

Abstract
Conventional small molecule drug-discovery approaches target protein pockets. However, the limited number of geometrically distinct pockets leads to widespread promiscuity and deleterious side-effects. Here, the idea of COmposite protein LIGands (COLIG) that interact with each other as well as the protein within a single ligand binding pocket is examined. As a practical illustration, experimental evidence that E. coli Dihydrofolate reductase inhibitors are COLIGs is presented. Then, analysis of a non-redundant set of all holo PDB structures indicates that almost 47–76% of proteins (based on different sequence identity thresholds) can simultaneously bind multiple, interacting ligands in the same pocket. Moreover, most ligands that are either Singletons and COLIGS bind at the bottom of ligand binding pocket and occupy 30% and 43% of the volume of the bottom of the pocket. This suggests the use of COLIGs as a potential new class of small molecule drugs.
Keywords: COLIG, pocket volume occupied by ligands, ligand-ligand vs ligand protein interactions, aromatic interactions, emergence of biochemical specificity
Graphical Abstract
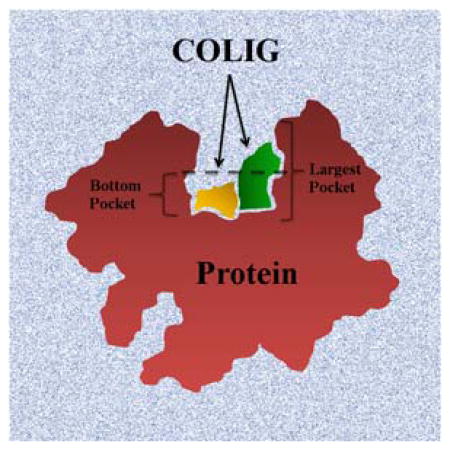
The canonical view of protein-ligand interaction is focused on one ligand per pocket at a time. Here, we present the concept of a COLIG (COmposite Protein LIGands), as multiple interacting ligands in the same pocket. Our analysis shows that majority of proteins can bind COLIGs and most ligands can form COLIGs. Finally, we find that ligands tend to bind in the deepest locations of the largest pocket of a protein.
Introduction
The field of small molecule drug discovery has to a large extent, been protein pocket centric. Pockets are surface concavities on proteins where substrates and allosteric modulators bind that can, in principle, be targeted for drug-discovery purposes1. The interactions of small molecules with functional groups in a protein’s pocket underlie most in silico approaches to drug discovery. Identification, comparison and analyses of binding pockets are pivotal for structure-based drug design endeavors, hit identification in virtual ligand screening2, detection of secondary binding sites3, annotation of functions in orphan hypothetical proteins4 and efforts at predicting drug side-effects by assessing off-target interactions5.
Mutations of critical amino acid residues in protein pockets have the potential for disrupting both the shape and chemical complementarity to the ligand of interest. The effect of these mutations on drug binding and drug response depends on the residue that gets mutated and its three-dimensional structural context6. Several examples have been presented in the literature which show that mutation of critical amino acid residues in the pocket leads to widespread drug-resistance7,8. For example, the T315I gatekeeper mutation in the Abl kinase domain acquired after treatment with ATP analogues leads to ~20% of patients having clinically acquired resistance9,10. Likewise, the T790M gatekeeper mutation in combination with the oncogenic L858R mutation in Epidermal growth receptor tyrosine in the tyrosine kinase Epidermal growth receptor accounts for resistance to gefitinib and erlotinib in approximately half of clinical cases 11,12. More generally, drug resistance acquisition by several protein targets from pathogenic microbes have been directly shown to arise from mutations critical for drug-binding13–15.
Overcoming the problem of drug resistance acquired through mutations on the protein target pockets is difficult, as such mutations spontaneously occur in a system under selection pressure. However, as the number of residues with which a given small molecule interacts grows, the relative effect on binding affinity of a given residue mutation diminishes 16. Thus, one strategy to overcome resistance is to introduce larger ligands. However, following Lipinski, these larger ligands might not be as bioavailable17. One way to surmount this difficulty is to find a pair of (or more) ligands that simultaneously interact with each other and the protein pocket; we term such ligands pairs COLIGS. Are there examples in nature of such composite protein-ligand interactions that can serve a role model for drug discovery?
Another reason to examine the role played by COLIGs is provided by recent work which shows that there are about 500 structurally distinct ligand binding pockets in nature18,19. While it is likely true that a given ligand binds to similar pockets in off-target proteins with varying specificities, such off target binding is one likely cause of drug side effects 20. Similar effects are likely to occur for endogenous metabolites21. Yet, cells have well defined biochemical pathways and biological functions22–24. Somehow biochemical control must emerge from the background of ligand-multiple protein target interactions. One way of reducing promiscuity could be the cooperative binding of multiple, interacting ligands in a given pocket; this possibility is explored here.
