

Abstract
NIMA-related kinase 2 (Nek2) is often upregulated in human cancer and is important in regulating the cell cycle and gene expression, and maintaining centrosomal structure and function. The present study aimed to investigate the expression pattern, clinical significance, and biological function of Nek2 in hepatocellular carcinoma (HCC). mRNA and protein levels of Nek2 were examined in HCC and corresponding normal liver tissues. The MTT and soft agar colony formation assays, and flow cytometry were employed to assess the roles of Nek2 in cell proliferation and growth. In addition, western blot analysis was performed to assess the expression of cell cycle- and proliferation-related proteins. The results revealed that Nek2 was upregulated in HCC tissues and cell lines. The clinical significance of Nek2 expression was also analyzed. Inhibiting Nek2 expression by siRNA suppressed cell proliferation, growth, and colony formation in hepatocellular carcinoma cell line HepG2 cells, induced cell cycle arrest in the G2/M phase by retarding the S-phase, and promoted apoptosis. Furthermore, Nek2 depletion downregulated β-catenin expression in HepG2 cells and diminished expression of Myc proto-oncogene protein (c-Myc), cyclins D1, B1, and E and cyclin-dependent kinase 1, whilst increasing protein levels of p27. This demonstrates that overexpression of Nek2 is associated with the malignant evolution of HCC. Targeting Nek2 may inhibit HCC cell growth and proliferation through the regulation of β-catenin by the Wnt/β-catenin pathway and therefore may be developed as a novel therapeutic strategy to treat HCC.
Keywords: hepatocellular carcinoma, Nek2, cancer proliferation, wnt/β-catenin
Introduction
Hepatocellular carcinoma (HCC) is the third most common cancer worldwide (1). Each year, >620,000 patients are diagnosed with HCC, and the 1-year survival rate remains <50% (2). Over the past few decades, HCC age-adjusted incidence rates have doubled (3), and primary liver cancer mortality rates have increased faster than the mortality rates of most other cancers (4).
Current therapeutic modalities for HCC include curative options, such as surgical resection, liver transplantation, and local ablation; or palliative procedures, such as catheter-directed therapies and systemic therapy (5). The main risk factors for HCC are viral hepatitis, alcohol abuse, aflatoxin B1-contaminated food, nonalcoholic fatty liver disease, and metabolic disorders (6). However, not all individuals exposed to these factors possess the same risk of developing HCC. It is a multifactorial disease, with a wide range of genetic and epigenetic alterations identified as contributory to the deregulation of key oncogenes and tumor-suppressor genes; however, genetic events in hepatic carcinogenesis are poorly understood (7). Previous studies undertaken by our group uncovered potential molecular targets for HCC treatment, by comparing the gene expression patterns of HCC with those of normal liver tissues using a cDNA expression microarray containing 1361 unique genes (8). Among the genes explored, NIMA-related kinase 2 (Nek2), also known as Serine/threonine-protein kinase 2, was identified as a potential target gene as it is highly expressed in HCC.
Nek2 is a member of the serine/threonine kinase family, and is important in regulating the cell cycle, gene expression, and maintaining centrosomal structure and function (9–11). Nek2 belongs to the never in mitosis A (NIMA)-related family of kinases, otherwise known as Neks (9). The Nek family comprises 11 members, all of which have been identified in mammals (9). Given the role of Nek2 in regulating centrosomal function during the S- and G2-phases, Nek2 regulation is strictly controlled throughout the cell cycle, with a hinged expression in such phases (12). Functional research has implicated Nek2 in the regulation of centrosome separation and spindle formation (9,13). Furthermore, high Nek2 expression levels have been reported in cell lines derived from breast, cervical, prostate, and colorectal cancers, as well as those from cholangiocarcinoma and lymphoma (14–17). A recent study involving a cohort of normal liver tissues, chronic liver disease induced by the hepatitis C virus, and HCC, identified Nek2 co-expression with several closely related genes (18).
The present study aims to investigate Nek2 expression and biological regulation in human HCC, analyze the association of Nek2 expression with the clinicopathological characteristics of HCC patients, and examine the Nek2-mediated control of cell growth and proliferation to explore the potential molecular mechanisms involved in the progress of HCC.
