

Abstract
The control and treatment of rheumatoid arthritis is a challenge in today's world. Therefore, the pursuit of natural disease-modifying antirheumatic drugs (DMRDs) remains a top priority in rheumatology. The present study focused on curcumin and its derivatives in the search for new DMRDs. We focused on prominent p38 mitogen-activated protein (MAP) kinase p38α which is a prime regulator of tumor necrosis factor-α (TNF-α), a key mediator of rheumatoid arthritis. In the present study, we used the X-ray crystallographic structure of p38α for molecular docking simulations and molecular dynamic simulations to study the binding modes of curcumin and its derivatives with the active site of p38α. The ATP-binding domain was used for evaluating curcumin and its derivatives. Molecular docking simulation results were used to select 4 out of 8 compounds. These 4 compounds were simulated using GROMACS molecular simulation platform; the results generated were subjected to molecular mechanics-Poisson Boltzmann surface area (MM-PBSA) calculations. The results showed cyclocurcumin as a potential natural compound for development of a potent DMRD. These data were further supported by inhibition of TNF-α release from lipopolysaccharide (LPS)-stimulated human macrophages following cyclocurcumin treatment.
Keywords: rheumatoid arthritis, MAP kinase, curcumin, docking, simulations, molecular mechanics-Poisson Boltzmann surface area
Introduction
Rheumatoid arthritis (RA), an autoimmune disorder, evolves over time causing progressive damage of synovial-lined joints and variable extra-articular manifestations (1). RA is the most common autoimmune disease, with an incidence higher than psoriasis, Crohn's disease, type I (insulin-dependent) diabetes, lupus and multiple sclerosis (2). The progression of RA is characterized by symmetric polyarticular inflammation of the synovium, resulting in pain and stiffness and over a period of time leads to joint damage resulting in loss of function (3). The etiology of the autoimmune disorder leading to RA remains unknown, yet the mechanisms of how it evolves have been decoded to a great extent (4–7). RA affects 1% of the world population; in 2010 the global prevalence of this disease was 0.24% and was found to be more common in females (8–10). In Australian, Western European and North American regions the modeled age-standardized prevalence was found to be highest and the lowest was found to be in Asia and North African regions (10). In the USA, RA affects 1.3 million people, making it the third common type of arthritis among Americans (11).
The pathophysiology of this disease has been linked to genetic and environmental factors. These factors lead to a clinical syndrome spanning several disease subsets that require inflammatory cascades (12–14). The overexpression of tumor necrosis factor-α (TNF-α) is a prime inflammatory cascade involved in RA. This cascade leads to the overexpression of cytokines such as interleukin-1 (IL-1), IL-6 and IL-10, which drives persistent inflammation and joint destruction (15–18). These components of the chronic inflammatory response have been used for designing RA management therapies. To date, the management of RA has been achieved using disease-modifying antirheumatic drugs (DMARDs). These drugs are either of biological origin or are synthetic chemical compounds and based on this classification are termed as biological (bDMARDs) and synthetic (sDMARDs), respectively. Both bDMARDs and sDMARDs are characterized by their capacity to reduce or reverse signs and symptoms of RA. They interfere with the pathophysiology of this disease, which leads to improvements in disability, quality of life, ability to work, and suppression of joint damage (19–21).
DMARDs include etanercept, infliximab, methotrexate (MTX), sulfasalazine, leflunomide, rituximab, abatacept and others. However, these DMARDs are associated with numerous side effects (20). In order to overcome this factor more and more research is being conducted on natural comp ounds (21). Curcumin (diferuloylmethane) extracted from Curcuma longa is one of the many natural products that have been reported to be potential bDMARDs (22–25). Here in this study, we employed in silico techniques to investigate the natural compound curcumin and its derivatives as possible bDMARDs. Molecular docking simulations of curcumin and its derivatives with p38α, one of the prominent p38 mitogen-activated protein (MAP) kinase that regulates the expression of TNF-α and thus plays a role in RA was performed (26). p38α is involved in the p38 MAP kinase pathway that regulates a number of cellular processes critical to the etiology of RA. The activation and infiltration of leukocytes as well as the production of inflammatory cytokines are p38-dependent processes. Targeting of p38α has demonstrated efficacy in animal models of RA and various p38α inhibitors are currently in phase II and I clinical trials for RA. Thus using p38α as a therapeutic target for the development of a novel compound is a viable option. The 4 top complexes generated by targeting p38α with curcumin and its derivatives, were subjected to molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) calculations. The top compound, cyclocurcumin, was further studied, and inhibition of the release of TNF-α from LPS-stimulated macrophages by cyclocurcumin treatment confirmed its role as a potent p38α inhibitor.