In this manuscript, we introduce the concept of COLIGs and show that a large fraction of PDB holo structures are bound to more than one ligand and that a substantial fraction of these structures have at least one pair of spatially proximate ligands that interact with each other as well as the residues in the protein pocket. First, we present experimental evidence for COLIGs in the biologically important protein, DHFR and show that a subset of folate analogues target the protein-NADPH COLIG pocket. Next, we systematically examine ligand-bound holo structures for the purpose of understanding druggable pockets with a substantial interaction surface contributed by a pair of small molecules. Finally, we examine the implications of our results for developing better drug discovery methods by reducing drug resistance and enhancing binding specificity.
Methods
Reagents
All reagents and chemicals, unless mentioned otherwise, were of high quality and procured from Sigma-Aldrich Co., USA, Amresco, or Fisher Scientific. E. coli dihydrofolate reductase was provided by Prof. Eugene Shakhnovich, Harvard University. The Developmental Therapeutics Program (DTP) of the National Cancer Institute (NCI) of the NIH provided the small molecules NSC309401, NSC339578, and NSC740.
Binding studies using differential scanning fluorimetry, DSF
Binding of the diaminopyrroloquinazoline and diaminopteridine classes of molecules to E. coli DHFR was tested by differential scanning fluorimetry, DSF. The experiments were carried out following previously reported protocols from our lab 1,2,4,25. Briefly, the reactions were carried out in 96 well plates on the RealPlex quantitative PCR instrument (Eppendorf, NY, USA). The reaction mixture contained 100 mM HEPES pH 7.3 and 150 mM NaCl with 5 X concentration of the reporter dye Sypro orange. Various compounds were tested for binding at 100 μM concentrations with or without the cofactor NADPH (100 μM) and 10 μM E. coli DHFR (EcDHFR).
Database preparation
The list of holo protein structures was obtained from Ligand Expo26 and their structures (92437 as of July 2016) downloaded from the Protein Data bank27. Holo structures whose ligands have less than six heavy atoms were excluded as well as those with “irrelevant” ligands (those used in crystallization solutions or any considered as L-peptide linking ligands by the PDB; see Supplementary Information for a list of irrelevant ligands). Next, each protein-ligand complex was assigned as a Singleton or COLIG. Singletons are those whose ligand is found in a protein pocket such that none of its heavy atoms are in contact (< 4.5 Å) with another ligand’s heavy atoms. Multi-Lig PDB structures are a subset of Singletons for which the protein binds multiple ligands, where each ligand binds to a different pocket. Thus, no pair of ligands has any of its ligand heavy atoms in contact. COLIGs are those cases when the protein is co-crystallized with multiple ligands that simultaneously bind in the same pocket with at least one heavy atom contact (< 4.5 Å) between ligands.
Pocket detection and volume calculation
For each protein structure, all of its pockets are detected by the CAVITATOR pocket detection algorithm as described in28. CAVITATOR assigns a residue to a pocket when there is at least one opposing residue across pocket. That is, residues associated with ledges are excluded.
The bottom residues in each pocket (Figure S1) are identified as follows: If there are less than 20 residues in the pocket, all are taken as bottom of pocket residues. If there are more than 20 residues in the pocket, the bottom 20 residues are obtained as follows: For each pocket, the pocket associated lattice sites on a cubic lattice whose grid spacing is 1 Å (solvent lattice sites lie between at least one pair of pocket residues) are determined. Then, the set of interfacial sites adjacent to the pocket associated sites are determined. Next, for all of the residues that line the pocket, the distance of each pocket residue to the each of the interfacial sites is calculated and ranked. The minimum distance of each residue to an interfacial site is calculated, and these minimum distances rank ordered. The subset of pocket lining residues whose minimum distance is the largest defines the bottom residues. Bottom pocket lattice sites are those within 5 Å of a bottom residue.
Pocket Occupancy (PO)
Ligands are mapped to the bottom lattice sites as follows: Each heavy atom is mapped to the closest cubic lattice grid point as all lattice sites within 2 Å. The PO is the fraction of bottom pocket sites occupied by the ligand(s) divided by the total number of bottom lattice sites.
Protein-ligand and ligand-ligand interaction calculations
LPC software29 was used to classify and assign the interactions to four major classes: Hydrogen bonds, Aromatic-Aromatic, Hydrophobic-Hydrophobic, Hydrophilic-Hydrophilic. To avoid over counting, the interactions for COLIG complexes were decomposed into protein-ligand1, protein-ligand2 and ligand1-ligand2.
Data sets
Non redundant data sets for the follow situations were prepared: a) proteins alone, b) ligands alone, c) Singletons (protein + one ligand per pocket) and d) COLIGs (protein + multiple ligands in each protein). Each of these cases is described below in detail:
Proteins alone: Protein clusters were obtained from RCSB statistics27 prepared using BLASTClust 30,31 at seven sequence identity cutoffs (100%, 90%, 50%, 40% and 30%).
