY, et al. Effect of high glucose levels on the calcification of vascular smooth muscle cells by inducing osteoblastic differentiation and intracellular calcium deposition via BMP-2/Cbfalpha-1 pathway. J Zhejiang Univ Sci B. 2010;11:905–911. doi: 10.1631/jzus.B1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boström KI, Jumabay M, Matveyenko A, et al. Activation of vascular bone morphogenetic protein signaling in diabetes mellitus. Circ Res. 2011;108:446–457. doi: 10.1161/CIRCRESAHA.110.236596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shioi A, Nishizawa Y, Jono S, et al. Beta-glycerophosphate accelerates calcification in cultured bovine vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1995;15:2003–2009. doi: 10.1161/01.atv.15.11.2003. [DOI] [PubMed] [Google Scholar]
- 27.Jono S, McKee MD, Murry CE, et al. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000;87:e10–e17. doi: 10.1161/01.res.87.7.e10. [DOI] [PubMed] [Google Scholar]
- 28.Watson KE, Bostrom K, Ravindranath R, et al. TGF-beta 1 and 25-hydroxycholesterol stimulate osteoblast-like vascular cells to calcify. J Clin Invest. 1994;93:2106–2113. doi: 10.1172/JCI117205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grainger DJ, Metcalfe JC, Grace AA, et al. Transforming growth factor-beta dynamically regulates vascular smooth muscle differentiation in vivo. J Cell Sci. 1998;111:2977–2988. doi: 10.1242/jcs.111.19.2977. [DOI] [PubMed] [Google Scholar]
- 30.Jian B, Narula N, Li QY, et al. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75:457–465. doi: 10.1016/s0003-4975(02)04312-6. discussion 465–456. [DOI] [PubMed] [Google Scholar]
- 31.Tintut Y, Patel J, Parhami F, et al. Tumor necrosis factor-alpha promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation. 2000;102:2636–2642. doi: 10.1161/01.cir.102.21.2636. [DOI] [PubMed] [Google Scholar]
- 32.Inoki K, Haneda M, Maeda S, et al. TGF-beta 1 stimulates glucose uptake by enhancing GLUT1 expression in mesangial cells. Kidney Int. 1999;55:1704–1712. doi: 10.1046/j.1523-1755.1999.00438.x. [DOI] [PubMed] [Google Scholar]
- 33.Inoguchi T, Battan R, Handler E, et al. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci U S A. 1992;89:11059–11063. doi: 10.1073/pnas.89.22.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inoguchi T, Xia P, Kunisaki M, et al. Insulin’s effect on protein kinase C and diacylglycerol induced by diabetes and glucose in vascular tissues. Am J Physiol. 1994;267:E369–E379. doi: 10.1152/ajpendo.1994.267.3.E369. [DOI] [PubMed] [Google Scholar]
- 35.Ishii H, Jirousek MR, Koya D, et al. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science. 1996;272:728–731. doi: 10.1126/science.272.5262.728. [DOI] [PubMed] [Google Scholar]
- 36.Koya D, Haneda M, Nakagawa H, et al. Amelioration of accelerated diabetic mesangial expansion by treatment with a PKC beta inhibitor in diabetic db/db mice, a rodent model for type 2 diabetes. FASEB J. 2000;14:439–447. doi: 10.1096/fasebj.14.3.439. [DOI] [PubMed] [Google Scholar]
- 37.Williams B, Schrier RW. Characterization of glucose-induced in situ protein kinase C activity in cultured vascular smooth muscle cells. Diabetes. 1992;41:1464–1472. doi: 10.2337/diab.41.11.1464. [DOI] [PubMed] [Google Scholar]
- 38.Kunisaki M, Bursell SE, Umeda F, et al. Normalization of diacylglycerol-protein kinase C activation by vitamin E in aorta of diabetic rats and cultured rat smooth muscle cells exposed to elevated glucose levels. Diabetes. 1994;43:1372–1377. doi: 10.2337/diab.43.11.1372. [DOI] [PubMed] [Google Scholar]
- 39.Lee TS, Saltsman KA, Ohashi H, et al. Activation of protein kinase C by elevation of glucose concentration: proposal for a mechanism in the development of diabetic vascular complications. Proc Natl Acad Sci U S A. 1989;86:5141–5145. doi: 10.1073/pnas.86.13.5141. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 40.Koya D, Jirousek MR, Lin YW, et al. Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J Clin Invest. 1997;100:115–126. doi: 10.1172/JCI119503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fulop T, Jr, Douziech N, Jacob MP, et al. Age-related alterations in the signal transduction pathways of the elastin-laminin receptor. Pathol Biol (Paris) 2001;49:339–348. doi: 10.1016/s0369-8114(01)00143-2. [DOI] [PubMed] [Google Scholar]
- 42.Urry DW. Neutral sites for calcium ion binding to elastin and collagen: a charge neutralization theory for calcification and its relationship to atherosclerosis. Proc Natl Acad Sci U S A. 1971;68:810–814. doi: 10.1073/pnas.68.4.810. [DOI] [PMC free article] [PubMed] [Google Scholar]