

Abstract
Following insights from recent crystal structures of the muscarinic acetylcholine receptor, binding modes of Positive Allosteric Modulators (PAMs) were predicted under the assumption that PAMs should bind to the extracellular surface of the active state. A series of well-characterized PAMs for adenosine (A1R, A2AR, A3R) and muscarinic acetylcholine (M1R, M5R) receptors were modeled using both rigid and flexible receptor CHARMM-based molecular docking. Studies of adenosine receptors investigated the molecular basis of the probe-dependence of PAM activity by modeling in complex with specific agonist radioligands. Consensus binding modes map common pharmacophore features of several chemical series to specific binding interactions. These models provide a rationalization of how PAM binding slows agonist radioligand dissociation kinetics. M1R PAMs were predicted to bind in the analogous M2R PAM LY2119620 binding site. The M5R NAM (ML-375) was predicted to bind in the PAM (ML-380) binding site with a unique induced-fit receptor conformation.
Keywords: ischemic, inosine, LUF6000, Cl-IB-MECA, MRS-542
Graphical abstract
A3R partial-agonist inosine, in the presence of positive allosteric modulator LUF6000, exhibits three-fold increased activity. Docking studies predict LUF6000 binding in a “capping” interaction through a hydrogen bond with the carbonyl group of inosine. The interaction between the molecular structures of LUF6000 and inosine may explain the increase in activity. This interaction is inosine-specific, and is not seen with radioligand MRS-542, which lacks a carbonyl group.
Introduction
G-protein coupled receptors (GPCRs) are the largest superfamily of membrane-bound receptors, with approximately 800 functioning proteins encoded by the human genome. These receptors are responsible for regulating a vast variety of physiological processes. Accordingly, they are targeted by over one-third of all marketed drugs.1 GPCRs primarily signal through G-proteins and β-arrestins. GPCRs are classically divided into 4 classes: A (rhodopsin-like), B (secretin-like), C (metabotropic glutamate receptors), and F (frizzled and smoothened receptors), or as 5 classes: glutamate, rhodopsin, adhesion, frizzled/taste2, and secretin (GRAFS).2 Class A is by far the largest class, accounting for 85% of the superfamily’s genes, and is the most well studied. Examples of class A rhodopsin-like GPCRs include the adrenergic, histaminergic, muscarinic acetylcholine, dopaminergic, and opioid receptors, all of which are targets of major FDA-approved drug classes essential to modern pharmacotherapy.
Classically, drug-targeting of GPCRs was focused on producing molecules that either mimic the activity of an endogenous ligand (agonism), or attenuate it (antagonism). Recent discoveries have led to the identification of “biased ligands” that can lead to activation of specific signaling pathways.3 For example, ligands that preferentially activate the classical G-protein signaling pathway at the expense of non-canonical signaling pathways are biased. However, these molecules focus on targeting the classic orthosteric binding site of the endogenous agonists. Advances in the understanding of the structure and function of GPCRs have led to the discovery of ligands that act at sites other than the orthosteric site. These allosteric ligands, termed ‘allosteric modulators’ can amplify (positive allosteric modulators [PAMs]) or attenuate (negative allosteric modulators [NAMs]) receptor activity. Endogenous compounds have been discovered that act as allosteric modulators, including cholesterol, arachidonic acid, magnesium, zinc, and sodium.4 These allosteric ligands may lead to more precise control over the physiologic effects of receptor activation. Allosteric modulators are being actively investigated in current drug discovery programs due to the potential for attractive attributes compared to orthosteric drugs, such as increased subtype selectivity (due to less conserved allosteric sites relative to the orthosteric site), wider therapeutic windows, and maximal ceiling effects.5 These attributes can provide a greater safety margin in the setting of an overdose.
The most widely-known, clinically useful allosteric modulators are the benzodiazepines, which are modulators of the ionotropic GABAA receptor. Additionally, GPCR allosteric modulators have been FDA-approved. Cinacalcet is used in the treatment of hyperparathyroidism by acting as a PAM for the calcium-sensing receptor.6 Maraviroc acts as a NAM at the CCR5 receptor, preventing viral entry of HIV.6 Plerixafor is a PAM for CXCR4, and is used for stem cell mobilization.6 Moving forward, GPCR allosteric modulators are being investigated for use in disorders such as Alzheimer’s disease, Parkinson’s disease, and schizophrenia.7,8
Potential allosteric binding sites on class A GPCRs include the extracellular domain (N-terminal and loop regions), membrane associated regions, intracellular domains, and within the transmembrane region.9,10 It is possible these allosteric sites are binding regions of undiscovered endogenous allosteric modulators, rather than cavities exclusively secondary to a structural role.4 Pepducins, a class of lipopeptides, have been found to act as intracellular allosteric modulators.11,12 Other classes of biomolecules, such as RNA aptamers, can bind both intracellularly and extracellularly.13 Numerous allosteric modulators appear to bind at or near the extracellular vestibule of the receptor, and this region in particular is of interest for druggability.14–16 Another class of ligands can span the orthosteric site and reach into the allosteric site, providing a dual mode of action.17 These are termed “bitopic ligands”, and bind to both the orthosteric and allosteric sites concurrently. They can help probe the allosteric site by competing with allosteric fragments of the bitopic ligand and by determining linker sizes that result in modulation. Our current work focuses on well-characterized allosteric modulators that bind to the GPCR extracellular surface and compete with agonist radioligand dissociation.
Recently, the M2 muscarinic acetylcholine receptor has been crystallized bound to PAM LY2119620 in complex with the orthosteric agonist iperoxo. This provides the first structural characterization of such a PAM extracellular surface binding site,16 where the PAM binds above the orthosteric agonist binding site Figure 1(A). Given this breakthrough in the structural knowledge of how a selective PAM binds to M2R, this knowledge can be extended to model the binding modes of other PAMs from the literature. As these PAM-bound GPCR complexes appear quite challenging to characterize using crystallography (due to modest PAM binding affinity), this research objective is important to the emerging field of the discovery and development of GPCR allosteric modulators. One of the most interesting conclusions from the analysis of the M2R crystal structure was that the agonist-bound active conformation of the receptor was very similar to the agonist-bound M2R structure in complex with the PAM iperoxo.16 This observation was in agreement with predictions from the Monod-Wyman-Changeux allosteric model, which is that the PAM should preferentially bind to and stabilize active conformations of the receptor.18 There were only very minor deviations in the backbone conformation, and few minor induced-fit changes in side chain conformations in the allosteric pocket (such as W422), which forms an important pi-stacking interaction with the bound PAM. Other parallel structural studies of active conformations of A2AR have also concluded that there is reasonable conservation of agonist-bound “active”, “partially active”, and “intermediately active” conformational states.19 These states are stabilized by combinations of agonists, protein engineering, and optimized nanobodies to stabilize the active conformation.19
Figure 1. Crystal structures of the active conformation of two representative class A GPCRs in which several chemical classes of Positive Allosteric Modulators (PAMs) have been reported.

(A) The active conformation of the M2 muscarinic acetylcholine receptor (M2R) bound to the PAM LY2119620 in complex with the agonist iperoxo (4MQT.pdb)16. (B) The structure of the active conformation of the A2A adenosine receptor (A2AR) bound to the endogenous agonist adenosine (2YDO.pdb). In each image, the GPCR transmembrane helices are colored by rainbow scanning from transmembrane helix 1 (TM1) in blue to (TM7) in orange. This coloring is utilized through the manuscript to show the extracellular receptor surface colored by residue topology. A transparent molecular surface is shown for the bound small-molecules particularly to show how LY2119620 binds to an allosteric site on the extracellular receptor surface “on top” of the bound agonist iperoxo in the orthosteric site.
However, these observations from recent X-ray crystal structures of agonist-bound conformational states of GPCRs only provide an experimental static snapshot of these flexible receptors. A growing body of evidence from biophysical studies also supports the viewpoint that these receptors exhibit significant flexibility and visit ligand-specific active conformational states.20–24 Complimentary to these experiments, there have been impressive advances in predicting the fluctuations of various ligand-stabilized conformational states from molecular dynamics simulations and studies utilizing advanced sampling strategies that aim to efficiently map the free energy landscapes of diverse receptor conformational states.25 One particularly relevant recent study compared the free energy landscape of M2R bound to full agonist iperoxo (IXO), the partial agonist arecoline (ARC) and the inverse agonist 3-quinuclidinyl-benzilate (QNB).26 The authors concluded that in the receptor-nanobody complex, compared to the partial agonist, the full agonist IXO binding resulted in increased fluctuations in the intracellular protein-coupling interface (TM5, TM6, and TM7) but not in the extracellular surface due to the stronger binding of the orthosteric agonist. Thus, when bound to the partial agonist, the extracellular surface exhibited enhanced fluctuations and flexibility in both the orthosteric pocket, the tyrosine lid, and ECL2 region of the allosteric pocket and the extracellular vestibule in general.26 This study provides evidence for distinct low-energy conformational states that are ligand-specific, two distinct conformations of the orthosteric pocket, where IXO binding shifts the receptor conformational equilibrium to the closed state of the tyrosine lid compared to the partial agonist.26 Previous studies of M2R binding to allosteric modulators also sampled various conformational states of the extracellular vestibule during binding and dissociation events in long-timescale MD simulations.27 Simulation evidence from both of these studies suggest that the extracellular vestibule is highly flexible, samples various conformational states, and that allosteric modulators may bind to and stabilize ligand-specific conformational states.26,27 Similar computational studies of ligand dissociation of the antagonist tiotropium bound to M3R have shown that orthosteric ligands may also transiently bind in the allosteric site during ligand dissociation.28 Experiments have shown that tiotropium dissociates more slowly from M3R compared to M2R29 and molecular dynamics simulations of tiotropium bound to the receptors show a corresponding increase in the flexibility of the ECL2 loop of M2R compared to M3R, which may also rationalize slower dissociation kinetics.28 Collectively these results from multiple simulation studies suggest that interactions that stabilize the conformational state of the dynamic extracellular vestibule may contribute to slower orthosteric ligand dissociation.
To our knowledge, no co-crystal structures of adenosine receptors bound to small-molecule PAMs and orthosteric agonists have yet been solved or published. However, numerous small-molecule PAMs of adenosine receptors have been characterized that compete with radioligand agonists and likely bind to an extracellular surface binding site in proximity to the orthosteric adenosine binding site.30 Previous A1R studies have found that residues on ECL2 are implicated in PAM activity31 and have also suggested that the allosteric binding site may be located in a pocket formed by the ECL2 loop bounded by residues S150 and M162.32 It is possible that Class A GPCR extracellular surface allosteric modulators share a common binding site or common molecular binding mechanisms. By predicting the consensus binding modes of several structurally related chemotypes of small-molecule PAMs for adenosine or muscarinic receptors, we provide a clear molecular picture of how PAMs bind to and stabilize agonist-bound conformational states. Our studies of adenosine receptors investigate the molecular basis of agonist radioligand probe-dependent effects, and predict specific interactions between PAM R-groups and reference agonist radioligand atoms. In performing our studies, we do not necessarily assume that structurally related compounds will always bind in the same binding mode, but we explore this possibility using free unrestrained docking in the presence of appropriate agonist radioligands. In general, our results from both flexible receptor docking and rigid receptor docking suggest a common molecular mechanism of how PAMs may slow the dissociation kinetics of pre-incubated agonist radioligands and have significant implications for the design of future experimental efforts to characterize PAM activity.
Methods
Although several previous computational investigations of the binding mode of GPCR PAMs have utilized advanced methodologies in which the receptor can be fully flexible and sample various conformational states,26,27,32,33 such methodologies may not be necessary at a first approximation. Based on the recent crystal structure of M2R PAM, it is possible that PAMs may bind to and stabilize the active conformation of the extracellular receptor surface. Muscarinic and adenosine receptors structures to date have exhibited minimal deviation from a similar active conformational state.16,19 Based on these recent observations from crystallography, in our initial work we utilized a CHARMM-based rigid receptor docking approach, assuming that PAMs preferentially bind to active conformation of GPCRs with minimal deviation in the backbone conformation of the extracellular vestibule. However, given a body of evidence from biophysical studies and from molecular dynamics simulations supporting a highly flexible active state of the extracellular vestibule, we also performed the docking using a more sophisticated flexible receptor docking approach. Therefore, our flexible receptor approach should reasonably explore the possibility that PAMs may bind to the receptor and induce novel activated state conformations that are on the order of 1.0–1.3 Å Cα RMSD from extracellular vestibule backbone conformations characterized by X-ray crystal structures of active conformations. We highlight the most noteworthy similarities and differences of docking results using both rigid and flexible receptor docking.