For Ligands, the similarity between two ligands was calculated using the Tanimoto coefficient obtained from Open Babel’s FP2 fingerprint32. Next, ligands were assigned to clusters using single-linkage clustering at five Tanimoto coefficient (TM) cutoffs: 1.0, 0.9, 0.8, 0.75, 0.7.
For Singletons (protein + one ligand per pockets), we combined the clustering results from a) and b). Two Singletons are considered similar only if both the sequence identity of the proteins and the TM of their ligands are more than the sequence identity and TM thresholds respectively. We used all possible combinations (25) of sequence identity and TM thresholds mentioned in a) and b).
We used the same metrics as Singletons to define similar COLIGs. Two COLIGs are similar if their respective protein and both ligands meet the criteria for sequence identity and TM thresholds respectively.
The full set of clustering results can be found at: http://cssb2.biology.gatech.edu/JCC-COLIG/
Results
As shown in Figure 1, there are three possible scenarios for ligand binding in a given pocket: There are Singletons, where a single ligand binds to a given pocket and there is only one ligand bound per protein. Since proteins can contain more than one ligand binding pocket whose shape and volume can accommodate at least a single ligand, there are Multi-Lig proteins where multiple ligands are bound. Yet each pocket contains a single ligand. For sufficiently large pockets, there is the case when it contains two or more ligands in the same pocket which interact with the protein, but not with each other. We term these Antisocial Ligands (but since these are like Singletons we do not focus on them here). Finally, there are COLIGS, where a given pocket contains two or more ligands that interact with each other and the protein.
Figure 1.

Cartoon representation of different members of holo protein database. In the Singleton (left) case, the protein (orange) binds one ligand (blue) in its pocket. Multi-Ligs (middle) are proteins having more than one Singleton ligand (cyan and green) bound to a protein (magenta). COLIGs (right) are a subset of holo PDBs that have at least two ligands (yellow and red) in the same pocket that are in contact with each other.
The canonical intuitive view is that most ligands interact with proteins as Singletons. However, there is also the recognition, particularly for enzymes that cofactors might be Multi-Ligs or implicitly COLIGS. But little attention has been paid in the past to COLIGs in general. Indeed, the motivation for their examination was provided by a thermodynamic analysis of the binding of methotrexate and other DHFR inhibitors to DHFR, the results of which we present next.
COLIGS of DHFR formed by folate analogues binding with NADPH
As shown in Figure 2, analysis of the structure of the ternary complex of E. coli DHFR bound to NADPH and methotrexate, MTX, indicates that that the inhibitor shares 34.5 Å2 of the binding surface with NADPH. This constitutes 7% of its total binding surface (contact surfaces with the protein and NADPH). Interestingly there are around a dozen cases in the PDB where MTX interacts with NAP/NDP; the resulting contact surfaces range between 30 to 100 Å2. Thus, it would not be unreasonable to expect that there is a favorable interaction free energy between MTX and NADPH that serves to further stabilize the ternary complex of protein, MTX and NADPH. This is seen Figure 3 for the thermal melting of the protein in the combined presence of both cofactor and the inhibitor. Not only is there an increased stability when MTX (NSC740) binds to the protein and NADPH as compared to MTX binding to 5h3protein alone (Figure 3A) but a similar effect is seen with other DHFR inhibitors (NSC309401), Figure 3B and NSC 339578, Figure 3C. The non-additive increase in melting temperature Tm is clearly shown in Table 1.
Figure 2.
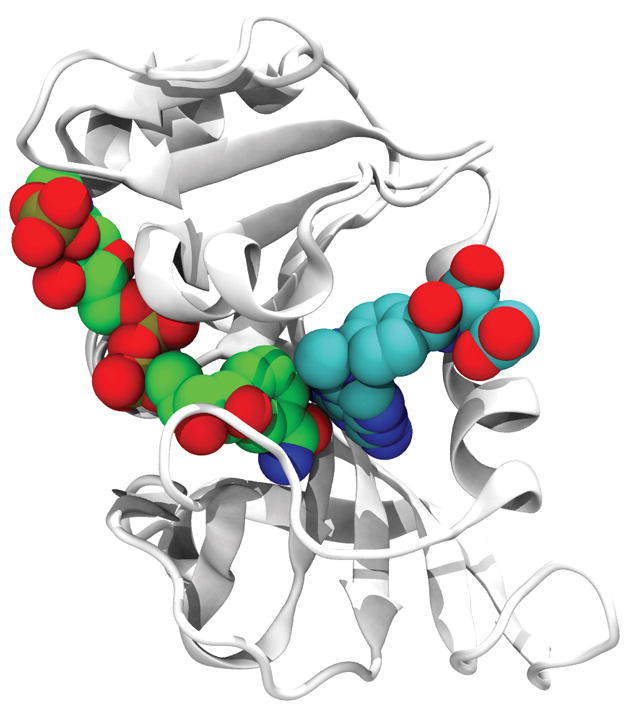
Structure of MTX (right) with NADPH (left) (PDB_ID: 4P68). The protein is shown in a cartoon representation and the ligands are shown in a space-filling representation.