Materials and methods
Patients and tissue samples
Patients were recruited from the First Municipal People's Hospital of Guangzhou, and clinical information was acquired from hospital registries. Primary tumor specimens were obtained from 52 patients diagnosed with HCC, all of whom provided informed consent for participation in the study. All 52 patients underwent routine resection in the First Municipal People's Hospital of Guangzhou between March 2010 and November 2012. HCC was diagnosed according to the World Health Organization histological classification of tumors of the liver and intrahepatic bile ducts (2000). All surgical specimens were immediately snap-frozen in liquid nitrogen and stored at −80°C until RNA and protein extraction. The samples were obtained from tumor tissues and corresponding adjacent healthy tissues 5–10 cm from each tumor <30 min following surgery. Clinical histopathological data including histopathological diagnosis, Edmondson-Steiner stage, and tumor grade were obtained from patient medical records. The present study was approved by the Ethics Committee of First Municipal People's Hospital of Guangzhou, Guangdong, China.
Cell culture and transfection
The human hepatocellular carcinoma cell lines of HepG2, Hep3B, BEL-7402, SMMC-7721, QCY-7701, PLC/PRF/5, and the human hepatocyte cell line HL-7702, were obtained from the cell repository of Sun Yat-sen University (Guangzhou, Guangdong, China). All cells were cultured at 37°C in a humidified atmosphere with 5% CO2 in Dulbecco's modified eagle medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal calf serum (Invitrogen; Thermo Fisher Scientific Inc.) and 100 IU/ml penicillin-streptomycin solution (Corning Inc., Corning, NY, USA). The cells were grown on sterilized culture dishes (Corning Inc.), harvested, and passaged with 0.25% trypsin-EDTA (Corning, Inc.) upon reaching 80% confluence. X-treme GENE siRNA Transfection Reagent (Roche Diagnostics GmbH, Mannheim, Germany) was used for siRNA transfection according to the manufacturer's instructions. Protein levels were assessed 72 h later by western blot analysis, and mRNA levels were quantified 48 h later by reverse transcription-quantitative polymerase chain reaction (RT-qPCR). The anti-Nek2 siRNA (Sc43960; Santa Cruz Biotechnology Inc., Dallas, TX, USA) and control siRNA (Sc37007; Santa Cruz Biotechnology, Inc.) were used to transfect HepG2 cells.
Immunohistochemistry (IHC)
IHC experiments were conducted on 5 µm formalin-fixed, paraffin-embedded sections of healthy tissue and tumor samples. The tissue sections were mounted on slides, treated with xylene, and rehydrated using alcohol-water mixtures before heat treatment in 10 mM citric acid (pH 6.0) for antigen retrieval. Endogenous peroxidase activity was stopped by the use of 0.3% H2O2 solution. After blocking with 10% bovine serum albumin (Beyotime Institute of Biotechnology, Haimen, China; ST023) the sections were incubated at 4°C overnight with primary human Nek2 antibodies (Abcam, Cambridge, UK; Ab55550; 1:400 dilution), rinsed, and incubated with biotinylated anti-mouse IgG (Dako North America Inc., Carpinteria, CA, USA; F0232; 1:100 dilution) for 30 min at room temperature. After rinsing, the samples were treated with streptavidin and biotinylated horseradish peroxidase (Dako North America Inc.). Counterstaining with hematoxylin was performed, and the sections were dehydrated in ethanol before mounting. The sections were analyzed using optical microscopy (Nikon Corp., Tokyo, Japan; magnification, ×100 or ×400). Staining specificity was confirmed using positive (gastric mucosa) and negative (heart muscle tissue) controls. Low Nek2 expression was defined as <10% and high expression as >10%.
RT-qPCR
Total RNA was extracted from HepG2 cells or tissues using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. Complementary DNA was synthesized from 1.5 µg of total RNA at 42°C for 60 min using the SYBR-Green PCR master mix (Thermo Fisher Scientific, Inc.; 4368577) and RT-qPCR was performed using the Brilliant II Sybr Green QPCR Master Mix (Agilient Technologies, Inc., Santa Clara, CA, USA). GAPDH was used as the reference gene. The primer sequences were as follows: Forward, 5′-CCAGCCCTGTATTGAGTG-3′ and reverse, 5′-ACTTCCGTTCCTTTAGCA-3′ for Nek2; forward 5′-GACCTGACCTGCCGTCTA-3′, and reverse, 5′-AGGAGTGGGTGTCGCTGT-3′ for GAPDH. The relative levels of gene expression were represented as ΔCq, and the fold-change of gene expression was calculated using the 2−ΔΔCq method.
Western blotting
The cells and tissue samples were lysed with 20 mmol/l Tris-HCl (pH 8.0), 137 mmol/l NaCl, 10% glycerol, 1% Triton X-100, and 2 mmol/l EDTA containing protease and phosphatase inhibitors at 4°C. Each lysate sample (20 µg) was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. The membranes were incubated overnight at 4°C with antibodies against Nek2 (Santa Cruz Biotechnology, Inc.; Sc55601; 1:1,000 dilution), β-catenin, C-Myc, CycD1, CycB1, CDK1, CycE, and P27 (Santa Cruz Biotechnology, Inc., 1:1,000 dilution). The membranes were developed using enhanced chemiluminescence. The Nek2 expression levels were normalized to that of GAPDH, and were detected using mouse anti-GAPDH antibody (Santa Cruz Biotechnology, Inc.; Sc365062; 1:2,000 dilution).