Homology Modeling & Refinement
Homology models of A1R and A3R were built using four crystal structures of A2AR as individual templates to generate an ensemble of receptor conformations. Three active conformations of A2AR were utilized: a thermostabilized receptor bound to the endogenous agonist adenosine (2YDO.pdb),19 Figure 1(B) , the receptor bound to the agonist NECA (2YDV.pdb),19 and the receptor bound to the agonist UK-432097 (3QAK.pdb).34 For A2AR it was not necessary to built a homology model as the thermostabilized A2AR receptor bound to the A2AR selective agonist CGS21680 (4UG2.pdb) was used.35 Finally, homology models for A1R and A3R were also built using an inactive conformation of A2AR bound to the antagonist ZM241385 (3EML.pdb).36 For each crystal structure, five homology models were constructed using Modeller with standard basic modelling protocol from a multiple sequence alignment.37,38 Homology models of M1R and M5R were built in a similar way using the crystal structure of the M2R complex simultaneously bound to the agonist iperoxo and the PAM LY2119620 (4MQT.pdb).16 Similarly, five models for M1R and M5R were constructed using Modeller with standard basic modelling protocol from a multiple sequence alignment.37,38
Supplementary Table 1. provides comparative values of initial homology model quality, including Modeler objective function, Modeler DOPE score, and a CHARMM minimum energy for the minimized complex with reference agonist. Homology models built for A1R using templates with ~51% sequence identity, and A3R using templates with ~42% sequence identity, resulted in similar ranges of Modeller objective function and DOPE scores. Resulting homology models for M1R using the template (4MQT.pdb) with 65% sequence identity result in a higher Modeller objective function values ranging from 4090-3910. Homology models for M5R using template (4MQT.pdb) with 68% sequence identity also resulted in a higher values of the Modeller objective function. In comparison of the Modeler scores and CHARMM minimized complex energy with reference agonists (Supplementary Table 1.) the majority of models have minimized complex energies that on the same order as the complex built from the crystal structure of the A2AR selective agonist CGS21680 (4UG2.pdb).35 The models with the least favorable complex energies were the M5R receptor models, but the reason for the difference in magnitude of the minimized potential energy was that the intracellular loop res: 212–445 was truncated from the model (which does not affect the flexible receptor model utilized). Upon visual inspection, all models exhibited reasonable structure-sequence-alignment in the extracellular region. Models were selected for further refinement and were then energy-minimized in CHARMM using a GBMV implicit membrane potential function.39–41 GBMV implicit membrane potentials were built for the receptors using CHARMM-GUI.42 N-terminal and C-terminal disordered regions of the models that were not fully resolved in the associated crystal structure template were truncated. This avoids artifacts and retains models that are as close to the template crystal structures as possible. In addition, poorly-modeled cytoplasmic loops were truncated and removed from the models, as they are irrelevant to the extracellular search space.
Molecular Docking
To investigate allosteric binding modes, we have used CHARMM-based docking methods for highly accurate predictions of small-molecule binding geometries to a rigid receptor surface. In a previously published assessment of docking accuracy, these CHARMM-based methods were shown to have the highest “discriminative power” to correctly predict binding geometries over diverse classes of protein-ligand interactions compared to other common scoring functions.43,44 After the GPCR complexes were initially minimized using a GBMV implicit membrane potential function, this implicit solvent representation was no longer used for docking. Molecular docking to the rigid receptor surface utilized the LPDB CHARMm force field, the Linear Interaction Energy (LIE) scoring approach and parameters as described in.44 The exact implementation of the GBMV implicit solvent (and the associated parameters) previously established for high docking accuracy was utilized.43,44 This approach should be sufficiently rigorous to model PAM binding to the extracellular receptor surface and some specific limitations of this approach are discussed when relevant in Results.
CHARMM-based Rigid Receptor Docking
Our approach to molecular docking uses the program CHARMM42 for an all-atom force field potential energy description of the protein-ligand complexes. A significant advantage of using CHARMM as an engine for molecular docking is that many different conformational searching strategies have been implemented, such as MD simulation techniques for flexible receptor docking.43 Docking consists of a sequence of independent trials that are composed of a large number of individual docking attempts (where a single conformation of a ligand is docked to the protein). An independent docking trial, initiated from a given ligand initial conformation at a specific reference ligand position, proceeds by generating a series of random initial ligand conformations, and then generating a series of random orientations that are docked to the protein binding site. Throughout this paper, docking was performed from 22 different reference positions on the GPCR extracellular surface. How these reference positions were determined is described in the next section. In our rigid receptor docking methods which have been described previously in,43,44 the CHARMm force field originally parameterized by Momany and Rone has been extended to describe ligands in the Ligand-Protein Database (LPDB) and was used to build potential energy functions for all ligands as previously described.43,44 For ligand poses that result from the simulated annealing conformational search the all-atom protein-ligand representation is then minimized, fixing the coordinates of the protein, using the standard hard-core repulsion for both VDW and electrostatics with a distance-dependent dielectric function (Rdie). Various components of the final minimized potential energy are employed to construct CHARMm-based LIE (Rdie) scoring function for ranking ligand poses from the conformational search. For each independent docking trial, the top-five lowest energy poses are then re-scored with the LIE scoring function using the GBMV implicit solvent, LIE (GBMV) as described in.44
CHARMM-based Flexible Receptor Docking
Our approach to flexible molecular docking uses the program CHARMM42 for an all-atom force field potential energy description and the dynamics of the protein-ligand complexes that is an extension of the previously described rigid receptor method, and ultimately uses the exact same LIE (GBMV) scoring function using the GBMV implicit solvent as described in.44
The model utilized here is analogous to previously described flexible receptor models using MD for conformational sampling utilizing the CHARMM consfix routine to create rigid segments of backbone and side chain residues. In table 1. of Armen et al.43 the type of flexible receptor model employed here for GPCRs is similar to what is denoted “MD model2” through “MD model4.” The model was developed such that the extracellular GPCR surface backbone and residues may exhibit flexibility ranging from minor side chain movements to backbone deviations on the order of 1.0 to 1.3 Å Cα RMSD. In the GPCR flexible receptor model, backbone and side chain flexibility is defined by four flexible segments and four rigid cluster segments using the CHARMM consfix routine. This effectively allows the extracellular residues of TM1 and each extracellular loop (ECL1, ECL2, and ECL3) to be appropriately flexible, yet the overall topology of the receptor is locked into an active conformation of the cytoplasmic side of the receptor. The extracellular flexible residue segments that define these flexible receptor models are defined in Supplementary Table 2. and also show that these models result in similar total to degrees of freedom for the conformational search space as similar previous models utilized in Armen et al.43 For example as shown in (Supplementary Table 2.) the A1R receptor, the following extracellular surface residues were defined as flexible segments (11–16, 64–85, 139–185, 249–276). For the M5R receptor, the following extracellular surface residues were defined as flexible segments (28–40, 86–109, 164–195, 460–483). To be perfectly clear, in the M5R receptor models, the intracellular loop res: 212–445 was truncated from the model, however, as these intracellular loop residues would be selected to be a rigid cluster of the receptor, the truncation of the loop does not affect the flexible receptor model.
For each independent docking trial, initiated from a given ligand initial conformation at a specific reference ligand position, 200 flexible ligand conformations are generated, and 200 diverse flexible conformations of the receptor in complex with appropriate radioligand agonists are generated using MD simulated annealing. After the newly generated flexible conformations of ligand and protein are combined, 200 new complexes are subjected to MD simulated annealing refinement with simultaneous ligand and receptor flexibility. For this conformational refinement of PAM GPCR binding modes, the receptor (backbone and side chain), as well as the bound agonist, and the PAM are all simultaneously flexible during the refinement. Exactly analogous to the rigid receptor docking, for each independent docking trial, the top-five lowest energy flexible-receptor ligand complexes according to the CHARMm-based LIE (Rdie) scoring function for ranking ligand poses from the conformational search. These top-five lowest energy complexes are then re-scored with the LIE scoring function using the GBMV implicit solvent, LIE (GBMV), as described in.44
GPCR Extracellular Surface Search Strategy
The following procedure was used to develop and establish a search strategy for the entire extracellular surface, and was eventually applied to each adenosine receptor PAM in this study. Towards this end, we were inspired to initiate our studies of adenosine receptor PAMs by docking the bitopic ligand 6 (LUF6258) to A1R, as was previously done by authors who studied the bitopic ligands as a function of linker length.45 Molecular docking was performed by generating 40 diverse molecular conformations of 6 using MarvinBeans.46 These ligand conformations of 6 were then used for 40 different initial conditions for independent docking into the active conformation models of A1R in the absence of any bound agonist. Iterative rounds of docking were performed with top ranked poses that had the adenosine moiety bound isosteric with the adenosine-bound A2AR crystal structure (2YDO.pdb).19 Supplementary Figure 1. depicts the 20 lowest energy conformations of the bitopic ligand bound to A1R showing diverse conformations of the PAM group binding to the receptor surface that were sampled. Given the large number of degrees of freedom for 6, rather than performing an exhaustive search of the bitopic conformational space, we utilized the preliminary results for the bitopic ligand to map the reasonable extracellular surface of the active conformational state of A1R where a PAM might bind. The coordinates of the aminothiophene PAM groups at the end of the bitopic linkers were used as new center-of-mass reference positions for docking. A total of 22 representative reference positions were identified that were used as starting points for numerous independent docking trials to search the extracellular receptor surface for low-energy allosteric PAM binding sites within reasonable agreement with the experimental data regarding the linker-length of 6 (9 carbon linker).
After constructing the receptor complex for all adenosine receptor models (A1R, A2AR, A3R) with the appropriate bound reference agonist (see next section in methods), the identical extensive search strategy for the entire extracellular receptor surface (described above) was applied for rigid receptor and flexible receptor docking of all adenosine receptor PAMs, including 3, 4, and 7–10. Figure 2. shows all of the adenosine receptor small-molecule PAMs used in our current study. For each small-molecule, once the lowest energy binding conformations are identified at particular “hot-spot” locations on the receptor surface from the extensive search, subsequent independent docking trials (additional sampling) was focused in proximity to these binding “hot-spots” initiated from different diverse initial conformations of the small-molecule (typically 20 to 40 conformations) generated with Marvin Beans.46 A similar overall procedure was performed for docking into the M1R and M5R models, although more focused sampling was appropriately performed in the proximity of the reference M2R PAM LY2119620 (again initiated typically from 20 to 40 diverse conformations of the small-molecule).16 For each individual homology model of a receptor conformation, top-ranked clusters were identified according to the LIE (GMBV) scoring function as described previously.44 Top-ranked “consensus clusters” were subsequently identified by comparing results of over an ensemble of receptor conformations (5 Modeller homology models of each crystal structure were utilized). A “consensus cluster” is defined as a top-ranked cluster conformation of similar geometry identified in multiple searches over different receptor conformations. In this way, the best predicted binding geometry is a commonly identified solution to the conformational search problem that is relatively robust over several molecular models of the receptor.
Figure 2.

Adenosine receptor PAMs used in this study.
Modeling Receptor Complexes with Specific Agonist Radioligands
In our studies of small-molecule PAMs that selectively bind to adenosine receptors, it was clear that it was important to use the most relevant agonist radioligands utilized in experimental studies to characterize the PAM activity. Starting with our models for A1R, as the radioligand [3H]CCPA was utilized in experimental studies of the bitopic ligands and to characterize PAM activity for all of our model A1R PAMs (1–5), it was selected as the common bound agonist in our A1R models Figure 3(A). For our studies of the A2AR selective PAM 8, docking was performed using the thermostabilized A2AR crystal structure bound with the selective agonist CGS21680 (4UG2.pdb)35, which was utilized in the experimental characterization of 8.47
Figure 3. Use of appropriate bound agonist radioligands in modeling PAM bound complexes of adenosine receptors.