Figure 3.

Differential scanning fluorimetry, DSF, curves and their first derivatives for E. coli DHFR in the presence inhibitors belonging to the pyrroloquinazoline and diaminopteridine class of compounds. (A) DSF curves for NSC740, MTX. (B) DSF curves for NSC309401, AMPQD. (C) DSF curves for NSC339578. The Gaussian fit of the first derivatives are given in (D)–(F).
Table 1.
Non-additive increase in thermal shifts of DHFR with inhibitor addition in the presence of NADPHa
| NSC ID | ΔTm with 100 μM NADPH | ΔTm with 100 μM inhibitor | ΔTm with 100 μM NADPH +inhibitor | Non-Additive ΔTm |
|---|---|---|---|---|
| NSC740 | 57.85 ± 0.25 | 64.40 ± 0.19 | 71.16 ± 0.18 | 3.36 |
| NSC309401 | 57.85 ± 0.25 | 64.33 ± 0.18 | 72.16 ± 0.15 | 1.98 |
| NSC339578 | 57.85 ± 0.25 | 61.95 ± 0.21 | 68.64 ± 0.17 | 0.84 |
The Tm for protein alone control is ~52 °C
How frequent are COLIGS?
As of July 2016, there were 92,43727 structures co-crystallized with at least one of the 21,166 ligands of the Ligand Expo26 dataset. After filtering the raw dataset as described in Methods, there 36,117 unique PDB-IDs (PDB_ID + chain_ID). Next, based on the total number of ligands in each PDB entry, we examined how many are Singletons, Multi-Ligs and COLIGS. Our analysis finds 32,347 Singletons, 2,135 Multi-Ligs and 3,900 COLIGs with unique PDB-IDs. To reduce bias due to over counting the same or highly similar homologs, we show the statistics for each case in Table 2. Irrespective of sequence identity, the probability of observing a COLIG in a holo protein is 0.12 to 0.16, when normalized by the total number of COLIGs plus Singletons. Considering the difficulty in co-crystallizing ligands and the bias towards solving Singleton structures, this is likely a lower bound for the fraction of COLIGs in nature.
Table 2.
Statistics of the number of PDB structures with Singletons, Multi-Ligs and COLIGs at different sequence identity thresholdsa
| Sequence Identity | <100% | <90% | <50% | <40% | <30% |
|---|---|---|---|---|---|
| Singleton | 14263 | 9287 | 7230 | 6227 | 5047 |
| Multi-Lig | 1199 | 967 | 846 | 783 | 696 |
| COLIG | 1954 | 1439 | 1227 | 1093 | 933 |
| Overlap of COLIG & Singletons | 913 (0.47) | 902 (0.63) | 848 (0.69) | 790 (0.72) | 704 (0.76) |
Numbers in parenthesis indicate the ratio of overlapped-COLIG-Singletons to the total number of COLIGs at the given sequence identity threshold.
Table 2 also shows the overlap between proteins that are both COLIG and Singletons. This overlap fraction varies anywhere from 0.48 and grows to 0.76 as the sequence threshold is reduced (when distant homologs are considered). This suggests that the majority of proteins can use their pocket to bind more than one ligand at the same time.
How many proteins that bind multiple ligands are COLIGS?
Another interesting question that can be answered using Table 2 is that given a protein structure solved with more than one ligand (Multi-Ligs + COLIGs), what would be the probability that the ligands bind in the same pocket and form COLIGs? The answer, which is the ratio (NCOLIGs/(NCOLIGs + NMulti-Ligs) varies between 0.57 to 0.62 based on the different sequence identity thresholds, a surprisingly non negligible number.
Are COLIGS restricted to enzymes and must one of the ligands in a COLIG be an enzyme cofactor?
It is well known that cofactors interact with their substrate and are often required for enzymatic catalysis33. However, our analysis clearly shows that we have non-cofactor complexes in non-enzymatic (no assigned EC number) proteins. In COLIGs, for 35% of the ligand-ligand complexes, neither of the ligands are cofactors nor nucleotides (the list of cofactors and nucleotides used in this analysis is provided in the Supplementary Information, Table S2). For Singletons, this percentage is 74%. With respect to PDB proteins that bind ligands, for COLIGs, 27% are not enzymes and for Singletons 39% are not enzymes. Thus we conclude that COLIGS are not restricted to enzymes and their associated cofactors; rather the phenomenon of multiple interacting ligands binding in the same pocket is more general.