Cell growth and proliferation
The cultured HepG2 cells were pelleted by centrifugation at 900 × g and re-suspended in phenol red-free medium supplemented with 10% charcoal-stripped fetal bovine serum (HyClone; GE Healthcare Life Sciences, Logan, UT, USA) and 2% L-glutamine. The HepG2 cells were then plated in triplicate, (1×103 cells/ml) in 96-well micro plates. This process was repeated six times. After seeding for 24 h, the HepG2 cells were transfected with either anti-Nek2 or control siRNAs (10 nM each) using the X-treme GENE siRNA Transfection Reagent (Roche Diagnostics GmbH) according to the manufacturer's protocol. The cells were counted daily over a 5-day period through the Cell Titer 96 Non-Radioactive cell proliferation assay (Promega Corp., Madison, WI, USA). Following this, 20 µl of 5 mg/ml MTS solution was added to each well, and the resulting mixture was incubated at 37°C for 4 h. Each solution was measured by a Bio-Rad micro plate reader (Bio-Rad Laboratories Inc.) at a test wavelength of 490 nm.
Cell colony formation
In the anchorage-independent colony growth assay, the HepG2 cells were transfected with siRNA for 48 h. A 12-well cell culture plate containing 0.5% agarose medium on top of regular medium containing 1% low-melting agar was seeded with ~500 transfected cells. After 15 days, the resulting colonies were stained with Giemsa and counted under a microscope.
Cell cycle and apoptosis analysis
HepG2 cells were seeded in 8 cm sterilized culture dishes for 12 h. The cells were then transfected with indicated amounts of anti-Nek2 or control siRNAs. At indicated time points, the cells were harvested, fixed with 1% paraformaldehyde, washed with phosphate buffer solution (PBS), and stained with 5 mg/ml propidium iodide (PI) in PBS supplemented with RNase A (Sigma-Aldrich, St. Loius, MO, USA) for 30 min at room temperature. Data were collected using a BD FACSCAlibur flow cytometer (BD Biosciences, San Jose, CA, USA). Apoptosis was measured using an Annexin V/FITC kit (Sigma-Aldrich) according to the manufacturer's protocol. Each assay was repeated in triplicate.
Statistical analyses
All statistical calculations were performed using SPSS 13.0 software (SPSS Inc., Chicago, IL, USA). A chi-square test was used to examine possible correlations between Nek2 expression and clinicopathological factors. The differences in the densitometry data on focus numbers between the control and Nek2-transfected cells were analyzed using Student's t-test. P<0.05 was considered to indicate a statistically significant result in both Student's t-test and the chi-square test.
Results
Nek2 expression in human HCC tissue and cell lines
Nek2 expression in resected human tissue specimens from healthy and cancerous livers was analyzed. Fifty-two HCC and normal samples were examined using Nek2-specific IHC staining antibodies (Fig. 1). The IHC results revealed clear differences in Nek2 expression between healthy and cancerous liver tissues. In the healthy and adjacent liver tissues, Nek2 was not expressed (Fig. 1A). By contrast, most HCC cases presented elevated protein levels in the nuclei (Fig. 1B-D). Expression of Nek2 was examined in 17 pairs of snap-frozen HCC and adjacent nonmalignant liver tissues by western blotting. Levels of Nek2 protein were significantly elevated in the HCC specimens (T) compared with those in adjacent nonmalignant tissues (N; Fig. 2A). Meanwhile, analysis of Nek2 transcription by RT-qPCR and western blot analysis demonstrated that HepG2 cells expressed significantly higher levels of Nek2 mRNA and protein compared to the other six cell lines (HL-7702, PLC/PRF/5, Hep3B, BEL-7402, SMMC-7721, and QCY-7701; Fig. 2B and C).
Figure 1.

Immunohistochemical analysis of Nek2 in hepatocellular carcinoma (HCC) cells and adjacent non-cancerous tissue. (A) Negative Nek2 expression in HCC tissues. (B) Strong nuclear staining of Nek2 in a case of HCC Edmondson I. (C) Strong nuclear staining of Nek2 in a case of HCC Edmondson IIa. (D) Strong nuclear staining of Nek2 in a case of HCC Edmondson III. Negative nuclear staining was observed in all non-cancerous liver cells. In neoplastic cells, elevated levels of Nek2 were observed in the nuclei.