Materials and methods
Data set
The X-ray crystallographic structure of p38α MAP kinase was downloaded from the RCSB Protein Data Bank (http://www.rcsb.org/pdb/home/home.do). The structure was cocrystallized with the pyrimidine-urea analogue 15 (PDB ID 2GHL, 2.10 Å X-ray resolution). The structure was stripped of the pyrimidine-urea analogue, where after removing water molecules, the charge and polar hydrogen atoms were added. The incomplete residues were corrected and the energy minimization of the modified structure was set to the default cut-off root mean square deviation (RMSD) value of 0.3 Å using OPLS 2001 force field by SPDB viewer (27). The structure of curcumin and its derivatives were retrieved from the PubChem database (Fig. 1).
Figure 1.

Two dimensional structures of curcumin and its derivatives used in the present study.
Molecular docking simulations
Molecular docking simulation was carried out using AutoDock 4.2.6 tool (28). This tool uses binding free energy evaluation for mining the top compound-protein pose. The tool uses binding site definition to create a rectangular grid. The execution of the docking runs are performed within the grid. The energy values calculated by this suite are the culmination of the intermolecular energy of the protein, the internal energy of the ligand and the torsional free energy. The default parameters were set, and the maximum evaluations were performed to elucidate the top 10 binding modes for each run. The best compound among curcumin and its derivatives was selected for further evaluation at the atomic level using molecular dynamics simulation. The results were analyzed using Discovery Studio Visualizer (29).
Molecular dynamics simulation
GROningen MAchine for Chemical Simulations GROMACS 4.5.6 atomic simulation tool was used to evaluate the ligand-p38α complexes over 100 nanoseconds (nsec) (30). GROMOS 43a1 force field was used for p38α and the force field for the top ligand was calculated using PRODRG webserver (31,32). The complexes were kept in a water bath, where water molecules were represented by simple point charge (SPC216) model. The system was ensured to be overall charge neutral before being subjected to energy minimization process, where position restrain procedure was performed with NVT and NPT ensembles, both of which follow Berendsen coupling scheme. Time duration of 1 nsec with coupling constant of 0.1 psec at a constant temperature of 300 K was used for NVT ensemble. This was followed by NPT ensemble with a time duration of 1 nsec; here a constant pressure of 1 bar was subjected to a coupling constant of 5 psec. The complexes were then subjected to simulation for 100 nsec at 300 K. For each run, the SHAKE algorithm for bond lengths and particle mesh Ewald method for long-range electrostatic interactions was employed, where a 14 Å cut-off for van der Waals interactions, a 12 Å cut-off for Coulomb interaction with updates every 2 steps (30,33). The trajectories generated were subjected to GROMACS inbuild tools for evaluation. The results generated were visualized using Pymol, Ligplus and VMD (34-36) and the graphs were plotted by using Grace program (37).
MM-PBSA calculations
The trajectories generated were used for MM-PBSA calculations (38,39). For each trajectory 1001 complexes were retrieved, each after 100 psec. Binding modes were analyzed using Grace program (37). The tool gave us the binding free energy of the four complexes. The binding free energy (ΔGbind) calculated by the tool includes the following species:
| (I) |
| (II) |
| (III) |
| (IV) |
In the equations Gcomplex, Gprotein and Gligand represent the free energy of the respective species, ΔEMM, ΔGsol and TΔS represent gas phase energy, solvation energy and an entropy term. The gas phase energy is the summation of the internal energy of the bonds, angle and torsion (ΔEval), electrostatic interaction energy (ΔEele), and van der Waals interaction energy (ΔEvdw). The salvation free energy comprises the polar salvation free energy (ΔGp), and the nonpolar salvation free energy (ΔGnp).
Principal component analysis
g_covar, g_anaeig in-build tools of GROMACS suite were used for indentifying the dominant motions of the selected complexes. PCA technique was used to trace these high-amplitude concerted motions in the p38α-curcumin-based complexes (40). Eigenvectors were generated from the covariance matrix generated by simple linear transformation. The first two eigenvectors (EV1 and EV2) having cosine content <0.2 were used to study free energy landscape (FEL). g_sham in-build tool was used to calculate FEL.
Cell culture
THP-1 monocytes were procured from American Type Culture Collection (ATCC; Manassas, VA, USA) and cultured in RPMI-medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% FBS and 100 U/ml PenStrep (Gibco, Grand Island, NY, USA). The cells were grown under standard culture conditions at 37°C under 5% CO2 in a humidified incubator. For macrophage differentiation, monocytes were cultured with macrophage CSF (M-CSF) at 100 ng/ml. Stock solution of 100 mM cyclocurcumin (Sigma-Aldrich) was prepared in RPMI-medium and diluted to different concentrations. Cells were stimulated with 10 ng/ml lipopoly-saccharide (LPS) (Sigma-Aldrich) for TNF-α production.