PAMs selected for use in this study have been shown to slow the dissociation of agonist radioligands in dissociation kinetics experiments. For each adenosine receptor the most appropriate agonist radiolabel is modeled bound to the receptor prior to docking PAMs to the complex: (A) for A1R the agonist radiolabel [3H]CPPA, (B) for A2AR the agonist radiolabel [3H]GGS-21680 and (C) for A3R the agonist MRS-524 was used which should also model the effects of the common reference agonist Cl-IB-MECA and the radioligand [125I]I-AB-MECA. Probe dependent molecular interactions are predicted between the carboxylic acid of [3H]CGS-21680 and the aryl urea of 8 when bound to AA2R as shown in (B).
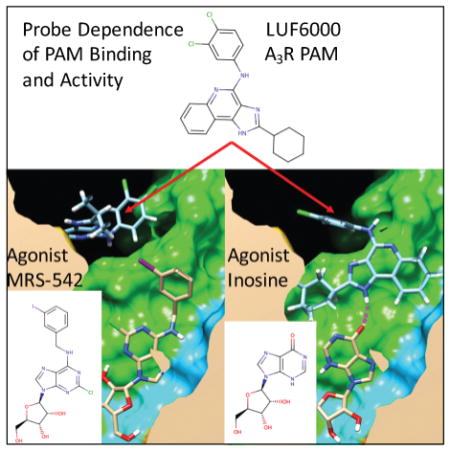
In other studies of the A3R selective PAMs, the allosteric modulation activity of 9 has been studied as a function of several A3R agonists and agonist radioligands of varying molecular structure.48 In this experimental study, the PAM activity of 9 was characterized using a series of four similar agonists (IB-MECA, Cl-IB-MECA, MRS-541, MRS-542) which all share a bulky m-iodobenzyl group. This common m-iodobenzyl group represents the largest common R-group of the agonists studied that might possibly participate in probe-dependent molecular interactions with the PAM binding modes and/or contribute to R-group specific enhanced stabilization of the activated state of the receptor. In these studies, 9 was shown to have increased PAM activity (Emax) for Cl-IB-MECA and other lower-efficacy agonists compared to more modest increases with high-efficacy agonists NECA, which does not share the bulky m-iodobenzyl group.48 As the agonist Cl-IB-MECA was one of the most commonly used agonist in functional studies and shows significant agonist-dependent PAM activity of 9 for A3R, we decided that it should be used as a reference ligand for docking of 9 and 10. However in practice, less atom clashes modeling into the homology models were produced using the very similar ligand, MRS-542, which shares the identical m-iodobenzyl group. The m-iodobenzyl group protrudes out of the orthosteric binding site Figure 3(C) and may form interactions with allosteric modulators as seen for the A2AR PAM 8 and agonist [3H]CGS21680 shown in Figure 3(B). For our goal of modeling PAMs onto the complex of the most relevant reference agonist, there should be no difference in using MRS-542 compared to Cl-IB-MECA, as the major difference in chemical structure between the two agonists forms interactions deep in the orthosteric pocket, not on the receptor surface. Molecular docking of 40 conformations of the agonist MRS-542 into the ensemble of A3R models resulted in the identification of a highly populated top-ranked cluster with nearly isosteric binding geometry of the adenosine moiety. This top-ranked cluster conformation was used to represent the binding mode of the bulky m-iodobenzyl group that is common to the series of reference agonists.
In strong contrast to adenosine receptors, in modeling the M1R and M5R receptors, probe dependence of different bound agonists was not investigated as the different agonists would remain bound in the orthosteric pocket below the closed aromatic “lid” of the receptor, and would therefore likely not form direct molecular interactions with the bound PAMs. Based on the crystal structure of the bound PAM,16 we assume that significant agonist probe-dependent molecular effects would likely result in minor conformational changes to the active conformation of the receptor itself. Both rigid and flexible receptor molecular docking was performed using the M1R and M5R homology models.
Results and Discussion
Adenosine Receptors
Adenosine is a nucleoside signaling molecule that modulates myriad physiological processes, with generally cytoprotective actions.49 These include inflammation, vasodilation, angiogenesis, myelination, and dopamine signaling. As a result of these properties, adenosine receptor ligands are being investigated for therapeutic uses such as multiple sclerosis, COPD, asthma, Parkinson’s disease, and oncology.50 Each adenosine subtype has unique pharmacological properties, different levels of tissue distribution, and may be implicated in different disease pathologies. Therefore, subtype-selective ligands and allosteric modulators are being pursued to minimize off-target effects. PAMs of adenosine receptors have shown promise as potential therapeutic agents, particularly in the context of tissues actively releasing adenosine in response to acute tissue injury. For example, PAMs of A1R have shown protective effects against ischemia reperfusion injury in several relevant tissues including the heart and brain, and have shown promise in animal models of neuropathic pain.51–55 An A2AR selective PAM was shown to attenuate inflammatory responses in a lipopolysaccharide-induced mouse model of inflammation, with implications for several chronic inflammatory diseases.56 Lastly, A3R selective PAMs have been proposed to be helpful in the treatment of ischemic diseases and other hypoxic conditions including angina, myocardial infarction, stroke and cancer.57
A1R
The very first allosteric modulators of A1R were identified more than 20 years ago using radioligand binding assays.58,59 Three amino-thiophene compounds, 1 (PD-71,605), 2 (PD-81,723) and 3 (PD-117,975), were found to increase the binding of agonist radioligands and slow the dissociation of radioligands such as the agonist [3H]N6-cyclohexyladenosine. Subsequent studies using radiolabeled ligand T-62 similar in structure to 1 have shown that these PAMs bind to A1R at a site that is distinct from the orthosteric binding site for classical agonists and antagonists. These amino-thiophene compounds (1–5) are classic model system A1R selective PAMs and have been characterized by numerous laboratories.60
A previous experimental study of bitopic ligands linking adenosine and a 3,4-dichloral derivative 5 (LUF5484) was able to provide significant insight into possible locations of the allosteric site.45 In these studies, bitopic ligands were characterized using equilibrium radioligand displacement assays with [3H]CCPA, both in the absence and in the presence of a model PAM 2. Although the bitopic ligand with a 6 carbon spacer exhibited the greatest affinity, bitopic ligand 6 (LUF6258) (with a 9 carbon linker) did not exhibit changes in either affinity or potency in the presence of 2, unlike compounds with shorter linkers. The authors interpreted the result to be due to bitopic ligand binding simultaneously at both the allosteric and orthosteric site. The authors concluded that the allosteric binding site should be within close proximity to the orthosteric agonist binding site on the extracellular surface of the receptor, possibly on the second extracellular loop (ECL2).45 In our own modeling of the bitopic ligand 6 shown in Supplementary Figure 1, a preliminary search of the bitopic conformational space was used to create a map of the extracellular surface of the active A1R where a PAM might bind. From this map of reference positions, a very thorough search of the extracellular conformational surface was performed for each subsequent compound investigated (See Methods and Figure 4).
Figure 4. Rigid receptor docking results for 3 (PD-117,975) from rigorous sampling of the entire extracellular surface.

(A) Surface is shown populated only by local minima structures (“top-5”) for 22 different reference positions for independent docking trials. (B) The all-atom structures of the top 5 lowest energy members of the “top-ranked cluster” where the heavy atom RMSD of all members is less than 1.5 Å from the minimum. The red circle indicates the yellow colored thiophene sulfur atom in (B) where this sulfur atom is depicted as red dots above in (D). (C) shows the 2nd lowest energy pose in comparison to the lowest energy cluster, where an important similarity is the orientation of the 1-amino and 2-carbonyl group of the thiophene ring. (D) A projection of the energy landscape showing only the local minima (“top-5”) structures where the X,Y coordinates of the thiophene sulfur atoms are plotted and colored according to the LIE (GBMV) scoring function, where the lowest energy poses are colored red and the highest energy local minima are shown in dark blue. (E) The same docking data plotting LIE (GBMV) by the heavy atom RMSD of the ligand atoms to the lowest energy pose. (F) Is the same plot zoomed in on a smaller scale closer to the minimum of the lowest energy cluster.
In subsequent docking of PAMs such as 2 (PD-81,723) and 3 (PD-117,975), significant attention was placed on sufficient sampling of the entire extracellular surface of the receptor to include the most distant possible PAM sites according to the previous preliminary bitopic ligand poses. Figure 4(A) shows the receptor surface covered with top-ranked local minimum binding poses of 3 where every pose shown must be a local minimum, within the top 5 lowest energy poses from an independent docking trial initiated at 22 different reference positions along the receptor surface. In this way, sufficient sampling of the extracellular surface of the receptor was achieved, where every pose shown in Figure 4(A) effectively represents a local minimum from one thousand docking attempts within an individual docking trial. Figure 4(D) is a representation of the energy landscape of the search space of 3, mapped onto an (x,y) scatterplot of the coordinates of the thiophene sulfur atom of 3 from top-ranked binding poses from independent docking trials sampling the receptor surface. Within the top 20 lowest energy poses out of more than 22,000 poses, 50% of top 20 poses are members of a “top-ranked” cluster with minimal deviation in predicted binding geometry shown in Figure 4(B). The top eight members of this cluster all are within 1.5 Å RMSD of the lowest energy pose. Statistics from the energy distributions show that the top-ranked poses from the search are on the order of Z score = −3.2, indicating a sufficient discrimination power of the scoring function to separate out the lowest energy solutions from the distribution of poses sampled on the receptor surface. Reasonable discrimination from the rest of the energy distribution, and tight conformational clustering (< 2.0 Å RMSD) of the top-ranked poses shown in Figures 4(D–F), provides confidence that, given our scoring function, this is the best prediction of ligand geometry.
Figure 4(B) shows the top-ranked consensus binding pose for 3 and (Supplementary Figure 2.) shows this pose in comparison to four other representative “hot-spots” on the extracellular receptor surface, where other low-energy poses and clusters of poses were identified. One of the hot-spots shown highlighted with an orange circle (Supplementary Figure 2.) is in the proximity to a ECL2 binding mode for 2 and structurally related PAMs that were recently predicted by Abagyan, Yeager and colleagues.32 The hot-spot circled in purple is in reasonable agreement with the model for 6 presented by the authors of the bitopic ligand study.45 Based on orientation and linker length, this result is logical. Yet, despite significant sampling in the proximity of these hot-spots, our consensus top-ranked binding mode is much more favorable in predicted free energy according to the LIE (Rdie) and LIE (GBMV) CHARMM-based scoring function (See Methods). Lastly, a binding pose between TM1 and TM7 was also found to be a common hot-spot and is highlighted with a red circle. This site has been observed to bind lipid and fatty acid like ligands such as (2S)-2,3-dihydroxypropyl (7Z)-tetradec-7-enoate in several GPCR structures such as β2AR,61,62 and has also been thought of as a possible site responsible for allosteric modulation by other endogenous lipids. In our studies of several other diverse PAM structural classes, some specific ligands form quite reasonable hydrophobic and “shape-fitting” complementary interactions with this site. We acknowledge that the implicit solvent model methodology currently used in our methods may be insufficient and/or somewhat inappropriate to evaluate ligand binding at this site rigorously, particularly in comparison to the other sites explored which should be modeled reasonably well using our current methods. Exploration of ligand binding to this specific site (between TM1 and TM7), may require more specific knowledge of receptor-receptor and receptor-lipid-PAM interactions, and explicit all-atom modeling of the membrane environment that is beyond the current scope of this work. From our extensive rigid receptor sampling of 3, we found that all of the lowest-energy poses, including our top-ranked cluster, involved specific interactions with ECL2 residues. Results from flexible receptor docking corroborated these observation of important specific hydrophilic interactions with ECL2 residues Figure 7 (E,F). The rigid receptor and flexible receptor binding modes of 3 and 4 modeled in complex with agonist radioligand [3H]CCPA, shown in Figure 5(A), Figure 6(E,F) and Figure 7(E,F) demonstrates specific protein-ligand interactions that are in agreement with compound derivative SAR data demonstrating that any modification of the amino group or the carbonyl group of the thiophene ring abrogates PAM activity.57,58
Figure 7. Flexible receptor consensus binding modes for adenosine receptor PAMs compared to rigid receptor docking results.