Figure 2.

Expression levels of Nek2 in hepatocellular carcinoma (HCC), adjacent non-cancerous tissue, and HCC cell lines. (A) Expression levels of Nek2 protein in HCC tissues (T) were higher than in adjacent non-cancerous tissues (N). (B) Protein expression levels of Nek2 in a variety of HCC cell lines. (C) Nek2 mRNA levels in HCC cell lines. Both protein and mRNA levels of Nek2 were significantly higher in HCC than those in control (HL-7702 cell line; *P<0.05, compared with HL-7702 cell line).
Associations between Nek2 expression and the clinicopathological parameters of HCC patients
Thirty (57.6%) out of 52 tumor tissues expressed Nek2 (Table I), and high levels of Nek2 were identified predominantly in the nuclei of cancerous liver cells. The correlation between Nek2 protein expression and the histological staging of liver cancer was analysed (Table I). Rates of Nek2 expression in tumors classed as Edmondson-Steiner stages I and II (Fig. 1B and C) were 52.38% (22/42, Table I) and those with Edmondson-Steiner stages III and IV (Fig. 1D) were 80% (8/10; Table I). The difference in Nek2 expression between samples from the two tumor classes was statistically significant (P<0.05). Among all the samples, those with concomitant portal venous tumor emboli exhibiting Nek2 expression comprised 90.0% (9/10), however, only 50.0% (21/42) without portal venous tumor emboli expressed Nek2 (Table I). A significant difference in Nek2 expression was noted in tumor malignancy (P<0.05). Nek2 expression was observed to be independent of episode age, tumor size, Hepatic Function and liver α-fetoprotein levels (Table I).
Table I.
Association of Nek2 expression with clinicopathological characteristics of patients with HCC.
| Characteristics | Patients, no. | Nek2 positive, no. (%) | χ2 | P-value |
|---|---|---|---|---|
| Age, years | ||||
| <60 | 17 | 11 (64.7) | 0.632 | 0.426 |
| ≥60 | 35 | 19 (54.3) | ||
| Tumor size, cm | ||||
| <5 | 28 | 17 (60.9) | 1.213 | 0.271 |
| ≥5 | 24 | 13 (54.2) | ||
| Hepatic function | ||||
| Child A | 15 | 9 (60) | 0.001 | 0.971 |
| Child B | 37 | 21 (56.7) | ||
| Edmondson stage | ||||
| Stage I and II | 42 | 22 (52.4) | 4.434 | 0.037a |
| Stage III and IV | 10 | 8 (80) | ||
| Portal vein tumor thrombus | ||||
| Positive | 10 | 9 (90) | 5.295 | 0.032a |
| Negative | 42 | 21 (50) | ||
| AFP | ||||
| Positive | 44 | 24 (54.5) | 1.160 | 0.281 |
| Negative | 8 | 6 (75.0) |
P<0.05. HCC, hepatocellular carcinoma; AFP, α-fetoprotein.
Inhibition of Nek2 suppresses HepG2 proliferation
Using siRNA as an efficient tool for Nek2 gene silencing in the HepG2 cell line, the role of Nek2 in HCC cell proliferation was analyzed. The proliferation rate was determined by MTS assay, assessing cell metabolic activity. A decrease in proliferation was observed in the HepG2 cells treated with anti-Nek2 siRNA compared with the cells transfected with the control siRNA and blank group (Fig. 3A). Consistent with the MTS result, the soft agar colony formation assays showed that Nek2 knockdown in HepG2 cells led to a significant decrease in focus number [1183±97 (Blank), 1232±28 (Control) vs. 160±14 (siNek2), P<0.05; Fig. 3B]. The cell viability of HepG2 cells treated with Nek siRNA decreased significantly compared with the control and blank groups (P<0.05; Fig. 3C).
Figure 3.

HepG2 cell proliferation and cell colony formation was down regulated by Nek2 inhibits. (A) Significant decreases in proliferation, (B) cell colony formation, and (C) cell viability were observed in HepG2 cells treated with Nek2 siRNA compared with cells transfected with control siRNA (control) and blank group (blank). *P<0.05, compared with siNek2 group.
Effect of Nek2 on cell cycle and apoptosis of HepG2
The cell cycle of HepG2 cells transfected with either control siRNA or anti-Nek2 siRNA was analysed using PI staining followed by fluorescence-activated cell sorting. This revealed that the proportion of cells transfected with anti-Nek2 siRNA in the G2/M phase decreased. Furthermore, the proportion of cells treated with anti-Nek2 siRNA in the S-phase increased, whereas the proportion progressing through the G1/S-phase decreased (Fig. 4A). These observations suggest that Nek2 knockdown by specific siRNA in HepG2 cells induces cell cycle arrest in the G2/M phase by retarding the S-phase. Moreover, apoptosis analysis revealed that HepG2 cells treated with anti-Nek2 siRNA exhibited a significantly higher apoptotic index than controls (Fig. 4B).