Cell proliferation assay
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay is a standard test to measure cell viability and is based on the conversion of MTT to formazan crystals by active mitochondrial dehydrogenases. Briefly, a cell suspension containing ~3×104 cells was plated into each well of a 96-well plate and allowed to attach for 24 h. Cyclocurcumin prepared in DMEM was added to the wells at a concentration of 10, 20, 40, and 60, 80 and 100 µM for 24 h. After treatment, 100 µl of MTT (Sigma-Aldrich) solution was added to each well at a concentration of 0.1 mg/ml (dissolved in PBS). After 4 h of incubation at 37°C in dark, the solution was removed and 100 µl of dimethyl sulfoxide (DMSO) was added to solubilize the produced formazan. Absorbance was measured at 570 nm using an automated microplate reader (BioTek Instruments, Winooski, VT, USA). The results are expressed as percentages relative to the controls. The percentage of proliferation inhibition was calculated as = (1 - ODsample/ODcontrol) ×100%.
TNF-α, enzyme-linked immunosorbent assay (ELISA)
Following treatment with cyclocurcumin and stimulation of human macrophages with LPS, the supernatants were harvested. Concentrations of TNF-α (BD Pharmingen, San Diego, CA, USA) were determined by ELISA, following the manufacturer's instructions. Absorbance was read at 450 nm on an ELISA plate reader (BioTek Instruments) using an in-built software program.
Statistical analysis
Statistical analysis was carried out by one-way ANOVA and the level of significance was tested at a P-value ranging from 0.01 to 0.001. Data are expressed as the mean ± SD of three independent experiments.
Results and Discussion
Molecular docking analysis
Molecular docking analysis was carried out using AutoDock 4.2 tool; 100 conformations for each compound were generated and the binding energy (kcal/mol) was set as the criteria for the cut-off for the molecular dynamics simulation analysis. Table I shows the results generated by docking curcumin and its selected derivatives against the active site of p38α. The top 4 compounds with the least binding energy were curcumin, bisdemethoxycurcumin, cyclocurcumin and demethoxycurcumin. Protein/ligand complexes from the molecular docking were subjected to post docking analysis, where weak interactions involved in the complex were studied (Fig. 2). Four complexes shortlisted for this analysis showed the favorable binding energy with a range -5 to -7 kcal/mol (Table II). The effective interactions studied here were supported by energies including final intermolecular energy, electrostatic energy and van der Waals energy. The effective interactions of the curcumin-p38α complex were evaluated using Discovery Studio Visualizer 4.5. The binding pocket of the complex involved GLY110, ASN115, ASP112, ASP168, VAL38, LEU104, GLU71, LYS53, VAL105, LEU75, LEU86, VAL30, THR106 and ALA111. The binding energy of curcumin-p38α was calculated to be the highest among the studied complexes, with no non-covalent interaction observed (Fig. 3A). The binding energy of the bisdemethoxycurcumin-p38α complex at -6.38 kcal/mol was the second highest calculated among the studied complexes. The bisdemethoxycurcumin-p38α complex formed one hydrogen bond (Fig. 4A). The interacting residue PHE169 of p38α is involved in active p38α inhibition. The oxygen atom of PHE169 formed a physical non-covalent interaction with bisdemethoxycurcumin, and the distance of the hydrogen bond formed was 2.36 A°. The binding pocket (Fig. 3B) of the bisdemethoxycurcumin-p38α complex involved LEU75, LEU74, THR106, ASP168, GLY170, GLU71, ILE84, PHE169, LEU167, MET109, GLY110, ASP112, ALA111 and ALA157. The third complex was that of cyclocurcumin with p38α protein kinase. The binding energy of the complex was calculated to be −6.12 kcal/mol. In the cyclocurcumin-p38α complex the results showed two hydrogen bonds (Fig. 3C) between the protein and ligand atoms. The interaction residues included LEU104 and VAL105 involved in active p38α inhibition. The binding pose of hit CID 69879809 clearly suggested effective inhibition. The atom 'O2' of the key residue LEU104 interacted with cyclocurcumin through hydrogen bond interactions (Fig. 4B). The key residue VAL105 of the active binding site cavity also interacted with cyclocurcumin oxygen atoms. Overall, the number of hydrogen bonds confirmed the high affinity of cyclocurcumin towards inhibition of p38α. In the case of the demethoxycurcumin-p38α complex, the results showed 3 hydrogen bonds between p38α and demethoxycurcumin (Fig. 3D). The interaction residues included SER154 and ASN155 (Fig. 4C). The interaction of an 'O' atom of SER154 and two 'O' atoms in the demethoxycurcumin aromatic ring showed a strong interaction pattern. The 'O' atom in both sides of the demethoxycurcumin aromatic ring was more potent in p38α inhibition.