We further surveyed Pfam34 to assess the protein families of COLIG proteins. As of September 2016 there were 7957 protein families recognized by Pfam. COLIG proteins covered 776 (10%) of these families while Singleton proteins covered 2779 (34%). Considering that the total number of Singleton proteins is seven times larger than COLIG proteins, it is not unreasonable to conclude that COLIG proteins cover an acceptable fraction (29%) of known holo protein families.
Are the ligands involved in COLIGS special?
Having shown that COLIGs are not limited to a group of proteins and are relatively abundant, we now examine if COLIGS involve a particular class of ligands. To address this issue, we counted the number of ligands with unique ligand IDs given by the PDB. There are 2,132 and 12,753 unique ligands in COLIGs and Singletons respectively. To remove contributions from very similar ligands, we measured the similarity between ligands as described in Methods and reported the numbers for various Tanimoto coefficient thresholds in Table 3.
Table 3.
Statistics of the number of Singleton and COLIG ligands at different Tanimoto coefficient thresholds a,b.
| Tanimoto coefficient | <1.0 | <0.9 | <0.8 | <0.75 | <0.70 |
|---|---|---|---|---|---|
| Singleton | 11262 | 9199 | 6950 | 5893 | 4834 |
| COLIG | 1838 | 1466 | 1056 | 881 | 705 |
| Overlap of COLIG & Singletonsb | 698 (0.38) | 586 (0.40) | 450 (0.43) | 381 (0.43) | 305 (0.43) |
| COLIG Pairs | 3364 | 3162 | 2913 | 2797 | 2649 |
COLIG pairs are assigned to the same cluster only if both ligands meet the similarity threshold with another COLIG pair.
The number in parentheses indicates the ratio of overlapped-COLIG-Singletons to the total number of COLIGs at the given TM threshold.
Irrespective of the Tanimoto threshold, there is always 0.38 to 0.43 overlap between Singletons and COLIG ligands. In other words, ~40% of the ligands have been observed in both Singletons and COLIGs. However, this significant between the overlapping and non-overlapping ligands resides in the number of PDB structures solved with that particular ligand. For overlapped cases, the average number of deposited structures (at different TM thresholds) are 24 ± 21, while the count for non-overlapping ones is 1.6 ± 0.2. This simply indicates that just as most proteins could bind as COLIGs, the majority of ligands can also serve as participant in COLIGs.
Examples of COLIGs in the PDB
In Figure 4, we present four examples of COLIGs taken from the PDB and discuss them in some detail. Figure 4A shows the structure of the Clostridium botulinum C3 Exoenzyme complex in contact with ADP and NIR (3-(aminocarbonyl)-1-((3r,4s,5r)-3,4-dihydroxy-5-methyltetrahydro-2-furanyl) pyridinium)35 with 20 heavy atom contacts between them. The deposited structure has one chain with only ADP and another with both ADP and NIR, indicating that part of the binding pocket for NIR is made as a result of ADP binding. Figure 4B presents the structure of farnesyltransferase in complex with benzofuran (NH7) inhibitor and Farnesyl Diphosphate (FPP)36. In this case, NH7 acts as the inhibitor and has 33 heavy atom contacts with FPP. Figure 4C shows the structure of E. coli XGPRT bound to CPRPP and Xanthine37 with 14 heavy atom contacts between the ligands. Xanthine and CPRPP are substrates of this protein which will turn into Guanosine-5′-monophosphate via an enzymatic reaction. This is a case where the protein is the enzyme and the COLIGs are its substrates. As we mentioned these cases are subset of COLIG classes and COLIGs are not only limited to enzymes.
Figure 4.

Examples of COLIGs in the PDB. A) Structure of the Clostridium Botulinum C3 Exoenzyme complex in contact with ADP (cyan) and NIR (green) (PDB_ID: 1GZF). B) Structure of farnesyltransferase (PDB_ID: 2ZIR) in complex with NH7 (green), FPP (cyan). C) Structure of E. Coli XGPRT (PDB_ID: 1A96) in complex with PCP (cyan) and XAN (green). D) Structure of human GARTfase (PDB_ID: 1RBY) in complex with KEU (green) and GAR (cyan).
Finally, Figure 4D shows the concept of a COLIG used by human GARTfase. The complex in this case is made of 10-(trifluoroacetyl)-5,10-dideazaacyclic-5,6,7,8-tetrahydrofolic acid (an experimental drug) and Glycinamide Ribonucleotide (GAR) sharing 20 heavy atom contacts.
What part of the pocket is occupied by Singletons and COLIGS?