Figure 4.

Effects of downregulating Nek2 on the cell cycle and apoptosis in HepG2 cells. (A) Nek2 knockdown increased the proportion of cells in the S-phase and decreased the proportion in the G2/M phase. Nek2 knockdown in HepG2 cells induces a cell cycle arrest in G2/M phase by slowing S-phase. (B) The apoptosis assay shows that HepG2 cells treated by Nek2 siRNA had a significantly higher apoptotic index than the control siRNA treated cells and blank cells. *P<0.05, compared with siNek2 group.
Nek2 regulates HCC growth through the β-catenin/Wnt pathway
The possible mechanisms of Nek2 involvement in HCC were detected by evaluating the protein levels of its established targets, including β-catenin, C-Myc, CycD1, CycB1, CDK1, CycE, and P27 genes. These are all involved in the β-catenin/Wnt pathway and are essential for cell growth and proliferation. Western blot analysis indicated that β-catenin protein expression was compromised in HepG2 cells treated with Nek2-specific siRNA (Fig. 5). Furthermore, Nek2 inhibition resulted in the diminished expression of C-Myc, CycD1, CycB1, CDK1, and CycE proteins. Treatment of HepG2 cells with control siRNA did not influence the protein expression of any of the tested targets. However, western blot analysis also revealed that Nek2 knock down in the HepG2 cells led to increased P27 protein expression. Overall, these results suggest that Nek2 expression induces cell cycle progression and proliferation by regulating the β-catenin/Wnt pathway.
Figure 5.
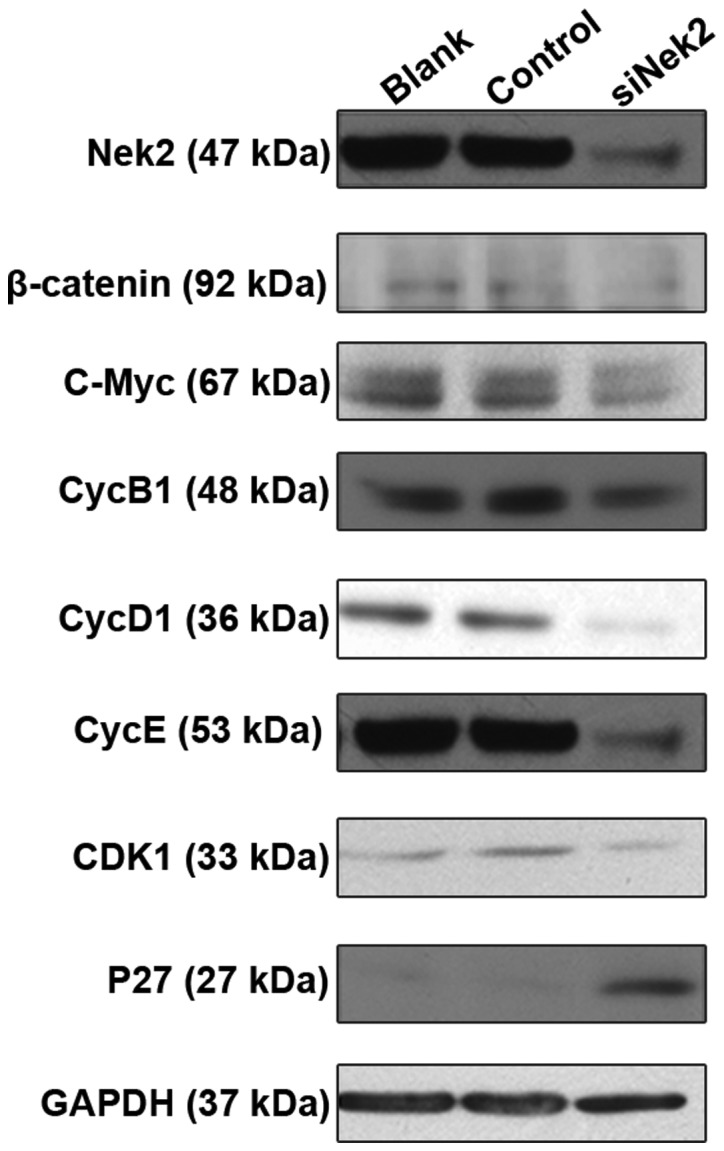
Western blot analysis of HepG2 cells. Western blotting revealed that Nek2 knockdown decreased protein levels of β-Catenin, c-Myc, CycD1, CycE, CycB1, CDK1, but increased p27 levels.