Table I.
Binding energy score of curcumin and its derivatives with p38α.
| Name | CID | ΔG kcal/mol |
|---|---|---|
| Curcumin | 969516 | −6.67 |
| Bisdemethoxycurcumin | 5315472 | −6.38 |
| Curcuminglucuronide | 71315012 | −3.15 |
| Cyclocurcumin | 69879809 | −6.12 |
| Demethoxycurcumin | 5469424 | −5.88 |
| Dihydrocurcumin | 10429233 | −4.48 |
| Hexahydocurcumin | 5318039 | −4.81 |
| Tetrahydrocurcumin | 124072 | −3.33 |
CID, PubChem compound identifier.
Figure 2.

Binding modes of the top four curcumin-based compounds with the active site of p38α.
Table II.
Autodock analysis of the top four curcumin-based compounds.
| Complexes | Ligand binding pocket | Interaction |
|---|---|---|
| Curcumin-p38α | GLY110, ASN115, ASP112, ASP168, VAL38, LEU104, GLU71, LYS53, VAL105, LEU75, LEU86, VAL30, THR106, ALA111 | Nil |
| Bisdemethoxycurcumin-p38α | LEU75, LEU74, THR106, ASP168, GLY170, GLU71, ILE84, PHE169, LEU167, MET109, GLY110, ASP112, ALA111, ALA157 | One Bisdemethoxycurcumin: H39 - A:PHE169:O 2.36 A° |
| Cyclocurcumin-p38α | ILE84, LEU75, LEU86, LYS53, VAL105, GLY85, THR106, LEU104, VAL52, GLU71, ASP168, ARG67, TYR35, PHE169 | Two Cyclocurcumin:H40 - A:LEU104:O 2.13 A° A:VAL105:N - Cyclocurcumin:O3 3.02 A° |
| Demethoxycurcumin-p38α | THR106, LYS53, LEU108, ILE84, LEU104, MET109, LEU167, VAL38, ASP112, HIS107, ASP168, TYR35, ASN155, SER154, GLU71, LEU75 | Three Demethoxycurcumin:H39 - A:SER154:OG 1.92 A° ASN155:ND2 - :Demethoxycurcumin:O1 3.04 A° A:SER154:OG - :Demethoxycurcumin:O2 2.67 A° |
Figure 3.

Two-dimensional docked conformation of (A) curcumin (B) bisdemethoxycurcumin (C) cyclocurcumin and (D) demethoxycurcumin with the ATP binding domain of p38α.
Figure 4.

Mapping of three compounds with their respective hits; green dotted lines represent hydrogen bonds. (A) Bisdemethoxycurcumin interacting with single amino acid PHE169. (B) The two hydrogen bond interactions between cyclocurcumin and LEU104 and VAL105. (C) Demethoxycurcumin forming three hydrogen bonds with SER154 and ASN155.
Molecular dynamic simulation analysis
After identification of the top 4 curcumin-based compounds from the molecular docking, molecular dynamic simulation (MDS) was employed to refine the top 4 complexes. MDS is a powerful technique to monitor protein-ligand complexes over a period of time. MDS run at a 100-nsec production was carried out under GROMOS force field. The analysis was evaluated by group properties, RMSD, hydrogen bond interactions and advance properties including PCA and MM/PBSA free energy calculations using trajectory files. All the 4 complexes were stable throughout the run. For each complex, RMSD was evaluated for the convergence of the protein structure towards an equilibrium state after lead binding. Fig. 5 shows the RMSD plot based on backbone atoms. Curcumin and bisdemethoxycurcumin in complex with p38α exhibited a jump at 30 nsec and equilibrated well and both of the complexes converged with an RMSD range almost >0.2 nm. The RMSD value of the bisdemethoxycurcumin-p38α complex was least among the complexes. With an RMSD of 0.3 nm, cyclocurcumin showed the highest deviation. From the RMSD plot of the demethoxycurcumin-p38α complex, the structure equilibrated at 30 nsec and then started to converge near 0.25 nm. The change in RMSD in all 4 complexes up to 30 nsec was due to ligand binding with the active site and after 30 nsec all of the structure obtained stability with less structural deviation at the end of the 100-nsec simulation. After ligand binding, the structural deviation of the bisdemethoxycurcumin-p38α complex was the most stable among the 4 under consideration. Inter-hydrogen bond interactions between protein and ligands were evaluated for the p38α-curcumin-based complexes (Fig. 6). In the case of the curcumin complex (Fig. 6A), NH plot results showed a range of 4–6 hydrogen bond interactions observed throughout the 100-nsec simulation and a maximum of 8 hydrogen bonds. NH analysis confirmed strong inhibition of p38α by curcumin in dynamic system similar to the docking results inferred with 6 hydrogen bonds. In the case of the bisdemethoxycurcumin complex (Fig. 6B), the results showed a range of 3–4 hydrogen bond interaction found throughout the 100-nsec simulation and a maximum of 6 hydrogen bonds similar to the docking results. In the case of the cyclocurcumin complex (Fig. 6C), the results showed a range of 1–5 hydrogen bond interactions found throughout the 100-nsec simulation and a maximum of 5 hydrogen bonds similar to the docking results. The inter-hydrogen bond interaction pattern suggested the plausible mode of the strong binding of the lead candidates with p38α which favored the inhibition mechanism.