This figure show the flexible receptor poses in magenta that correspond to Figure 6 along with their induced fit protein-ligand interactions. Poses shown in cyan are superimposed consensus binding modes from rigid receptor docking. The heavy atom RMSD between the two ligand poses is shown for each structure. Black arrows are shown to visualize how the rigid receptor pharmacophore groups may undergo minor rotations towards the flexible receptor results to highlight similarities and differences in the predicted poses. For example, for (A) and (D), only one of the three pharmacophore groups was not in the same orientation for both the rigid and flexible docking result. Supplementary Figure 4. shows the rigid receptor docking poses on the rigid receptor surface, exactly as they were submitted in the original draft of the manuscript.
Figure 5. Flexible docking results for adenosine receptor PAMs and specific protein-ligand interactions with salt-bridge residues.

Top-ranked poses from flexible docking are shown for (A) Top view of 4 bound to A1R in complex with CPPA showing specific electrostatic interactions of the 1-amino group of the thiophene ring with E170 and the 2-carbonyl group of the thiophene ring with K173 (shown in magenta). The charged basic amine forms strong electrostatic interactions with E172 (B) Top view of 8 bound to A2AR in complex with GGS-21680, highlighting that the PAM binds on the extracellular surface “on top” of the salt bridge formed by residues H264 and E169. The aryl amine group of 8 forms a hydrogen bond to the charged side chain of E169 while still forming a salt-bridge (E169 and H264). The hydrogens from the urea group of 8 form probe-dependent electrostatic interactions with the carboxylic acid group of GGS-21680 (C) Top view of 9 bound to A3R in complex with MRS-542 showing specific hydrogen bonding interactions of the aryl amine with S170 (in close proximity to E156) and the imidazole nitrogen with Q167. The other imidazole nitrogen of 9 is within 3.8 Å of the carboxylic acid OE1 atom of E258.
Figure 6. Flexible receptor consensus binding modes for adenosine receptor PAMs showing the receptor surface and orientations of 3-point pharmacophores.

Following extensive sampling of the entire extracellular receptor surface for each compound, top-ranked clusters from flexible docking were identified supporting a consensus binding mode over several chemical series. The binding modes of azaadenines 7 and 8 are very similar to that of 9 (LUF6000) and 10 (LUF6096). The A2AR and A3R selective PAMs 8–10 are compared to the A1R PAMs 3 and 4 and similar pharmacophore features from the common binding modes are mapped onto 2D structures. For example, the aryl amines of 7–10 form similar binding interactions with the ECL2 loop, similar to the 1-amino group of the thiophene in 3 and 4. For simplicity, the compounds are all shown on the receptor surface. Supplementary Figure 3. shows the exact same poses with corresponding protein-ligand interactions with specific amino acids.
This has been widely interpreted to infer that these groups participate in direct hydrogen bonding or electrostatic interactions with the receptor. In the consensus binding mode, the amino group or the carbonyl group of the thiophene ring participate in strong protein-ligand interactions. Larger R-group substituents at the 4 and 5 positions of the thiophene ring were well tolerated in SAR studies.57,58 Compound 4 is a structurally-related derivative of 3 that has been shown to have increased PAM activity (~7 fold increase in Bmax) compared to 2 in competition assays with the radioligand [3H]CCPA.63 In comparing the predicted binding mode of 3 and 4, the R-group extension leading to improved potency is rationalized by complementary hydrophobic interactions with the hydrophobic patch on the receptor surface shown in Figure 5 (A).
The binding mode for 3, shown in Figure 6(E), demonstrates electrostatic interactions that are strong determinants of molecular binding geometry. The amino group of the thiophene forms strong electrostatic interactions with the carboxylic acid side chain of residues E170 and E172 and the carbonyl group of the thiophene forms strong electrostatic interaction with the NZ primary amine of residue K173. These two strong electrostatic interactions are formed directly below the indole side chain of W156 in both rigid receptor and flexible receptor top-ranked poses, providing a partially buried environment for these important interactions. The overall binding mode of the PAM forms strong protein-ligand electrostatic interactions to stabilize the ECL2 loop conformation, yet also forms hydrophobic and electrostatic contacts with TM6 (res:T257, A258) and the ECL3 loop between TM6 and TM7 (res:P261, H264, P266). The charged tertiary amine of 3 and 4 is within proximity to form electrostatic interactions with both E172 and E153. The PAM sits on the extracellular surface of the receptor, on top of a strong electrostatic interaction (a salt-bridge) between the imidazole side chain of H264 (TM6) and the carboxylic acid side chain of E172 (ECL2). Interestingly, the hydrophobic cyclopentyl group of the agonist radioligand [3H]CCPA sits in a very favorable hydrophobic pocket buried directly below this salt-bridge residue pair H264 (TM6) and E172 (ECL2) shown in Figure 7(B). The carboxylic acid of E172 also forms a strong electrostatic interaction with the amino group of the [3H]CCPA ligand. For A2AR, it has been shown that disruption of the analogous extracellular salt bridge above the orthosteric binding pocket accelerates the dissociation of ligands from A2AR, while stabilization slows it.64 In A2AR, this salt bridge is part of a “triad” of hydrogen bonding interactions that acts as a “cap” over the orthosteric site, preventing influx of water into the receptor that accelerates dissociation.64 This “cap” characteristic is also found in other GPCRs, such as the muscarinic receptors. M2R and M3R both share an analogous network of residues that buffer the orthosteric site from water.16 The β2-adrenergic receptor has an analogous salt bridge on the extracellular surface that has been implicated in receptor activation.65
We propose that the predicted binding mode for 4 shown in Figure 5(A) offers two mechanisms to rationalize slow dissociation kinetics of agonist radioligands such as [3H]CCPA by (1) forming new specific protein-ligand interactions connecting ECL2 loop to the ECL3 loop, thus stabilizing the overall receptor conformation via multiple topological points of contact and (2) by PAM ligand atom density in direct contact with the salt bridge pair and effectively being a “cap” over an electrostatic side chain conformational change required for radioligand [3H]CCPA dissociation.
While this manuscript was in review, to comprehensively investigate the differences between our docking modes and that of Kennedy et al,32 we performed additional targeted flexible docking for 3 and 4 into A1R in an attempt to recapitulate similar induced-fit low energy binding modes.32 While we were able to obtain a similar induced binding pocket in the ECL2 region using a flexible receptor approach, this binding mode was predicted to have less-favorable energy compared to flexible docking poses in the extracellular region that were similar to those obtained with a rigid docking methodology (Supplementary Figure 5.). Our best flexible receptor docking poses were on the order of 4.3 Å and 4.8 Å for 3 and 4 respectively from the rigid receptor results (Figure 7). For homology models, this is not bad in comparison to the best flexible receptor docking poses for 8 docking to A2AR modeled from the crystal structure of the A2AR selective agonist CGS21680 (4UG2.pdb).33 Presumably the closer agreement (3.6 Å) between the rigid and flexible receptor results for A2AR compared to A1R were that the complexes for A2AR were built from crystal structures in complex with the reference agonist, rather than from homology models. While this manuscript was in review, a paper by Nguyen et al.66 was published that investigated the effects of alanine-scanning mutagenesis and radioligand binding assays to determine their influence on allosteric ligand affinity, cooperativity, and efficacy of PAMs PD-81,723 and VCP171 on A1R.66 The authors hypothesized that the allosteric site is most likely located within a hydrogen bonding network within the extracellular vestibule.66 This binding site is analogous to the allosteric binding site observed in the activated M2R crystal structure in complex with iperoxo and PAM LY2119620. Our results from both flexible and rigid receptor docking demonstrate that A1R PAMs also bind in this extracellular vestibule utilizing important interactions with E172 and K173. In the new paper by Nguyen et al., residues E172 and K173 were experimentally verified to mediate PAM activity of PD-81,723 and VCP171 for A1R in agreement with our models.66 In addition, W156A was found to not affect allosteric ligand affinity, but did, probe-dependently, reduce cooperativity.66
A2AR
In a study of a chemical series of adenine and 8-azaadenine derivatives, A1R PAM compound 7 was selective for A1R, while 8 was identified as a PAM with improved selectivity for A2AR.47 Compound 8 was found to act as a PAM in A2AR in equilibrium displacement studies using both radioligand [3H]ZM241385 and [3H]CGS21680. Compound 8 was also shown to act as a PAM of agonist CGS21680 in functional assays. This is also an ideal model PAM for our molecular docking studies as a crystal structure of A2AR bound to it had recently been solved.35 Molecular docking results for this receptor highlight the importance of modeling PAM binding to the receptor in the presence of the experimentally relevant radioligand, as shown in Figure 3(B). In all of the lowest energy poses of compound 8, the aryl urea group forms favorable electrostatic interactions with the charged carboxylic acid of the reference agonist CGS21680 that protrudes from the agonist pocket out of the receptor surface Figure 5(B) and Figure 6(A). This binding mode may explain why the urea functional group was favorable for PAM activity in R-group modifications. The carbonyl group of the aryl urea also forms an electrostatic interaction with K153. In 8 a longer and bulkier group containing the urea linkage was found to enhance A2AR PAM activity in comparison to 7, where smaller para nitro and para amino group derivatives retained greater activity for A1R.47 It is not immediately apparent as to why the para amino N6 derivative is more selective for A1R. In the predicted binding mode, the [1,2,3]triazolo-pyrimidin-7-amine core of 8 forms several electrostatic interactions with ECL2 residues shown in Figure 5(A). The 7-amino group (aryl amine) of the core forms complementary electrostatic interactions with E169 where the dipole of the aryl amine group is nicely accommodated by close proximity to the residue pairs. The 5-phenyl substituent of 8 is contained by a shallow hydrophobic pocket on the surface formed by three residues (M174, F257, P260) from TM5 and TM6. Similar to our A1R binding mode for 3, the PAM [1,2,3]triazolo-pyrimidin-7-amine core of 8 was found to sit directly on the A2AR extracellular surface on top of the salt bridge of H264 (TM6) and E169 (ECL2). In the crystal structure of the agonist CGS21680, E169 also forms very strong electrostatic interactions with the 7-amino group of the adenosine agonist. Therefore, similar to our proposed PAM binding mode for A1R, for A2AR the dissociation of [3H]CGS21680 in the presence of 8 will require a side chain rearrangement at least involving the E169 side chain.
A3R
The first selective A3R PAMs were several 3-(2-pyridinyl)isoquinoline derivatives of the prototype PAM, VUF-5455, that slowed the dissociation of the agonist radioligand [125I]-AB-MECA.67,68 In a subsequent study of structural modifications of VUF-5455 and DU-124183, the compound 9 (LUF6000), exhibited the greatest PAM activity of the series using the agonist Cl-IB-MECA.48,69 A structurally similar quinoline derivative 10 (LUF6096) with similar cyclohexyl and 3,4-dichloral-phenyl R-group substitutions was also identified as the most potent PAM in a series of derivatives with minimal orthosteric site binding.70 Therefore, we selected 9 and 10 as our model system A3R PAMs to focus on, utilizing molecular docking to predict the allosteric binding site.
The observed consensus binding mode for 9 in complex with MRS-542 is shown in Figure 6(B). The binding interaction specificity appears to be strongly determined by burial of the hydrophobic surface and by hydrophobic shape complementarity of the rigid tricyclic ring and the 2-position cyclohexyl group. The cyclohexyl group binds to a large hydrophobic patch of the extracellular A3R surface defined by residues V169, I253, V256. In our model, the protonated imidazole nitrogen in the tricyclic ring, the electropositive NH hydrogen, is in the vicinity of E258 from top of TM6. Although not close enough to hydrogen bond, water-mediated hydrogen bonds may be possible. The hydrophobic surface formed by residues V169, M174, I253 and V256 in the A3R receptor is different compared to the A1R and A2AR structures. In our binding mode, these extensive residue differences may account for the selectivity of 9 for PAM activity at A3R. In this binding mode, the 4-amino aryl amine NH group of 9 has its dipole oriented towards the receptor hydroxyl groups in close proximity, provided by S170, which is closely interacting with E156. Interestingly, in the majority of low-energy poses from flexible docking, the 4-amino group is oriented towards the more hydrophilic residues of ECL2, and the third aromatic ring of the tricyclic structure is more oriented towards I253 (TM6). The 3,4-dichloral-phenyl group is oriented to form interactions involving the ECL2 and TM5 residues R173 and M174 Fig 7(B) and Supplemental Fig 3(B).