Discussion
The role of Nek2 in several human cancers has been explored previously. Nek2 expression is increased in breast, cervical, and prostate carcinoma cell lines, as well as in numerous cell lines established from lymphomas (19). An assessment of ~1,700 genes revealed marked up-regulation of Nek2 in cell lines derived from Ewing's tumor and apediatric osteosarcoma (20); and an increase in Nek2 mRNA levels was subsequently reported in non-Hodgkin lymphoma (21). Furthermore, in breast cancer models, Nek2 plays a role in tumorigenesis and drug resistance (22). However, though Nek2 is well established as an oncogene, its expression pattern and clinical significance remains poorly defined in human HCC.
The present study demonstrated that the cellular location of Nek2 was notably variable, with increased cytoplasmic expression with or without nuclear Nek2 accumulation. These differential results may be explained by Nek2's varying isoform expression, as its bipartite nuclear localization sequence spans the splice site (23). Isoform-specific antibodies may enable the expression and localization of specific Nek2 splice variants for further investigation in HCC tissues.
In the present study, Nek2 expression was increased in human HCC tissues, which has not been reported previously. HCC cells expressed elevated levels of Nek2, compared to the negligible levels detected in normal tissue cells. This may be due to the low Nek2 mRNA copy numbers in healthy tissue. High Nek2 expression was detected in 22 (52.4%) Edmondson stages I and II cases, and in eight (80%) of the Edmondson stages III and IV cases, suggesting that Nek2 is positively associated with more advanced HCC. Furthermore, Nek2 expression in low-stage HCCs was lower than that in high-stage HCCs. Meanwhile, other clinical association analyses demonstrated that expression of Nek2 was positively correlated not only with tumor histological grade and tumor metastasis, but also with hepatitis C viral infection, as has been reported previously (18). Thus, Nek2 may play an important role in HCC progression and serve as a potential biomarker for the biological behavior of HCC cells.
To elucidate the function of Nek2 in HCC cells, anti-Nek2 siRNA was transfected into the HepG2 cell line, which exhibited the highest Nek2 expression among all the seven HCC cell lines tested. Nek2 knockdown decreased the proliferation rate and colony formation ability in HepG2 cells, suggesting that decreased Nek2 activity in HCC cells may decrease cancer cells' aggressive growth and increased proliferative properties. The latter hypothesis is supported by the established role of Nek2 in cell proliferation. In A549 lung adenocarcinoma cells and human embryonic kidney HEK293T cells, Nek2 overexpression led to the activation of the Akt pathway and an increase in β-catenin protein levels (24). β-Catenin binds to and is phosphorylated by Nek2, and exists in a complex with Rootletin (25). In addition, the expression of CycD1, CycE, and C-Myc genes, which are required for cell proliferation and regulated by Wnt/β-catenin signaling, is controlled by Nek2 (24,26). Consistent with these data, blocking Nek2 expression decreased levels of C-Myc, CycD1, and CycE1. The analysis of the underlying mechanisms of Nek2-mediated control of cell proliferation revealed that blocking Nek2 affected the cell cycle by retarding G2/M transition. This result was congruent with the expanded role of Nek2 as a regulator of CycB1, CDK1 and P27, all of which promote cell cycle progression and stimulate the G2/M transition.
Nek2 regulates the capacity for microtubule organization of the centrosome at the time of mitotic entry (26) and is involved in the regulation of chromatin condensation, mitotic checkpoint signaling, and cytokine activity (27). It may trigger centrosome separation at the onset of mitosis through the phosphorylation of multiple linker components.
Furthermore, Nek2 expression is positively correlated with human proliferative disease. Nek2 regulates β-catenin during centrosome separation by phosphorylation. In the cell cycle, cytoplasmic and nuclear β-catenin levels increase in the S-phase, peak at late G2/M, and abruptly decrease as the cells re-enter G1 after mitosis (28). Manipulating β-catenin levels, particularly in G2, affects centrosome maturation and function, which highlights the role of β-catenin in the centrosome (29). When cytoplasmic β-catenin is stabilized, it can translocate into the nucleus, where it interacts with a family of T cell factor/lymphocyte enhancer factor transcription factors to drive cell proliferation by directly inducting cell cycle regulators, such as c-Myc, CycB1, and CycD1 (30,31). However, high β-catenin levels may act via the Wnt signaling pathway to stimulate cellular proliferation by dysregulating the cell cycle (high C-Myc, CycD1, and CycE expression) and inhibit tumor suppresion p16, p27, and wild-type p53; (32,33).