Figure 5.
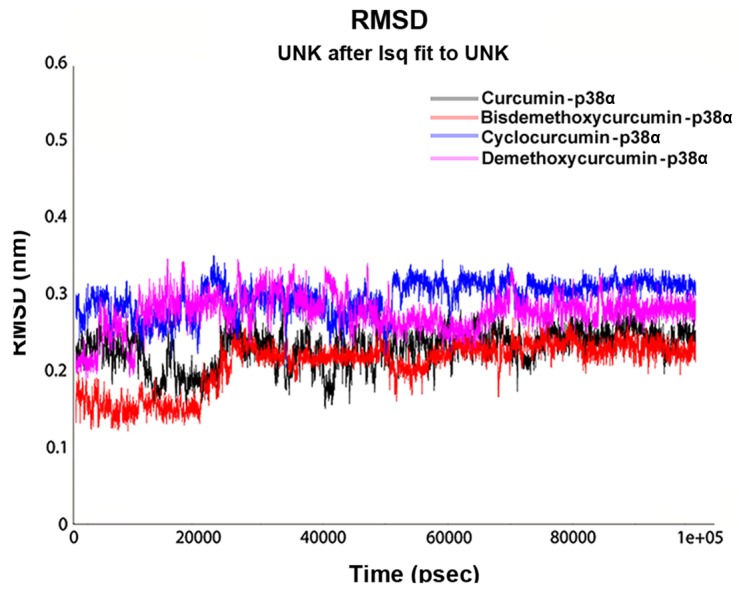
Root mean square deviation (RMSD) of the top four p38α-cur-cumin-based complexes calculated over the backbone atoms as a function of time with respect to the starting conformation.
Figure 6.

Hydrogen bond pattern over time for the three physically interacting curcumin-based compounds with p38α. (A) curcumin, (B) bisdemethoxycurcumin and (C) cyclocurcumin.
MM/PBSA calculation
The best 4 ligands in complex with p38α from MDS were used to calculate the binding free energy using MM-PBSA method. Snapshots were extracted at every 10-psec of stable intervals from 100-nsec MD trajectory and served as input for calculation. The binding free energy and its corresponding components obtained from the MM/PBSA calculation of the p38α curcumin-based complexes are listed. The results indicated that 4 curcumin-based compounds possessed a negative binding free energy of −141.727, −132.758, −151.455 and −133.56 kcal/mol (Table III). Moreover, van der Waals, electrostatic interactions and non-polar solvation energy negatively contribute to the total interaction energy while only polar solvation energy positively contributes to total free binding energy. The relative binding free energies of the 4 p38α-curcumin-based complexes supported the strong binding in the dynamic system. The interaction with the active site of p-p38α is shown in Fig. 7.
Table III.
MM-PBSA score of the top four curcumin-based compounds.
| Complex | ΔGbind (kcal/mol) |
|---|---|
| Curcumin-p38α | −141.727 |
| Bisdemethoxycurcumin-p38α | −132.758 |
| Cyclocurcumin-p38α | −151.455 |
| Demethoxycurcumin-p38α | −133.56 |
MM-PBSA, molecular mechanics Poisson-Boltzmann surface area.
Figure 7.

Residue wise calculation of the binding mode interaction between the top 4 curcumin-based compounds and the active site of p38α.
Principal component analysis
Principal component analysis or essential dynamics was performed on the cyclocurcumin complex with p38α. The analysis was used to monitor the overall strenuous motions of the complex. The results showed that the first 6 eigenvectors formed 90% of the total motion of the complex. The first 2 components of the eigenvectors were projected at 300 K and analysis of this revealed the clusters of stable states for the complex (41). The FEL was plotted over this projection, which provided a clear description of the stability of the complex (Fig. 8).
Figure 8.

Projection of the motion along the first and second principal eigenvectors in phase space plotted against each other for the cyclocurcumin-p38α complex.