It is well known that ligands often bind to concave surfaces, viz., pockets. However, where does the ligand bind within this concave surface? To answer this question, we defined the bottom of pockets as described in Methods. Our results show that ligands mainly bind at the pocket bottom. 93% and 83% of COLIGs and Singletons, respectively bind to the bottom of the pocket. This the region that ligand binding prediction should focus on. Another interesting observation is that ligands tend to bind to the largest pocket. In 76% of the COLIGs and 64% of Singletons, ligands chose the pocket with the largest volume to bind to. Our results suggest that these pocket properties (Pocket rank of pocket based on size and the deepest part of the pocket) can play an essential role in detecting druggable pockets.
Fraction of the volume of the pocket bottom occupied by Singletons and COLIGS
To address this issue, we introduce the concept of pocket occupancy (PO) as described in Methods. Our results (Figure 6A) show that for the majority (~80%) of cases less than 40% of the pocket is utilized by Singletons, with an average PO of 0.30 ± 0.23. Figure 6B shows an example in which only 22% of the pocket is used by the ligand. For COLIGs the PO shifts toward higher occupancy values, and on average, 0.43 ± 0.27 of pockets are occupied by the ligands. Now let’s focus on the absolute value of pocket volume (PV), we find that the PV for COLIGs and Singletons have very similar values: 756 ± 451 Å3 and 774 ± 86 Å3, respectively. Therefore, the difference in PO is the result of different absolute values of occupied volumes in COLIGs (281 ± 151 Å3) and Singletons (170 ± 107 Å3) and not the difference in the volumes of COLIG and Singleton pockets. Finally, considering the average PO for Singletons and COLIGs, we estimate that more than 80% of proteins have a pocket that satisfies the volume requirement to form COLIGs. We would like to point out that the results remain the same at different similarity threshold for both ligands and proteins (Figure S2).
Figure 6.

(A) Probability density distribution and cumulative fraction of bottom pocket occupancy, PO, for COLIGs (magenta) and Singletons (green). (B) Example from the Singleton (PDB_ID: 2A3A) set whose PO is 22%.
Comparison of ligand-ligand and ligand-protein interaction modes
To address this issue, as described in Methods, we examined the nature of the ligand-ligand and ligand-protein contacts for COLIGs. Figure 7A shows the distribution of the contact surface areas. In order to have comparisons of relatively similar sizes, we looked at the ligand-residue interaction and compared it to ligand-ligand interactions. The contact surface area between ligands, ligand-ligand, (55 ± 33 Å2) is on average twice as large as that between ligand-residue surfaces (24 ± 19 Å2). Considering that bottom pockets are comprised of 10–20 amino acids the total surface area of ligand-protein interactions will be roughly 5–10 times more than ligand-ligand. Although the ligand-ligand contact surface is almost twice of an average ligand-residue, still the majority of ligand’s contact surface comes from ligand-protein interactions. Next, we categorized the interactions into four major groups: Hydrogen bond (HB), Aromatic-Aromatic contacts (Arom), Hydrophobic (Phob) and destabilizing hydrophilic-hydrophilic contacts (DC). The sub-plot of Figure 7B shows the fraction of each of these interactions types for ligand-ligand and ligand-protein interactions. COLIGS clearly show a factor of 3–4 times enrichment for aromatic-aromatic interactions for ligand-ligand interactions compared to ligand-residue ones. We have further analyzed the interaction types in detail by considering all the combinations of the mentioned four interactions (Figure 7B). The interaction decomposition again shows that aromatic-aromatic interactions are more favored in ligand-ligand interactions independent of other interaction types. This suggests the importance of aromatic-aromatic interactions.
Figure 7.

A). Probability density distribution and cumulative fraction of contact surface for ligand-residue (blue) and ligand-ligand (orange) contacts. B) Fraction of each interaction type; Hydrogen Bond (HB), Aromatic (Arom), Hydrophobic (Phob) and destabilizing hydrophilic-hydrophilic contacts (DC) for ligand-residue (blue) and ligand-ligand (orange). A four bit vector represents the combinations of these contacts, 1 (0) indicating the presence (absence) of a particular interaction type. The order of the interactions from left to right is HB, Arom, Phob and DC.
Conclusions
In this study, we have introduced the concept of COLIGs whereby at least two interacting ligands bind to the same pocket of a protein. We began by providing experimental results which show that Folate analogues which bind to the NADPH-bound binary form of the enzyme are COLIGs. Analysis of the ternary complex structure of E. coli DHFR with methotrexate and NADPH shows that the contact surface of 34.5 Å2 (7% of the total contact surface) significantly stabilizes the enzyme. It is interesting to point out that more than 80% of COLIGs have greater than 34.5 Å2 ligand-ligand surface area.
Based on the particular sequence identity threshold, between 47–76% of ligands in the current holo protein database form COLIGS. That is, COLIGS are surprisingly prevalent. Since on average only 30% of a pocket is utilized and considering the average PO of COLIGs, 85% of pockets meet the volume requirement to form a COLIG pocket. Thus, COLIGs may even be more prevalent in nature that the estimates that are provided here.