In conclusion, Nek2 is frequently upregulated in HCC compared to healthy tissues, and is associated with HCC stages. The present study demonstrates that blocking Nek2 by siRNA inhibits HCC cell proliferation in vitro. This inhibition can be traced back to the attenuation of the expression of proteins controlling cell growth and proliferation. These results are a promising starting point for the elucidation of the functional importance of Nek2 in HCC progression. Further studies may allow Nek2 to serve as a molecular target closely associated with cell proliferation for the future development of HCC therapeutics.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (grant no. 81270037).
References
- 1.Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 3.Bodzin AS, Busuttil RW. Hepatocellular carcinoma: Advances in diagnosis, management, and long term outcome. World J Hepatol. 2015;7:1157–1167. doi: 10.4254/wjh.v7.i9.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 5.Slotta JE, Kollmar O, Ellenrieder V, Ghadimi BM, Homayounfar K. Hepatocellular carcinoma: Surgeon's view on latest findings and future perspectives. World J Hepatol. 2015;7:1168–1183. doi: 10.4254/wjh.v7.i9.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oda K, Uto H, Mawatari S, Ido A. Clinical features of hepatocellular carcinoma associated with nonalcoholic fatty liver disease: A review of human studies. Clin J Gastroenterol. 2015;8:1–9. doi: 10.1007/s12328-014-0548-5. [DOI] [PubMed] [Google Scholar]
- 7.Choi KJ, Baik IH, Ye SK, Lee YH. Molecular targeted therapy for hepatocellular carcinoma: Present status and future directions. Biol Pharm Bull. 2015;38:986–991. doi: 10.1248/bpb.b15-00231. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y. Guangzhou Medical University; 2009. Identification of differentially expressed genes in hepatocellular carcinoma by laser capture microdissection and microarray. [Google Scholar]
- 9.Fry AM. The Nek2 protein kinase: A novel regulator of centrosome structure. Oncogene. 2002;21:6184–6194. doi: 10.1038/sj.onc.1205711. [DOI] [PubMed] [Google Scholar]
- 10.Schultz SJ, Fry AM, Sütterlin C, Ried T, Nigg EA. Cell cycle-dependent expression of Nek2, a novel human protein kinase related to the NIMA mitotic regulator of Aspergillus nidulans. Cell Growth Differ. 1994;5:625–635. [PubMed] [Google Scholar]
- 11.Uto K, Sagata N. Nek2B, a novel maternal form of Nek2 kinase, is essential for the assembly or maintenance of centrosomes in early Xenopus embryos. Embo J. 2000;19:1816–1826. doi: 10.1093/emboj/19.8.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fry AM, Schultz SJ, Bartek J, Nigg EA. Substrate specificity and cell cycle regulation of the Nek2 protein kinase, a potential human homolog of the mitotic regulator NIMA of Aspergillus nidulans. J Biol Chem. 1995;270:12899–12905. doi: 10.1074/jbc.270.21.12899. [DOI] [PubMed] [Google Scholar]
- 13.Fletcher L, Cerniglia GJ, Nigg EA, Yend TJ, Muschel RJ. Inhibition of centrosome separation after DNA damage: A role for Nek2. Radiat Res. 2004;162:128–135. doi: 10.1667/RR3211. [DOI] [PubMed] [Google Scholar]
- 14.Bowers AJ, Boylan JF. Nek8, a NIMA family kinase member, is overexpressed in primary human breast tumors. Gene. 2004;328:135–142. doi: 10.1016/j.gene.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Kokuryo T, Senga T, Yokoyama Y, Nagino M, Nimura Y, Hamaguchi M. Nek2 as an effective target for inhibition of tumorigenic growth and peritoneal dissemination of cholangiocarcinoma. Cancer Res. 2007;67:9637–9642. doi: 10.1158/0008-5472.CAN-07-1489. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki K, Kokuryo T, Senga T, Yokoyama Y, Nagino M, Hamaguchi M. Novel combination treatment for colorectal cancer using Nek2 siRNA and cisplatin. Cancer Sci. 2010;101:1163–1169. doi: 10.1111/j.1349-7006.2010.01504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsunoda N, Kokuryo T, Oda K, Senga T, Yokoyama Y, Nagino M, Nimura Y, Hamaguchi M. Nek2 as a novel molecular target for the treatment of breast carcinoma. Cancer Sci. 2009;100:111–116. doi: 10.1111/j.1349-7006.2008.