The salt bridge described in A1R (H264, E172) and A2AR (H264, E169) does not exist in the A3R structure, due to glutamate being replaced by valine. This is one of many sequence differences that result in a more hydrophobic extracellular receptor surface for A3R. As A2AR residue H264 becomes E258 in the A3R models, this E258 residue extends out into solvent rather than being buried in the exact location of the H264 residue in A2AR. This was the case for the E258 conformation in all of the models built by Modeller from all template pdb files (2YDO.pdb, 3QAK.pdb, 4UG2.pdb, 3EML.pdb)19,32–34. This can be rationalized in two ways. First, there is a minor change in backbone conformation. Secondly, in the A3R structure, the environment where the A2AR H264 imidazole side chain is buried is a very hydrophobic environment (V159, L164, I149, I253) in the A3R structure, and there are no complementary electrostatic or polar interactions for the E258 side chain carboxylic acid. In the study where 9 was identified as having increased PAM activity in both radioligand dissociation and functional assays (Cl-IB-MECA), SARs were determined at both the 4-amino and the 2-positions. A 3,4-dichloro-phenyl was found to be optimal at the 4-amino position and a cyclohexyl group was found to be optimal at the 2-position. SAR studies revealed that, while keeping the 2-position fixed as a cyclopentyl group, varying the 4-amino position favored the 3,4 dichloro-phenyl, particularly in comparison with fused indoles, indazoles, and more hydrophobic 2,3-dihydro-1H-indenes.69 In a similar way, when the optimal 3,4-dichloro-phenyl was fixed at the 4-amino position, the 2-position cyclohexyl was most favorable, and smaller groups (hydrogen) or an aromatic furan ring, were found to have the lowest PAM activity and were the least favorable. Our binding modes seem to be in reasonable agreement with these SAR observations. The cyclohexyl group appears more favorable on the flat hydrophobic surface (V169, I253, V256) than a furan or certainly a hydrogen would be. In our binding model, the 3,4-dichloro-phenyl group is participating in interactions with the R173 and M174 side chains. Although these interactions do not immediately rationalize all SAR substitutions, the model does provide evidence for specific interactions that may reasonably have an effect on SAR variants such as the fused ring substituents that were found to reduce activity compared to the 3,4-dichloro-phenyl groups.
We also performed docking of model A3R PAMs 9 and 10 into A3R models in complex with endogenous agonist adenosine only (rather than the bulkier agonists Cl-IB-MECA and MRS-542). Results in the absence of the bulkier m-iodobenzyl group show that both 9 and 10 may be able to bind slightly deeper into the orthosteric pocket and effectively form a “capping” interaction with the endogenous agonist. Our top-ranked poses for 9 and 10 both bind with the 3,4-dichloro-phenyl pointed “down” deeper in the agonist pocket, while the cyclohexyl group remained in contact with the hydrophobic patch (data not shown). Our top-ranked binding poses in complex with endogenous adenosine were similar to results from another recent molecular dynamics (MD) simulation study where 9 (LUF6000) was shown to act as a cap over the orthosteric site of A3R.33 In that study, LUF6000 transiently complexed to a meta-binding site located in the ECL2 region before descending deeper into the receptor. Similar results were seen with 10 (LUF6096).70 Predicted CHARMM-based LIE (GBMV) free energies suggest that these “capping” interaction binding modes should be more thermodynamically favorable even compared to the consensus binding modes of 9 and 10 in the presence of the bulkier agonists Cl-IB-MECA and MRS-542.
In cyclic AMP functional assays, for PAM 9 the maximum enhancement of the efficacy of the full agonist NECA was only 16%, where it was shown to be 50% for Cl-IB-MECA, over 200% for other partial agonists, and 300% of control for the efficacy of endogenous partial agonist inosine.71 To investigate the molecular basis for this observation, we also performed docking of 9 into A3R models in complex with inosine (rather than the bulkier agonist Cl-IB-MECA). Shown in Supplementary Figure 6, 9 was predicted to bind in a “capping” interaction binding mode with favorable hydrophobic interactions at the top of the agonist pocket, where the indole hydrogen of 9 forms a hydrogen bond with the carbonyl group of the agonist inosine. This specific and complementary interaction between the molecular structure of the agonist and the bound PAM demonstrates a reasonable model for the molecular basis of agonist probe-dependent PAM activity, similar to our findings for the A2AR selective PAM 8, where the aryl urea group forms favorable electrostatic interactions with the charged carboxylic acid of the reference agonist CGS21680 shown in Figure 5(B). The strong PAM activity of 9 for the endogenous partial agonist inosine in comparison to adenosine is relevant to the potential therapeutic use of 9, as the concentration of inosine and adenosine increase approximately 30 fold in the brain and in the heart under ischemic conditions.72 As inosine itself was found to reduce ischemic brain injury in rats through an A3R dependent mechanism,73 the increased PAM activity of 9 for inosine in comparison to adenosine should be very relevant to the in vivo mechanism of action of 9.
Muscarinic Receptors
The muscarinic acetylcholine receptors (M1-M5) have an important role in parasympathetic neurotransmission and in the regulation of several major physiological processes such as cardiac inotropy/chronotropy, smooth muscle contraction and glandular secretion.74 Traditional orthosteric antagonists are a major class of FDA approved drugs used for treating a variety of conditions, including COPD, urinary incontinence, urinary bladder spasms, and Parkinson’s disease.75,76 However, the vast majority of the traditional muscarinic agonist and antagonists lack sufficient selectivity for any one specific muscarinic receptor, often resulting in undesirable side effects in specific tissues.77 The recent discovery of subtype selective muscarinic receptor PAMs offers new possibilities for exploring the pharmacology and the potential therapeutic use of selective muscarinic receptor modulators.78
M1R
As investigational drugs, M1R selective agonists and PAMs have been investigated as potential cognition-enhancing agents for improving the symptoms of Alzheimer’s disease.79 M1R knockout mice show cognitive deficits,80,81 and some compounds that activate M1R have shown improvements in cognitive effects in preclinical work.82–84 However, these results were all limited due to non-selective side effects. Even the current standard of care for Alzheimer’s disease, acetylcholinesterase inhibitors, result in limiting gastrointestinal side effects from non-selective activation of nicotinic and muscarinic acetylcholine receptors. Several recent studies of the Merck M1R PAM candidate 12 (PQCA) in rodent and primate cognition animal models,85 have shown promising improvements in cognition with the absence of gastrointestinal side effects.86,87 On the other hand, recent preclinical studies by Pfizer on their M1R PAM preclinical candidate PF-06767832, which exhibits optimal physicochemical properties as a CNS drug, have shown that selective M1R activation may still result in adverse cholinergic gastrointestinal and cardiovascular side effects.8 More clinical research in this area is needed to ultimately determine if M1R selective PAMs will be a successful therapeutic strategy. Recent work has also established preliminary proof-of-concept that an M1R or dual M1R / M4R selective agonist,88,89 or PAM may be effective in the treatment of drug addiction,90,91 or schizophrenia with a “dopamine hypersensitivity phenotype”.92–94 Several classes of M1R PAMs have been characterized that are structurally distinct from LY2119620. Figure 8. shows all the muscarinic receptor PAMs and NAMs used in this study. Our results in this study show that the diverse chemotypes of M1R PAMs bind in very reasonable isosteric modes compared to the crystal structure of M2R PAM LY2119620,16 exhibiting partial overlap of some specific functional group pharmacophore features, suggesting a common mechanism of PAM binding to stabilize the active conformation of the receptor. Interestingly, while there was reasonable similarity in our results for rigid receptor docking and flexible receptor docking for the adenosine receptor PAMs, the flexible receptor results for the M1R receptor were in greater consensus agreement with experimental data from mutation studies and SAR studies. Thus, for clarity only flexible receptor results will be presented for M1R and M5R. In retrospect, the comparison of rigid docking to flexible receptor docking showed that minor deviations in backbone conformation were required to adequately identify consensus low-energy poses that are in superior agreement with experimental observations to date.
Figure 8.

Muscarinic receptor PAMs and NAMs used in this study.
11 (BQCA) is a model M1R PAM that has been the subject of numerous studies and investigations.95 We investigated a potent derivative, 12 (PQCA).95 The top-ranked binding mode for 12 predicted from flexible receptor docking is shown in Figure 9(A). The carboxylic acid group interacts with lysine K392 (where the PAM is under the hydrophobic part of the K392 side chain) and the lysine is forming another salt-bridge-like electrostatic interaction with residue Y179 (ECL2). The carboxylic acid is interacting with both Y179 and K392, which form a salt bridge above the ligand, which is presumably important in stabilizing the active conformational state of the receptor. The binding mode of 12 is also compared to the reference M2R PAM LY2119620 in Figure 9(A). The carboxylic acid group of 12 forms an important protein-ligand interaction, where the orientation of the carboxylic acid is analogous to the ether of LY2119620 bound to M2R, where the ether oxygen atom of LY2119620 is 3.3 Å from the amide nitrogen atom (ND2) of residue N410 of M2R.16 In M1R, the equivalent residue of N410 is S388 where the shorter serine side chain is unable to interact with the PAM carboxylic acid of 12. Instead, a very important interaction is formed to the primary amine of the K392, which interacts with Y179 as mentioned above.
Figure 9. Flexible receptor predicted binding modes for M1R PAMs and comparison to LY2119620.

(A) Crystallographic binding geometry of M2R PAM LY2119620 and comparison to the binding mode of 12 (PQCA) bound to M1R shown from a top view of the receptor surface showing specific interactions. Binding geometry of 17 (B), 18 (C), and 19 (D), respectively.
Very detailed mutational studies have been performed to map the M1R binding site of 11.96,97 Our predicted binding modes for 12, a potent derivative, are in agreement with data for residues that perturb 11 receptor affinity and PAM activity. Residues Y179 and W400 form the most important contacts with all of our predicted binding modes for 12, in agreement with the observation that they had the greatest effect in abrogating binding and PAM activity of 11. 12 also forms close contact with Y106, Y157, Y381, which are other residues where mutation was found to diminish PAM activity.96,97 The predicted bound conformation of 12 also has minimal contact with residues E397 or E401, and forms no strong or complimentary protein-ligand interactions with these residues. This is also in agreement with the observation that mutation of these residues does not affect binding of 11. 96,97
Our predicted modes for 12 are also in agreement with the experimental observation that mutation of Y82 and Y85 do not affect 11 binding, but result in a 10-fold loss of affinity for 17.85 Isosteric changes to the carboxylic acid group of 11 and 12 have shown reductions in PAM activity and binding.98 We conclude that our binding mode is also in agreement with these observations from the SAR.98 In the binding mode for 12, the carboxylic acid forms favorable electrostatic interactions with Y179 and K392, which form a salt bridge above the PAM. These side chains are requisite for ligand dissociation and by interacting with these side chains, we provide a mechanism to explain slower radioligand dissociation kinetics. The crystal structure of LY2119620 bound to the iperoxo M2R complex itself provides strong structural evidence for such a molecular interpretation. However, modeling of these PAM chemotypes to various receptors has broadened this observation to several specific PAM-receptor interactions.
As shown in Figure 9(C), the binding mode of 14 shows similar hydrophobic interactions as 17, with the methyl-hydroxybenzimidazole in a deep hydrophobic region bound by L102 and W101. Additionally, the morpholine group forms close hydrophobic interactions with the Y85 and L86 side chains, interactions that are also seen with the cyclohexanol groups of 17, 18, and 19.99,100 This site, featuring Y85 and L86, was frequently seen as a binding site for the hydrophobic moieties on allosteric modulators in this study. The carbonyl group of 14 forms a hydrogen bond with the backbone peptide bond amide of E397, which also brings the side chain of E397 in closer proximity to the side chain of Q177.