We would also like to point out that more than half (35%) of COLIG complexes are formed without the presence of a known cofactor In addition, not all of the proteins that have COLIGs are enzymes. In fact, 27% of them are not (e.g. transferases). This number for the Singleton set was 39%. As might be expected there is a bias in holo structures towards proteins that are enzymes, with COLIGS being somewhat enriched relative to Singletons.
From the ligand perspective, (based on different Tanimoto coefficient clustering thresholds) 42–48% of ligands are found in both the Singleton and COLIG sets. Thus, there is nothing unique about the ligands that participate in a COLIG. One possible explanation why the remaining 52–58% of ligands are not also found as Singletons might be because of the small number of deposited holo structures in the PDB. However, based on our analysis of the nature of the driving forces to form COLIGS, we find that aromatic-aromatic interactions dominate. Ligand pairs lacking the possibility of forming stabilizing aromatic interactions might be underrepresented in COLIGs. This important issue needs to be further examined in the future.
Previous studies have shown that one protein-one ligand is an overly simplified explanation 20,38,39. Here, we would like to take one step further and suggest that the notion of one pocket-one ligand is at best only partly true. One can imagine that in vivo under the crowded conditions COLIGs might be even more frequent. In fact, nature might have used COLIGs to enhance interaction specificity. They might play a significant role in the regulation of biochemical processes. COLIGs also represent a promising approach to drug discovery as targeting COLIGs could provide an extra level of specificity. Knowing that certain metabolites are over expressed in a tissue (e.g. cancer cells), one can design drugs that form COLIGs with those metabolites, therefore increasing their binding affinity only in these over expressed tissues. Finally, since COLIGs are significantly stabilized by ligand-ligand contacts and contact additional protein residues relative to Singletons, their binding might be more resistant to protein mutations. These and related issues will be explored in greater detail the near future.
Supplementary Material
Figure 5.

(A) 2D-histogram of full pocket occupancy versus bottom pocket occupancy (B) Fraction of ligands binding to pockets ranked by their volume size for Singletons (green) and COLIGs (magenta).
Acknowledgments
This project was funded by GM-118039 of the Division of General Medical Sciences of the NIH. The authors wish to thank Prof. Eugene Shakhnovich, Harvard University, for providing purified E. coli DHFR protein. We would also like to thank the Developmental Therapeutics Program of the National Cancer Institute for providing the small molecules used in this study, and Dr. Mu Gao for his insightful suggestions.
Footnotes
((Additional Supporting Information may be found in the online version of this article.))
References and Notes
- 1.Roy A, Srinivasan B, Skolnick J. J Chem Inf Model. 2015;55(8):1757–1770. doi: 10.1021/acs.jcim.5b00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Srinivasan B, Zhou H, Kubanek J, Skolnick J. J Cheminform. 2014;6:16. doi: 10.1186/1758-2946-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Srinivasan B, Forouhar F, Shukla A, Sampangi C, Kulkarni S, Abashidze M, Seetharaman J, Lew S, Mao L, Acton TB, Xiao R, Everett JK, Montelione GT, Tong L, Balaram H. The FEBS journal. 2014;281(6):1613–1628. doi: 10.1111/febs.12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Srinivasan B, Kempaiah Nagappa L, Shukla A, Balaram H. Experimental parasitology. 2015;151–152:56–63. doi: 10.1016/j.exppara.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 5.Zhou H, Gao M, Skolnick J. Sci Rep. 2015;5:11090. doi: 10.1038/srep11090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao M, Zhou H, Skolnick J. Structure. 2015;23(7):1362–1369. doi: 10.1016/j.str.2015.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cencic R, Hall DR, Robert F, Du Y, Min J, Li L, Qui M, Lewis I, Kurtkaya S, Dingledine R, Fu H, Kozakov D, Vajda S, Pelletier J. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(3):1046–1051. doi: 10.1073/pnas.1011477108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kempf M, Baraduc R, Bonnabau H, Brun M, Chabanon G, Chardon H, Croize J, Demachy MC, Donnio PY, Dupont P, Fosse T, Gibel L, Gravet A, Grignon B, Hadou T, Hamdad F, Joly-Guillou ML, Koeck JL, Maugein J, Pechinot A, Ploy MC, Raymond J, Ros A, Roussel-Delvallez M, Segonds C, Vergnaud M, Vernet-Garnier V, Lepoutre A, Gutmann L, Varon E, Lanotte P. Microbial drug resistance. 2011;17(1):31–36. doi: 10.1089/mdr.2010.0031. [DOI] [PubMed] [Google Scholar]