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drozdov I, Bornschein J, Wex T, Valeyev NV, Tsoka S, Malfertheiner P. Functional and topological properties in hepatocellular carcinoma transcriptome. Plos One. 2012;7:e35510. doi: 10.1371/journal.pone.0035510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu Z, Zhou W, Huang J, Yang Y, Wendlandt E, Xu H, He X, Tricot G, Zhan F. Nek2 is a novel regulator of B cell development and immunological response. Biomed Res Int. 2014;2014:621082. doi: 10.1155/2014/621082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wai DH, Schaefer KL, Schramm A, Korsching E, Van Valen F, Ozaki T, Boecker W, Schweigerer L, Dockhorn-Dworniczak B, Poremba C. Expression analysis of pediatric solid tumor cell lines using oligonucleotide microarrays. Int J Oncol. 2002;20:441–451. [PubMed] [Google Scholar]
- 21.Andreasson U, Dictor M, Jerkeman M, Berglund M, Sundström C, Linderoth J, Rosenquist R, Borrebaeck CA, Ek S. Identification of molecular targets associated with transformed diffuse large B cell lymphoma using highly purified tumor cells. Am J Hematol. 2009;84:803–888. doi: 10.1002/ajh.21549. [DOI] [PubMed] [Google Scholar]
- 22.Marina M, Saavedra HI. Nek2 and Plk4: Prognostic markers, drivers of breast tumorigenesis and drug resistance. Front Biosci (Landmark Ed) 2014;19:352–365. doi: 10.2741/4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu W, Baxter JE, Wattam SL, Hayward DG, Fardilha M, Knebel A, Ford EM, da Cruz e Silva EF, Fry AM. Alternative splicing controls nuclear translocation of the cell cycle-regulated Nek2 kinase. J Biol Chem. 2007;282:26431–26440. doi: 10.1074/jbc.M704969200. [DOI] [PubMed] [Google Scholar]
- 24.Das TK, Dana D, Paroly SS, Perumal SK, Singh S, Jhun H, Pendse J, Cagan RL, Talele TT, Kumar S. Centrosomal kinase Nek2 cooperates with oncogenic pathways to promote metastasis. Oncogenesis. 2013;2:e69. doi: 10.1038/oncsis.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mbom BC, Nelson WJ, Barth A. β-catenin at the centrosome: Discrete pools of β-catenin communicate during mitosis and may co-ordinate centrosome functions and cell cycle progression. Bioessays. 2013;35:804–969. doi: 10.1002/bies.201300045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rapley J, Baxter JE, Blot J, Wattam SL, Casenghi M, Meraldi P, Nigg EA, Fry AM. Coordinate regulation of the mother centriole component nlp by nek2 and plk1 protein kinases. Mol Cell Biol. 2005;25:1309–1324. doi: 10.1128/MCB.25.4.1309-1324.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayward DG, Fry AM. Nek2 kinase in chromosome instability and cancer. Cancer Lett. 2006;237:155–166. doi: 10.1016/j.canlet.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 28.Mbom BC, Siemers KA, Ostrowski MA, Nelson WJ, Barth AI. Nek2 phosphorylates and stabilizes β-catenin at mitotic centrosomes downstream of Plk1. Mol Biol Cell. 2014;25:977–991. doi: 10.1091/mbc.E13-06-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olmeda D, Castel S, Vilaro S, Cano A. β-catenin regulation during the cell cycle: Implications in G2/M and apoptosis. Mol Biol Cell. 2003;14:2844–2860. doi: 10.1091/mbc.E03-01-0865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gehrke I, Gandhirajan RK, Kreuzer KA. Targeting the WNT/β-catenin/TCF/LEF1 axis in solid and haematological cancers: Multiplicity of therapeutic options. Eur J Cancer. 2009;45:2759–2767. doi: 10.1016/j.ejca.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 31.Sekiya T, Nakamura T, Kazuki Y, Oshimura M, Kohu K, Tago K, Ohwada S, Akiyama T. Overexpression of Icat induces G(2) arrest and cell death in tumor cell mutants for adenomatous polyposis coli, β-catenin, or Axin. Cancer Res. 2002;62:3322–3326. [PubMed] [Google Scholar]
- 32.Chou J, Lin YC, Kim J, You L, Xu Z, He B, Jablons DM. Nasopharyngeal carcinoma-review of the molecular mechanisms of tumorigenesis. Head Neck. 2008;30:946–963. doi: 10.1002/hed.20833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salomon A, Keramidas M, Maisin C, Thomas M. Loss of β-catenin in adrenocortical cancer cells causes growth inhibition and reversal of epithelial-to-mesenchymal transition. Oncotarget. 2015;6:11421–11433. doi: 10.18632/oncotarget.3222. [DOI] [PMC free article] [PubMed] [Google Scholar]