17 was found to exhibit a 50-fold improvement in affinity for the same allosteric site as 11 while retaining a similar level of positive cooperativity with acetylcholine.96 Our binding mode for 17 was found to be in good agreement with LY2119620, as shown in Figure 9(B). The pyrazole on 17 was shown to form a ring stack with Y179 without breaking Y179’s electrostatic interaction with K392. The docking also showed the ligand’s cyclohexanol group forming a hydrogen bond to E397. Additionally, the residues I180, L102, W101, L86, and the hydrophobic tail of E401 formed a hydrophobic pocket that interacted mainly with the tricyclic ring and cyclohexyl group of 17, similar to the binding of the methyl-hydroxybenzimidazole of 14. The methyl-pyrazole-pyridine moiety of 17 is underneath, and interacting with a hydrogen bond network formed by Y179, K392, and S184. These observations and the docking pose for 17 seem to be in good agreement with another docking and mutational study.96
Figure 10(A,B) shows two predicted binding modes for 13. The binding pose shown in Figure 10(A), which was found to be the lowest energy conformation is similar to that of a docking poses reported from the group who identified the molecule.95 This binding mode for 13 is also similar to our prediction for compound 14, with the carbonyls of 13 in the same region as that of 14. Like 17, there is ring-stacking interactions with W400. The methyl-phenyl-pyrazole region wraps around the hydrophobic portion of E397, into the hydrophobic site formed by L86 and Y85, as was seen with 14. The second binding mode, which was only slightly lower in energy, shown in Figure 10(B), shows a “flipped” conformation, with the carbonyl of the isatin-like group interacting with E397 and Y85. The benzene region of this group is interacting with what the pyrazole region was formerly interacting with: the L86, Y85 hydrophobic site. The pyrazole region of this second binding mode goes deep into the hydrophobic binding region formed by L102, I180, and W101.
Figure 10. Flexible receptor predicted binding modes for M1R PAMs and comparison to LY2119620.

(A) Crystallographic binding geometry of M2R PAM LY2119620. and comparison to the top-ranked binding mode of 13 (ML-137) bound to M1R shown from a top view of the receptor surface showing specific interactions. (B) Binding geometry of the 2nd ranked binding mode of 13, that is quite similar in energy to (A). Binding geometry of 14 (GSK-1804165) (C) and 15 (D), respectively.
Compound 15 is an optimized derivative of truncations of 16, aimed at showing what substituents were more important for activity.95 The binding mode for 15, is very similar to that of the compound 13 second binding pose, as shown in Figure 10(B). The carbonyl of 13 hydrogen bonds with T398, and with the pyrazole region going into the deep hydrophobic region mentioned earlier. The fluoro-benzene forms extensive pi-pi interactions with W400, Y85, and Y179. The predicted binding mode of 18 shown in Figure 9(C) shares similarities with the earlier compounds. The methyl-phenyl-pyrazole portion binds similarly to that of 17, forming ring stacking interactions with W400, and Y179, underneath a salt bridge formed by K392 and Y179. The ether group of these compounds is bound deep in the hydrophobic pocket formed by L102, I180, and W101. It also forms additional pi-pi interactions with Y85. The cyclohexanol of 18 has hydrophobic interactions with L86, as well as a hydrogen bond with the carbonyl backbone of G89. The binding mode of 19, shown in Figure 9(B), forms very similar interactions as 18, with some minor, but significant differences. Like 18, the methyl-phenyl-pyrazole region binds underneath a salt bridge (in this case formed by K392 and Y179) with ring stacking interactions with W400 and Y179.99,100 The oxygen of the diphenyl ether forms an electrostatic interaction with Q177, breaking a salt bridge that is seen with E397, possibly contributing to the “capping” function of PAM 19. E397 is instead interacting with the cyclohexanol, forming a hydrogen bond. The superpositions of these diverse chemotypes and their literature-associated SAR, suggests that the most important pharmacophores for potent PAM activity is the (1) R-group of hydrophobic fused rings (interactions with Y82, W101, L102, I180, L86), (2) R-groups with electrostatic interactions with K392 and Y179 salt-bridge residues, as well as those that may affect the salt bridge of Q177 and E397, and (3) ring stacking with aromatic residues along the extracellular vestibule, such as W400 and Y179.
M5R
M5R knockout mice models suggest that selective M5R antagonists or selective NAMs have reasonable proof-of-concept potential for treating addiction.101 For the M5R we compared the docking the M5R selective NAM 20 (ML-375) and the M5R selective PAM 21 (ML-380). PAM 21 bound to the M5R in a very similar mode, shown in Figure 11(B) to the other M1R PAMs initially shown in Figure 9 and 10. In superposition of the binding modes of 21 and 17 shown in Figure 11(E), similarities are seen in the location of the tricyclic groups of 21 and 17. There is also similar ligand density in the location of the di-fluorobenzene and the cyclohexanol group.
Figure 11. Flexible receptor M5R NAM unique binding mode.

Predicted lowest energy binding modes for an M5R selective NAM 20 (ML-375) (A) and PAM 21 (ML-380) (B) binding to M5R. (C) Superposition of (A) and (B) from a side view of the M5R surface shows how the p-chloro group of NAM forms unique interactions with the receptor compared to the PAM mode. (D) Only the superposition of the NAM and PAM is shown to highlight how there is minimal overlap in ligand density for the NAM (p-chloro group) with the superimposed bound PAM. (E) NAM binding mode compared with a superposition of 17 as a representative of the tricyclic M1R PAM (F) Specific residues of M5R that participate in protein-ligand interactions with the NAM, where induced-fit side chain rearrangements of Y87 and H478 are key to the interactions with the NAM (p-chloro group).
The trifluoromethyl-phenyl group of 21 binds in the same mode as that of the methyl-hydroxybenzimidazole. The sulfonamide forms strong electrostatic interactions with R95 and K470 which can should contribute to the stability of the PAM-bound complex. While PAM 21 was predicted to bind to M5R in a mode that is very similar and analogous to the other M1R PAMs chemotypes, NAM 20 was found to bind in different mode that is not exactly isosteric with other M1R PAMs or 21. These binding modes are shown in Figure 11 (A,C,F), illustrating how 20 binds in the same common allosteric pocket, but displays a unique mode of binding that may account for NAM-specific activity.
The p-chloro-benzene moiety of 20, uniquely, has no overlap with 21, suggesting that the activity of this moiety may contribute specifically to NAM activity. The p-chloro-benzene participates in ring stacking interactions with Y87 and Y90. The p-chloro-benzene disrupts a Y87 mediated hydrogen bond that is normally found in all M1 and M5 PAM-bound structures observed. In both the crystal structure of M3R bound to the antagonist tiotropium and in molecular dynamics simulations by the authors the conformation of (F124 in M3, Y80 in M2, and Y87 in our M5 models) was found to be very important as it directly interacts with TM7 and influences TM7 conformation and residue Y529 M3R of the Tyrosine lid.28 Interestingly, residues in the Y87 position have been implicated in side chain mediated conformational changes linked to the functional activation and selective ligand binding to M3R F124A,28,102 and M1R Y82A mutant.103,104 We speculate that the specific induced-fit conformational changes involving the equivalent M5R residues Y87 (M1R Y82) and Y481 (M1R Y404) residues that form direct contacts with the bound NAM p-chloro-benzene functional group, may provide a structural explanation as to how M5R selective NAM is able to preferentially stabilize inactivated conformational states compared to PAMs.
Gentry et al. determined that the (S)-enantiomer of this 20 functions as a NAM, but the (R)-enantiomer is inactive, suggesting that this site is critical to NAM activity.105 This study determined that the SAR is quite shallow, and that there was a cooperative relationship between benzamide and phenyl substituents. However, the chlorine substituent was universally more active than the other replacements (methyl, fluoro). The unique induced-fit conformational change in our models involving M5R residue Y87 that directly contacts the NAM p-chloro-benzene, may explain NAM activity. A very recent experimental study suggests that the PAM 21 binding site does overlap with the NAM 20 site in M5R, as the NAM 20 was found to compete with the PAM activity of 21.106 Both of our proposed binding modes are consistent with this experimental observation,106 and illustrate how a NAM and PAM may bind in a way that they share two of 3-point pharmacophore interactions within the same overlapping binding site, while the difference in the binding interaction of the NAM (p-chlorobenzene) may be responsible for NAM activity.
Other recent computational investigations using MD simulations have simulated binding of PAMs and NAMs to M2R and hypothesized that two mechanisms contributed to allosteric modulator effect.27 The first was electrostatic repulsion between ligand and modulator, with repulsion leading to negative modulation. The second mechanism found was a coupling of the two sites to influence each other’s shape, with PAMs keeping the receptor in a state more amenable to orthosteric binding of the affected ligand. 27 As NAM 20 does not exhibit an overall positive charge (both nitrogens are neutral tertiary amides) the proposed electrostatic repulsion mechanism does not seem to be applicable to NAM 20. Our predicted NAM 20 binding mode utilizes binding interactions that are distinct from the binding of PAM 21. It is reasonable to expect that these different NAM/PAM binding modes should stabilize different conformational states of the receptor, as the extracellular surface of the receptor may partially collapse in response to bound small-molecule NAMs and PAMs, resulting in the corresponding modulation of receptor activity.
Conclusions
The results of our study demonstrate that there were several major similarities between the results of rigid and flexible receptor docking. There was a greater similarity in results for the adenosine receptors, and this may be due to the majority of the PAM ligand density still located on the extracellular surface. In the comparison for the M1R, it was clear that the flexible receptor results were in better agreement with experimental observations. We suspect that the reason for this difference is likely due to M1R PAMs binding deeper into the extracellular surface, which requires minor backbone conformational changes. In addition, interesting conclusions may also be drawn from comparing the predicted 3D conformations of tricyclic muscarinic receptor PAMs and tricyclic adenosine receptor PAMs. In results for docking of 9 (LUF6000) to endogenous adenosine-bound A3R and for endogenous inosine, the tricyclic core does not always bind on top of the surface as it does in complex with MRS-542 with the bulky m-iodobenzyl R-group, summarized in Supplementary Figure 5. These proposed “capping” interaction binding modes for 9 and 10 in the presence of endogenous agonists are more related in 3D conformational space to the bound conformation of M1R PAM tricyclics. Figure 12 shows 17 (Benzoquinazolinone 12) and 16 (DBPQ) compared to A3R tricyclic 9 (LUF6000) and quinazoline 10 (LUF6096). This 2D/3D structural comparison may illuminate previously unrecognized and unappreciated 2D structural similarities between these related PAM chemotypes for the adenosine and muscarinic receptors. These types of structural insights into PAM binding are informative for the design of enriched libraries of small-molecules that may exhibit PAM activity at similar receptors.
Figure 12. Similar M1R and A3R tricyclic chemotypes.

Similar 3-point pharmacophore groups are mapped to the structurally related tricyclic M1R PAMs (A) 17 (benzoquinazolinone 12) and (B) 16 (DBPQ) from (C) the superposition of the two bound to M1R. Similar 3-point pharmacophore groups are mapped onto A3R PAMs (D) 9 (LUF6000) and (E) 10 (LUF6096). M1R PAM 17 is shown in (F) bound to M1R illustrating the superposition with 9 binding to A3R in a “capping” interaction binding mode in complex with endogenous adenosine or inosine.