- 9.Fei F, Stoddart S, Groffen J, Heisterkamp N. Molecular cancer therapeutics. 2010;9(5):1318–1327. doi: 10.1158/1535-7163.MCT-10-0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lahti JL, Tang GW, Capriotti E, Liu T, Altman RB. Journal of the Royal Society, Interface/the Royal Society. 2012;9(72):1409–1437. doi: 10.1098/rsif.2011.0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kosaka T, Yatabe Y, Endoh H, Yoshida K, Hida T, Tsuboi M, Tada H, Kuwano H, Mitsudomi T. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12(19):5764–5769. doi: 10.1158/1078-0432.CCR-06-0714. [DOI] [PubMed] [Google Scholar]
- 12.Balak MN, Gong Y, Riely GJ, Somwar R, Li AR, Zakowski MF, Chiang A, Yang G, Ouerfelli O, Kris MG, Ladanyi M, Miller VA, Pao W. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12(21):6494–6501. doi: 10.1158/1078-0432.CCR-06-1570. [DOI] [PubMed] [Google Scholar]
- 13.Hyde JE. The FEBS journal. 2007;274(18):4688–4698. doi: 10.1111/j.1742-4658.2007.05999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peterson DS, Walliker D, Wellems TE. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(23):9114–9118. doi: 10.1073/pnas.85.23.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lambert PA. Advanced drug delivery reviews. 2005;57(10):1471–1485. doi: 10.1016/j.addr.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Kim R, Skolnick J. Journal of computational chemistry. 2008;29(8):1316–1331. doi: 10.1002/jcc.20893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Deliv Rev. 2001;46(1–3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 18.Gao M, Skolnick J. PLoS Comput Biol. 2013;9(10):e1003302. doi: 10.1371/journal.pcbi.1003302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skolnick J, Gao M. Proc Natl Acad Sci U S A. 2013;110(23):9344–9349. doi: 10.1073/pnas.1300011110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou H, Gao M, Skolnick J. Nature Scientific Reports. 2015;5:11090. doi: 10.1038/srep11090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skolnick J, Gao M, Zhou H. F1000Res. 2016:5. doi: 10.12688/f1000research.7374.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y. Nucleic acids research. 2008;36(Database issue):D480–484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. Nucleic acids research. 2010;38(Database issue):D355–360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanehisa M, Limviphuvadh V, Tanabe M. Neuroproteomics. 2010 [Google Scholar]
- 25.Srinivasan B, Tonddast-Navaei S, Skolnick J. Eur J Med Chem. 2015;103:600–614. doi: 10.1016/j.ejmech.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng Z, Chen L, Maddula H, Akcan O, Oughtred R, Berman HM, Westbrook J. Bioinformatics. 2004;20(13):2153–2155. doi: 10.1093/bioinformatics/bth214. [DOI] [PubMed] [Google Scholar]
- 27.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao M, Skolnick J. Bioinformatics. 2013;29(5):597–604. doi: 10.1093/bioinformatics/btt024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M. Bioinformatics. 1999;15(4):327–332. doi: 10.1093/bioinformatics/15.4.327. [DOI] [PubMed] [Google Scholar]
- 30.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 31.Mount DW. CSH Protoc. 2007;2007 doi: 10.1101/pdb.top15. pdb top17. [DOI] [PubMed] [Google Scholar]
- 32.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Journal of cheminformatics. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walsh C. Nature. 2001;409(6817):226–231. doi: 10.1038/35051697. [DOI] [PubMed] [Google Scholar]
- 34.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer EL, Tate J, Punta M. Nucleic Acids Res. 2014;42(Database issue):D222–230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Menetrey J, Flatau G, Stura EA, Charbonnier JB, Gas F, Teulon JM, Le Du MH, Boquet P, Menez A. J Biol Chem. 2002;277(34):30950–30957. doi: 10.1074/jbc.M201844200. [DOI] [PubMed] [Google Scholar]
- 36.Asoh K, Kohchi M, Hyoudoh I, Ohtsuka T, Masubuchi M, Kawasaki K, Ebiike H, Shiratori Y, Fukami TA, Kondoh O, Tsukaguchi T, Ishii N, Aoki Y, Shimma N, Sakaitani M. Bioorg Med Chem Lett. 2009;19(6):1753–1757. doi: 10.1016/j.bmcl.2009.01.074. [DOI] [PubMed] [Google Scholar]
- 37.Vos S, Parry RJ, Burns MR, de Jersey J, Martin JL. J Mol Biol. 1998;282(4):875–889. doi: 10.1006/jmbi.1998.2051. [DOI] [PubMed] [Google Scholar]
- 38.Xie L, Xie L, Kinnings SL, Bourne PE. Annual review of pharmacology and toxicology. 2012;52:361–379. doi: 10.1146/annurev-pharmtox-010611-134630. [DOI] [PubMed] [Google Scholar]
- 39.Zhao Z, Xie L, Xie L, Bourne PE. Journal of medicinal chemistry. 2016;59(9):4326–4341. doi: 10.1021/acs.jmedchem.5b02041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.