Cyclocurcumin inhibits the release of TNF-α from human macrophages
TNF-α is a key factor in a variety of inflammatory diseases. Keeping in view the role of p38α in regulating TNF-α expression, we evaluated its expression in the culture supernatants of human macrophages (which constitute the major TNF-α-producing cells in highly relevant inflammatory disorders) treated with non-cytotoxic doses of cyclocurcumin. In order to determine the suitable dosage of cyclocurcumin that result in less cytotoxicity, macrophages (obtained with treatment of THP-1 monocytes with CSF), were treated with different concentrations of cyclocurcumin (10–100 µM) for 24 h, and the viability was measured by MTT assay. The results demonstrated that treatment with cyclocurcumin at concentrations such as 10, 20 and 40 µM exhibited the least cytotoxic effects (Fig. 9). Therefore, these concentrations of cyclocurcumin, without cytotoxic effects on macrophages were used. To test the effect of cyclocurcumin, macrophages were treated with the indicated concentrations of cyclocurcumin or 10 µm SB203580, a selective inhibitor of p38 MAPK. This was followed by stimulation with 10 ng/ml LPS for 18 h to induce the production of TNF-α. It was observed that treatment of the LPS-stimulated macrophages with cyclocurcumin led to significant inhibition in the release of TNF-α in a dose-dependent manner, similar to the inhibition caused by SB203580 (Fig. 10). Thus, these data confirm the role of cyclocurcumin in overcoming p38α-induced production of TNF-α and hence can be used as a therapeutic agent to target RA.
Figure 9.

Evaluation of the least cytotoxic doses of cyclocurcumin on macrophages by MTT assay. Results are expressed in terms of percentage inhibition of cell proliferation. Data are expressed as mean ± SD of results obtained from at least three independent experiments. Statistical analysis by one-way ANOVA, **P≤0.01 and ***P≤0.001.
Figure 10.

Effect of cyclocurcumin on tumor necrosis factor-α (TNF-α) production in macrophages. Cells were treated with the indicated concentrations of cyclocurcumin or 10 µm SB203580 or left untreated and then stimulated with 10 ng/ml lipopolysaccharide (LPS) for 18 h. The culture supernatants were then removed and assayed for TNF-α by enzyme-linked immunosorbent assay (ELISA). Data are expressed as mean ± SD of results obtained from at least three independent experiments. **P<0.01 and ***P<0.001 compared to the control.
In conclusion, natural products have been described as a gold mine for arthritis treatment. These products can suppress the expression of TNF-α, IL-1β, cyclooxygenase-2, lipooxygenase, matrix metalloproteinases or adhesion molecules, or suppress the activation of nuclear factor-κB (NF-κB). All are factors linked to the treatment of RA (42). Curcumin has long been reported to have an antirheumatoid effect. Initial studies have linked it with the downregulation of NF-κB (43). A plethora of research on curcumin has given us an opportunity to investigate it and its derivatives as potential kinase inhibitors for p38α. To the best of our knowledge, our study is the first to use an advanced computational approach to identify the possible DMRDs. MM-PBSA is a potent tool in the field of drug design (44) and this approach was employed in this study for short listing of compounds. From the molecular docking and molecular dynamic simulation studies, the potential binding mode of cyclocurcumin with p38α with stability was revealed as a top compound for the treatment of RA. Finally, inhibition of the release of TNF-α from LPS-stimulated macrophages by cyclocurcumin treatment confirms its role as a potent p38α inhibitor.
Acknowledgments
The authors would like to acknowledge the funding support for this study by the Shandong Province Important Research and Development Plan (no. 2015GSF118002), the Shandong Province Natural Science Foundation of China Youth Fund Projects (no. ZR2010HQ016) and the Natural Science Foundation of China Youth Fund Projects (no. 81202824/H1008).