In this study we extend recent structural insights from the recently solved crystal structure of M2R in complex with a bound PAM. Results over a series of well-characterized model system PAMs for adenosine receptors (A1R, A2AR, A3R) and muscarinic receptors (M1R, M5R) provide a reasonably clear molecular picture of how small-molecules PAMs bind to and stabilize agonist-bound conformational states. Our docking results for several structurally related chemotypes such as 3 (PD-117,975) and 9 (LUF6000) binding to different adenosine receptor subtypes showed remarkable similarities in the lowest free energy binding region identified, as well as the specific protein-ligand interactions of related pharmacophore groups. Using both a rigid receptor model and flexible receptor model to compare related series, there is a clear consensus in what is the most reasonable low-energy location for PAM binding, which is utilizing residues from both ECL2 and ECL3. A comparison of the rigid receptor and flexible receptor results are shown in Figure 7. All of the adenosine receptor PAMs were predicted to have important electrostatic polar interactions with residues in ECL2, while having favorable hydrophobic contacts with residues from ECL3. This was not necessarily expected prior to docking the PAMs, as there are possibilities for hydrophobic and hydrophilic interactions with both ECL2 and ECL3. Thermodynamically favorable “capping” interactions such as those proposed for 9 bound to A3R in complex with inosine may rationalize large probe-dependent increases in PAM activity for specific agonists (300% of control for the efficacy of inosine).71
A weakness of the methodology employed by our study is that we were unable to adequately evaluate the possibility of binding modes in other regions of the GPCR, particularly as the TM1/TM7 domain. Both rigid and flexible receptor approaches utilized within this study suggest that the PAM binding modes we present that interact between ECL2 and ECL3 are more favorable than modes between TM1/TM7. A more rigorous exploration of ligand binding at this site should be explored in future studies. Although the vast majority of experimentally identified PAMs discovered to date have been shown to bind to the extracellular vestibule of the adenosine and muscarinic receptors, this does not exclude the possibility that other PAMs discovered in the future may bind to other regions of a GPCR.
The proposed binding modes provide a structural rationalization of how PAMs for each receptor may slow specific agonist radioligand dissociation kinetics. PAM ligand binding may form direct molecular interactions with the agonists as predicted for some specific adenosine receptor PAMs. On the other hand, a common mechanism to both the adenosine and muscarinic receptors is through PAM binding with direct molecular interaction with specific residue side chains that are involved in necessary side-chain rearrangements required for agonist ligand dissociation. Namely, (1) the adenosine receptors (A1R, A2AR) PAMs were predicted to interact directly with the specific salt bridge residue pairs that have been implicated in ligand dissociation kinetics,63,64 and (2) muscarinic receptors PAMs may form direct molecular interaction with analogous salt bridge residues, such as 12 shown in Figure 9(A), where the carboxylic acid group interacts with lysine K392, forming a salt-bridge electrostatic interaction with residue Y179 (ECL2). Induced fit docking results also show that compounds 13, 14 and 19 may also bind and induce new analogous side chain interactions mediated by Q177 (ECL2) that would stabilize the receptor. In total, these proposed binding modes provide a structural basis for how PAMs may slow agonist radioligand dissociation kinetics. These slowed kinetics may underpin the augmentative activity PAMs appear to provide in concert with orthosteric agonists. This structural rationalization provides important insights that may provide a venue for further research and development into drug discovery and design of future novel, subtype specific PAMs for clinical applications.
Supplementary Material
Acknowledgments
We are thankful for Dr. Jeffery L. Benovic for reading the manuscript and offering suggestions. LAS, KZR and RSA designed research. LAS, KZR, RSA performed molecular docking. LAS, KZR, and RSA analyzed data and wrote the manuscript. RSA initiated and directed the entire project. This research was supported by NIH funding P01-HL114471 (to JLB and RSA).
Footnotes
Additional Supporting Information in the form of Supplementary Figures 1–6 and Supplementary Tables 1 and 2 may be found in the online version of this article. These are currently placed at the end of the manuscript after the Graphical Abstract Figure.
References and Notes
- 1.Rask-Andersen M, Almén MS, Schiöth HB. Nat Rev Drug Discovery. 2011 doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- 2.Schiöth HB, Fredriksson R. Gen Comp Endocrinol. 2005;1–2:94–101. doi: 10.1016/j.ygcen.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 3.Kenakin T, Christopoulos A. Nat Rev Drug Discov. 2013;3:205–16. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- 4.van der Westhuizen ET, Valant C, Sexton PM, Christopoulos A. J Pharmacol Exp Ther. 2015;2:246–60. doi: 10.1124/jpet.114.221606. [DOI] [PubMed] [Google Scholar]
- 5.May LT, Leach K, Sexton PM, Christopoulos A. Annu Rev Pharmacol Toxicol. 2007:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- 6.Mohr K, Schmitz J, Schrage R, Tränkle C, Holzgrabe U. Angew Chem Int Ed Engl. 2013;2:508–16. doi: 10.1002/anie.201205315. [DOI] [PubMed] [Google Scholar]
- 7.Tison F, Keywood C, Wakefield M, Durif F, Corvol J-CC, Eggert K, Lew M, Isaacson S, Bezard E, Poli S-MM, Goetz CG, Trenkwalder C, Rascol O. Mov Disord. 2016 doi: 10.1002/mds.26659. [DOI] [PubMed] [Google Scholar]
- 8.Tison F, Keywood C, Wakefield M, Durif F, Corvol J-CC, Eggert K, Lew M, Isaacson S, Bezard E, Poli S-MM, Goetz CG, Trenkwalder C, Rascol O. Mov Disord. 2016 doi: 10.1002/mds.26659. [DOI] [PubMed] [Google Scholar]
- 9.Yanamala N, Klein-Seetharaman J. Pharmaceuticals (Basel) 2010;10:3324–3342. doi: 10.3390/ph3103324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lazareno S, Popham A, Birdsall NJ. Mol Pharmacol. 2002;6:1492–505. doi: 10.1124/mol.62.6.1492. [DOI] [PubMed] [Google Scholar]
- 11.Carr R, Du Y, Quoyer J, Panettieri RA, Janz JM, Bouvier M, Kobilka BK, Benovic JL. J Biol Chem. 2014;52:35668–84. doi: 10.1074/jbc.M114.618819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carr R, Benovic JL. Biochem Soc Trans. 2016;2:555–61. doi: 10.1042/BST20150230. [DOI] [PubMed] [Google Scholar]
- 13.Kahsai AW, Wisler JW, Lee J, Ahn S, Cahill TJ, Iii, Dennison SM, Staus DP, Thomsen AR, Anasti KM, Pani B, Wingler LM, Desai H, Bompiani KM, Strachan RT, Qin X, Alam SM, Sullenger BA, Lefkowitz RJ. Nat Chem Biol. 2016 doi: 10.1038/nchembio.2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silvano E, Millan MJ, Mannoury la Cour C, Han Y, Duan L, Griffin SA, Luedtke RR, Aloisi G, Rossi M, Zazzeroni F, Javitch JA, Maggio R. Mol Pharmacol. 2010;5:925–34. doi: 10.1124/mol.110.065755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao ZGG, Kim SKK, Gross AS, Chen A, Blaustein JB, Jacobson KA. Mol Pharmacol. 2003;5:1021–31. doi: 10.1124/mol.63.5.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hübner H, Pardon E, Valant C, Sexton PM, Christopoulos A, Felder CC, Gmeiner P, Steyaert J, Weis WI, Garcia KC, Wess J, Kobilka BK. Nature. 2013;7478:101–6. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lane JR, Sexton PM, Christopoulos A. Trends Pharmacol Sci. 2013;1:59–66. doi: 10.1016/j.tips.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 18.Gentry PR, Sexton PM, Christopoulos A. J Biol Chem. 2015;32:19478–88. doi: 10.1074/jbc.R115.662759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, Tate CG. Nature. 2011;7352:521–5. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, Lerch MT, Kobilka TS, Thian FS, Hubbell WL, Prosser RS, Kobilka BK. Cell. 2015;5:1101–11. doi: 10.1016/j.cell.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kahsai AW, Xiao K, Rajagopal S, Ahn S, Shukla AK, Sun J, Oas TG, Lefkowitz RJ. Nat Chem Biol. 2011;10:692–700. doi: 10.1038/nchembio.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu JJ, Horst R, Katritch V, Stevens RC, Wüthrich K. Science. 2012;6072:1106–10. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rahmeh R, Damian M, Cottet M, Orcel H, Mendre C, Durroux T, Sharma KS, Durand G, Pucci B, Trinquet E, Zwier JM, Deupi X, Bron P, Banères J-LL, Mouillac B, Granier S. Proc Natl Acad Sci USA. 2012;17:6733–8. doi: 10.1073/pnas.1201093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sounier R, Mas C, Steyaert J, Laeremans T, Manglik A, Huang W, Kobilka BK, Déméné H, Granier S. Nature. 2015;7562:375–378. doi: 10.1038/nature14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miao Y, McCammon JA. Curr Opin Struct Biol. 2016:83–89. doi: 10.1016/j.sbi.2016.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miao Y, McCammon JA. Proc Natl Acad Sci USA. 2016;43:12162–12167. doi: 10.1073/pnas.1614538113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC, Arlow DH, Canals M, Lane JR, Rahmani R, Baell JB, Sexton PM, Christopoulos A, Shaw DE. Nature. 2013;503:295–9. doi: 10.1038/nature12595. [DOI] [PubMed] [Google Scholar]
- 28.Kruse AC, Hu J, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E, Green HF, Liu T, Chae PS, Dror RO, Shaw DE, Weis WI, Wess J, Kobilka BK. Nature. 2012;282:552–556. doi: 10.1038/nature10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barnes PJ. Chest. 2000;117:63S–66S. [Google Scholar]
- 30.Göblyös A, Ijzerman AP. Biochim Biophys Acta. 2011;5:1309–18. doi: 10.1016/j.bbamem.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 31.Peeters MC, Wisse LE, Dinaj A, Vroling B, Vriend G, Ijzerman AP. Biochem Pharmacol. 2012;84:76–87. doi: 10.1016/j.bcp.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 32.Kennedy DP, McRobb FM, Leonhardt SA, Purdy M, Figler H, Marshall MA, Chordia M, Figler R, Linden J, Abagyan R, Yeager M. Mol Pharmacol. 2014;2:301–9. doi: 10.1124/mol.113.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deganutti G, Cuzzolin A, Ciancetta A, Moro S. Bioorg Med Chem. 2015;14:4065–71. doi: 10.1016/j.bmc.2015.03.039. [DOI] [PubMed] [Google Scholar]
- 34.Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZGG, Cherezov V, Stevens RC. Science. 2011;6027:322–7. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lebon G, Edwards PC, Leslie AG, Tate CG. Mol Pharmacol. 2015;6:907–15. doi: 10.1124/mol.114.097360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaakola VPP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. Science. 2008;5905:1211–7. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sali A, Blundell TL. J Mol Biol. 1993;3:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 38.Martí-Renom MA, Stuart AC, Fiser A, Sánchez R, Melo F, Sali A. Annu Rev Biophys Biomol Struct. 2000:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 39.Lee MS, Feig M, Salsbury FR, Jr, Brooks CL., 3rd J Comp Chem. 2003;24:1348–56. doi: 10.1002/jcc.10272. [DOI] [PubMed] [Google Scholar]