Abbreviations
- RA
rheumatoid arthritis
- MAP
mitogen-activated protein
- MDS
molecular dynamic simulation
- RMSD
root mean square deviation
- MM-PBSA
molecular mechanics-Poisson Boltzmann surface area
- FEL
free energy landscape
References
- 1.Grassi W, De Angelis R, Lamanna G, Cervini C. The clinical features of rheumatoid arthritis. Eur J Radiol. 1998;27(Suppl 1):S18–S24. doi: 10.1016/S0720-048X(98)00038-2. [DOI] [PubMed] [Google Scholar]
- 2.Jacobson DL, Gange SJ, Rose NR, Graham NM. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol. 1997;84:223–243. doi: 10.1006/clin.1997.4412. [DOI] [PubMed] [Google Scholar]
- 3.Kahlenberg JM, Fox DA. Advances in the medical treatment of rheumatoid arthritis. Hand Clin. 2011;27:11–20. doi: 10.1016/j.hcl.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Demoruelle MK, Deane KD, Holers VM. When and where does inflammation begin in rheumatoid arthritis? Curr Opin Rheumatol. 2014;26:64–71. doi: 10.1097/BOR.0000000000000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nielen MM, van Schaardenburg D, Reesink HW, Twisk JW, van de Stadt RJ, van der Horst-Bruinsma IE, de Gast T, Habibuw MR, Vandenbroucke JP, Dijkmans BA. Increased levels of C-reactive protein in serum from blood donors before the onset of rheumatoid arthritis. Arthritis Rheum. 2004;50:2423–2427. doi: 10.1002/art.20431. [DOI] [PubMed] [Google Scholar]
- 6.Masson-Bessière C, Sebbag M, Durieux JJ, Nogueira L, Vincent C, Girbal-Neuhauser E, Durroux R, Cantagrel A, Serre G. In the rheumatoid pannus, anti-filaggrin autoantibodies are produced by local plasma cells and constitute a higher proportion of IgG than in synovial fluid and serum. Clin Exp Immunol. 2000;119:544–552. doi: 10.1046/j.1365-2249.2000.01171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snir O, Widhe M, Hermansson M, von Spee C, Lindberg J, Hensen S, Lundberg K, Engström A, Venables PJ, Toes RE, et al. Antibodies to several citrullinated antigens are enriched in the joints of rheumatoid arthritis patients. Arthritis Rheum. 2010;62:44–52. doi: 10.1002/art.25036. [DOI] [PubMed] [Google Scholar]
- 8.Murray CJ, Vos T, Lozano R, Naghavi M, Flaxman AD, Michaud C, Ezzati M, Shibuya K, Salomon JA, Abdalla S, et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2197–2223. doi: 10.1016/S0140-6736(12)61689-4. [DOI] [PubMed] [Google Scholar]
- 9.Gibofsky A. Overview of epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis. Am J Manag Care. 2012;18(Suppl 13):S295–S302. [PubMed] [Google Scholar]
- 10.Cross M, Smith E, Hoy D, Carmona L, Wolfe F, Vos T, Williams B, Gabriel S, Lassere M, Johns N, et al. The global burden of rheumatoid arthritis: Estimates from the global burden of disease 2010 study. Ann Rheum Dis. 2014;73:1316–1322. doi: 10.1136/annrheumdis-2013-204627. [DOI] [PubMed] [Google Scholar]
- 11.Clough J. The Cleveland Clinic Guide to Arthritis. Kaplan Publishing; New York, NY: 2009. [Google Scholar]
- 12.van Oosterhout M, Bajema I, Levarht EW, Toes RE, Huizinga TW, van Laar JM. Differences in synovial tissue infiltrates between anti-cyclic citrullinated peptide-positive rheumatoid arthritis and anti-cyclic citrullinated peptide-negative rheumatoid arthritis. Arthritis Rheum. 2008;58:53–60. doi: 10.1002/art.23148. [DOI] [PubMed] [Google Scholar]
- 13.van der Helm-van Mil AH, Huizinga TW. Advances in the genetics of rheumatoid arthritis point to subclassification into distinct disease subsets. Arthritis Res Ther. 2008;10:205. doi: 10.1186/ar2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010;376:1094–1108. doi: 10.1016/S0140-6736(10)60826-4. [DOI] [PubMed] [Google Scholar]
- 15.Eastgate JA, Symons JA, Wood NC, Grinlinton FM, di Giovine FS, Duff GW. Correlation of plasma interleukin 1 levels with disease activity in rheumatoid arthritis. Lancet. 1988;2:706–709. doi: 10.1016/S0140-6736(88)90185-7. [DOI] [PubMed] [Google Scholar]
- 16.Feldmann M, Brennan FM, Maini RN. Brennan, FM, and Maini, RN Rheumatoid Arthritis. Cell. 1996;85:1277–1289. doi: 10.1016/S0092-8674(00)81109-5. [DOI] [PubMed] [Google Scholar]
- 17.Choy EH, Isenberg DA, Garrood T, Farrow S, Ioannou Y, Bird H, Cheung N, Williams B, Hazleman B, Price R, et al. Therapeutic benefit of blocking interleukin-6 activity with an anti-interleukin-6 receptor monoclonal antibody in rheumatoid arthritis: A randomized, double-blind, placebo-controlled, dose-escalation trial. Arthritis Rheum. 2002;46:3143–3150. doi: 10.1002/art.10623. [DOI] [PubMed] [Google Scholar]
- 18.van Roon JA, van Roy JL, Gmelig-Meyling FH, Lafeber FP, Bijlsma JW. Prevention and reversal of cartilage degradation in rheumatoid arthritis by interleukin-10 and interleukin-4. Arthritis Rheum. 1996;39:829–835. doi: 10.1002/art.1780390516. [DOI] [PubMed] [Google Scholar]
- 19.Smolen JS, Landewé R, Breedveld FC, Buch M, Burmester G, Dougados M, Emery P, Gaujoux-Viala C, Gossec L, Nam J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying anti-rheumatic drugs: 2013 update. Ann Rheum Dis. 2014;73:492–509. doi: 10.1136/annrheumdis-2013-204573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smolen JS, van der Heijde D, Machold KP, Aletaha D, Landewé R. Proposal for a new nomenclature of disease-modifying antirheumatic drugs. Ann Rheum Dis. 2014;73:3–5. doi: 10.1136/annrheumdis-2013-204317. [DOI] [PubMed] [Google Scholar]
- 21.Smolen JS, Aletaha D, Koeller M, Weisman MH, Emery P. New therapies for treatment of rheumatoid arthritis. Lancet. 2007;370:1861–1874. doi: 10.1016/S0140-6736(07)60784-3. [DOI] [PubMed] [Google Scholar]
- 22.Dcodhar S, Sethi R, Srimal R. Preliminary study on anti-rheumatic activity of curcumin (diferuloyl methane) Indian J Med Res. 1980;71:632–634. [PubMed] [Google Scholar]
- 23.Kuttan G, Hari Kumar K, Guruvayoorappan C, Kuttan R. Antitumor, anti-invasion, and antimetastatic effects of curcumin. In: Aggarwal BB, Surh YJ, Shishodia S, editors. The Molecular Targets and Therapeutic Uses of Curcumin in Health and Disease. Springel; New York, NY: 2007. pp. 173–184. [DOI] [PubMed] [Google Scholar]
- 24.Lev-Ari S, Strier L, Kazanov D, Elkayam O, Lichtenberg D, Caspi D, Arber N. Curcumin synergistically potentiates the growth-inhibitory and pro-apoptotic effects of celecoxib in osteoarthritis synovial adherent cells. Rheumatology (Oxford) 2006;45:171–177. doi: 10.1093/rheumatology/kei132. [DOI] [PubMed] [Google Scholar]
- 25.Joe B, Rao UJ, Lokesh BR. Presence of an acidic glycoprotein in the serum of arthritic rats: Modulation by capsaicin and curcumin. Mol Cell Biochem. 1997;169:125–134. doi: 10.1023/A:1006877928703. [DOI] [PubMed] [Google Scholar]
- 26.Newton R, Holden N. Inhibitors of 38 mitogen-activated protein kinase: Potential as anti-inflammatory agents in asthma? BioDrugs. 2003;17:113–129. doi: 10.2165/00063030-200317020-00004. [DOI] [PubMed] [Google Scholar]
- 27.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 28.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Discovery Studio, version 3.5 . Accelrys Inc; San Diego, CA, USA: 2012. [Google Scholar]
- 30.Hess B, Bekker H, Berendsen HJ, Fraaije JG. LINCS: A linear constraint solver for molecular simulations. J Comput Chem. 1997;18:1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- 31.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 32.Schüttelkopf AW, van Aalten DM. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 33.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- 34.DeLano WL. The PyMOL Molecular Graphics System. World Wide Web; 2002. http://www.pymol.org. [Google Scholar]
- 35.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–28. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 36.Laskowski RA, Swindells MB. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 37.Turner P. XMGRACE, version 5.1.19. Center for Coastal and Land-Margin Research. Oregon Graduate Institute of Science and Technology; Beaverton, OR: 2005. [Google Scholar]
- 38.Kumari R, Kumar R, Lynn A. Open Source Drug Discovery Consortium: g_mmpbsa - a GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 39.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amadei A, Linssen AB, Berendsen HJ. Essential dynamics of proteins. Proteins. 1993;17:412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- 41.Chikan NA, Vipperla B. KAISO inhibition: An atomic insight. J Biomol Struct Dyn. 2015;33:1794–1804. doi: 10.1080/07391102.2014.974072. [DOI] [PubMed] [Google Scholar]
- 42.Khanna D, Sethi G, Ahn KS, Pandey MK, Kunnumakkara AB, Sung B, Aggarwal A, Aggarwal BB. Natural products as a gold mine for arthritis treatment. Curr Opin Pharmacol. 2007;7:344–351. doi: 10.1016/j.coph.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 43.Singh S, Aggarwal BB, Aggarwal B. Activation of transcription factor NF-kappa B is suppressed by curcumin (diferuloylmethane) [corrected] J Biol Chem. 1995;270:24995–25000. doi: 10.1074/jbc.270.42.24995. [DOI] [PubMed] [Google Scholar]
- 44.Zhao FL, Yang GH, Xiang S, Gao DD, Zeng C. In silico analysis of the effect of mutation on epidermal growth factor receptor in non-small-cell lung carcinoma: from mutational analysis to drug designing. J Biomol Struct Dyn. 2017;35:427–434. doi: 10.1080/07391102.2016.1146165. [DOI] [PubMed] [Google Scholar]