- 40.Chen J, Im W, Brooks CL. J Am Chem Soc. 2006;11:3728–36. doi: 10.1021/ja057216r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feig M, Im W, Brooks CL. J Chem Phys. 2004;2:903–11. doi: 10.1063/1.1631258. [DOI] [PubMed] [Google Scholar]
- 42.Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, Jo S, Pande VS, Case DA, Brooks CL, MacKerell AD, Klauda JB, Im W. J Chem Theory Comput. 2016;1:405–13. doi: 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Armen RS, Chen J, Brooks CL. J Chem Theory Comput. 2009;10:2909–2923. doi: 10.1021/ct900262t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rahaman O, Estrada TP, Doren DJ, Taufer M, Brooks CL, Armen RS. J Chem Inf Model. 2011;9:2047–65. doi: 10.1021/ci1003009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Narlawar R, Lane JR, Doddareddy M, Lin J, Brussee J, Ijzerman AP. J Med Chem. 2010;8:3028–37. doi: 10.1021/jm901252a. [DOI] [PubMed] [Google Scholar]
- 46.http://www.chemaxon.com
- 47.Giorgi I, Biagi G, Bianucci AM, Borghini A, Livi O, Leonardi M, Pietra D, Calderone V, Martelli A. Eur J Med Chem. 2008;8:1639–47. doi: 10.1016/j.ejmech.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 48.Gao Z-GG, Ye K, Göblyös A, Ijzerman AP, Jacobson KA. BMC Pharmacol. 2008:20. doi: 10.1186/1471-2210-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jacobson KA, Gao ZGG. Nat Rev Drug Discov. 2006;3:247–64. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caruso M, Alamo A, Crisafulli E, Raciti C, Fisichella A, Polosa R. Expert Opin Ther Targets. 2013;7:761–72. doi: 10.1517/14728222.2013.795220. [DOI] [PubMed] [Google Scholar]
- 51.Mizumura T, Auchampach JA, Linden J, Bruns RF, Gross GJ. Circ Res. 1996;79:415–23. doi: 10.1161/01.res.79.3.415. [DOI] [PubMed] [Google Scholar]
- 52.Mizumura T, Auchampach JA, Linden J, Bruns RF, Gross GJ. Circ Res. 1996;79:415–23. doi: 10.1161/01.res.79.3.415. [DOI] [PubMed] [Google Scholar]
- 53.Daval JL, Von Lubitz DK, Deckert J, Redmond DJ, Marangos PJ. Brain Res. 1989;2:212–26. doi: 10.1016/0006-8993(89)90058-9. [DOI] [PubMed] [Google Scholar]
- 54.Li X, Conklin D, Ma W, Zhu X, Eisenach JC. Pain. 2002;1–2:117–25. doi: 10.1016/s0304-3959(02)00011-8. [DOI] [PubMed] [Google Scholar]
- 55.Li X, Conklin D, Pan HLL, Eisenach JC. J Pharmacol Exp Ther. 2003;3:950–5. doi: 10.1124/jpet.102.047951. [DOI] [PubMed] [Google Scholar]
- 56.Welihinda AA, Emento EP. J Inflamm (Lond) 2014;11:37. doi: 10.1186/s12950-014-0037-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao ZG, Kim SG, Soltysiak KA, Melman N, IJzerman AP, Jacobson KA. Mol Pharmacol. 2002;62:81–9. doi: 10.1124/mol.62.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bruns RF, Fergus JH, Coughenour LL, Courtland GG, Pugsley TA, Dodd JH, Tinney FJ. Mol Pharmacol. 1990;38:950–8. [PubMed] [Google Scholar]
- 59.Bruns RF, Fergus JH. Mol Pharmacol. 1990;38:939–49. [PubMed] [Google Scholar]
- 60.Goblyos A, Ijzerman AP. Purinergic Signal. 2009;5:51–61. doi: 10.1007/s11302-008-9105-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, Kobilka BK. Nature. 2013;502:575–9. doi: 10.1038/nature12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lane JR, Beukers MW, Mulder-Kreiger T, Ijzerman AP. Biochem Pharmacol. 2010;79:48–56. doi: 10.1016/j.bcp.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 63.Romagnoli R, Baraldi PG, Carrion MD, Cara CL, Cruz-Lopez O, Iaconinoto MA, Preti D, Shryock JC, Moorman AR, Vincenzi F, Varani K, Andrea Borea P. J Med Chem. 2008;51:5875–9. doi: 10.1021/jm800586p. [DOI] [PubMed] [Google Scholar]
- 64.Segala E, Guo D, Cheng RK, Bortolato A, Deflorian F, Sore AS, Errery JC, Heitman LH, Ijzerman AP, Marshall FH, Cooke RM. J Med Chem. 2016;59:6470–9. doi: 10.1021/acs.jmedchem.6b00653. [DOI] [PubMed] [Google Scholar]
- 65.Bokoch MP, Zou Y, Rasmussen SG, Liu CW, Nygaard R, Rosenbaum DM, Fung JJ, Choi HJ, Thian FS, Kobilka TS, Puglisi JD, Weiss WI, Pardo L, Prosser RS, Mueller L, Kobilka BK. Nature. 2010;463:108–12. doi: 10.1038/nature08650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nguyen A, Vecchio E, Thomas T, Nguyen T, Aurelio L, Scammells P, White P, Sexton P, Gregory K, May L, Christopoulos A. Mol Pharmacol. 2016 doi: 10.1124/mol.116.105015. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 67.Gao ZG, Van Muijlwijk-Koezen JE, Chen A, Muller CE, Ijzerman AP, Jacobson KA. Mol Pharmacol. 2001;60:1057–63. [PMC free article] [PubMed] [Google Scholar]
- 68.Van Mijlwijk-Koezen JE, Timmerman H, Link R, van der Goot H, Ijzerman AP. J Med Chem. 1998;41:3987–93. doi: 10.1021/jm980036q. [DOI] [PubMed] [Google Scholar]
- 69.Goblyos A, Gao ZG, Brussee J, Connestari R, Santiago SN, Ye K, Ijzerman AP, Jacobson KA. J Med Chem. 2006;49:3354–61. doi: 10.1021/jm060086s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heitman LH, Goblyos A, Zweemer AM, Bakker R, Mulder-Krieger T, van Veldhoven JP, de Vries H, Brussee J, Ijzerman AP. J Med Chem. 2009;52:926–31. doi: 10.1021/jm8014052. [DOI] [PubMed] [Google Scholar]
- 71.Gao ZG, Verzijl D, Zweemer A, Ye K, Goblyos A, Ijzerman AP, Jacobson KA. Biochem Pharmacol. 2011;82:658–68. doi: 10.1016/j.bcp.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Backstrom T, Goiny M, Lockowandt U, Liska J, Franco-Cereceda A. J Appl Physiol. 2003;94:1122–8. doi: 10.1152/japplphysiol.00138.2002. [DOI] [PubMed] [Google Scholar]
- 73.Shen H, Chen GJ, Harvey BK, Bickford PC, Wang Y. Stroke. 2005;36:654–9. doi: 10.1161/01.STR.0000155747.15679.04. [DOI] [PubMed] [Google Scholar]
- 74.Greig NH, Reale M, Tata AM. Recent Pat CNS Drug Discov. 2013;8:123–41. doi: 10.2174/1574889811308020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Karmarkar R, Khullar V. Expert Opin Emerg Drugs. 2015;20:613–24. doi: 10.1517/14728214.2015.1086995. [DOI] [PubMed] [Google Scholar]
- 76.Roth M. Drugs. 2015;75:1–8. doi: 10.1007/s40265-014-0319-0. [DOI] [PubMed] [Google Scholar]
- 77.Matera C, Tata AM. Recent Pat CNS Drug Discov. 2014;9:85–100. doi: 10.2174/1574889809666141120131238. [DOI] [PubMed] [Google Scholar]
- 78.Kruse AC, Kobilka BK, Gautam D, Sexton PM, Christopoulos A, Wess J. Nat Rev Drug Discov. 2014;13:549–60. doi: 10.1038/nrd4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Melancon BJ, Tarr JC, Panarese JD, Wood MR, Lindsley CW. Drug Discov Today. 2013;18:1185–24. doi: 10.1016/j.drudis.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Medeiros R, Kitazawa M, Caccamo A, Baglietto-Vargas D, Estrada-Hernandez T, Cribbs DH, Fisher A, LaFeria FM. Am J Pathol. 2011;179:980–91. doi: 10.1016/j.ajpath.2011.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM, Silva AJ. Nat Neurosci. 2003;6:51–8. doi: 10.1038/nn992. [DOI] [PubMed] [Google Scholar]
- 82.Sedman AJ, Bockbrader H, Schwarz RD. Life Sci. 1995;56:877–82. doi: 10.1016/0024-3205(95)00023-y. [DOI] [PubMed] [Google Scholar]
- 83.Hatcher JP, Loudon JM, Hagan JJ, Clark MS. Psychopharmacology (Berl) 1998;138:275–82. doi: 10.1007/s002130050672. [DOI] [PubMed] [Google Scholar]
- 84.Wienrich M, Meier D, Ensinger HA, Gaida W, Raschig A, Walland A, Hammer R. Life Sci. 2001;68:2593–600. doi: 10.1016/s0024-3205(01)01057-8. [DOI] [PubMed] [Google Scholar]
- 85.Uslaner JM, Eddins D, Puri V, Cannon CE, Sutcliffe J, Chew CS, Pearson M, Vivian JA, Chang RK, Ray WJ, Kuduk SD, Wittmann M. Psychopharmacology (Berl) 2013;225:21–30. doi: 10.1007/s00213-012-2788-8. [DOI] [PubMed] [Google Scholar]
- 86.Andersen MB, Croy CH, Dencker D, Werge T, Bymaster FP, Felder CC, Fink-Jensen A. PLoS One. 2015;10:e0122722. doi: 10.1371/journal.pone.0122722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lange HS, Cannon CE, Drott JT, Kuduk SD, Uslaner JM. J Pharmacol Exp Ther. 2015;355:442–50. doi: 10.1124/jpet.115.226712. [DOI] [PubMed] [Google Scholar]
- 88.Mckinze DL, Bymaster FP. Handb Exp Pharmacol. 2012;213:233–65. doi: 10.1007/978-3-642-25758-2_9. [DOI] [PubMed] [Google Scholar]
- 89.Bodick NC, Offen WW, Levey AI, Cutler NR, Gauthier SG, Satlin A, Shannon HE, Tollefson GD, Rasmussen K, Bymaster FP, Hurley DJ, Potter WZ, Paul SM. Arch Neurol. 1997;54:465–73. doi: 10.1001/archneur.1997.00550160091022. [DOI] [PubMed] [Google Scholar]
- 90.Thomsen M, Conn PJ, Lindsley C, Wess J, Boon JY, Fulton BS, Fink-Jensen A, Caine SB. J Pharmacol Exp Ther. 2010;332:959–69. doi: 10.1124/jpet.109.162057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thomsen M, Lindsley CW, Conn PJ, Wessell JE, Fulton BS, Wess J, Caine SB. Psychopharmacology (Berl) 2012;220:673–85. doi: 10.1007/s00213-011-2516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wess J, Eglen RM, Gautam D. Nat Rev Drug Discov. 2007;6:721–33. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- 93.Shekhar A, Potter WZ, Lightfoot L, Lienemann J, Dube S, Mallinckrodt C, Bymaster FP, McKinze DL, Felder CC. Am J Psychiatry. 2008;165:1033–9. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- 94.Dencker D, Wortwein G, Jeon J, Thomsen M, Sager TN, Mork A, Woldbey DP, Wess J, Fink-Jensen A. J Neurosci. 2011;31:5905–8. doi: 10.1523/JNEUROSCI.0370-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Han C, Chatterjee A, Noetzel MJ, Panarese JD, Smith E, Chase P, Hodder P, Niswender C, Conn PJ, Lindsley CW, Stauffer SR. Bioorg Med Chem Lett. 2015;2:384–8. doi: 10.1016/j.bmcl.2014.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Abdul-Ridha A, Lane JR, Mistry SN, Lopez L, Sexton PM, Scammells PJ, Christopoulos A, Canals M. J Biol Chem. 2014;289:33701–11. doi: 10.1074/jbc.M114.604967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Abdul-Ridha A, Lopez L, Keov P, Thal DM, Mistry SN, Sexton PM, Lane JR, Canals M, Christopoulos A. J Biol Chem. 2014;289:6067–79. doi: 10.1074/jbc.M113.539080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kuduk SD, Beshore DC. Curr Topics in Med Chem. 2014;14:1738–1754. doi: 10.2174/1568026614666140826120224. [DOI] [PubMed] [Google Scholar]
- 99.Mistry SN, Jörg M, Lim H, Vinh NB, Sexton PM, Capuano B, Christopoulos A, Lane JR, Scammells PJ. J Med Chem. 2016;1:388–409. doi: 10.1021/acs.jmedchem.5b01562. [DOI] [PubMed] [Google Scholar]
- 100.Kuduk SD, Chang RK, Di Marco CN, Pitts DR, Greshock TJ, Ma L, Wittmann M, Seager MA, Koeplinger KA, Thompson CD, Hartman GD, Bilodeau MT, Ray WJ. J Med Chem. 2011;54:4773–80. doi: 10.1021/jm200400m. [DOI] [PubMed] [Google Scholar]
- 101.Kruse AC, Kobilka BK, Gautam D, Sexton PM, Christopoulos A, Wess J. Nat Rev Drug Discov. 2014;13:549–60. doi: 10.1038/nrd4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li B, Scarselli M, Knudsen CD, Kim SK, Jacobson KA, McMillin SM, Wess J. Nat Methods. 2007;4:169–174. doi: 10.1038/nmeth990. [DOI] [PubMed] [Google Scholar]
- 103.Lebon G, Langmead CJ, Tehan BG, Hulme EC. Mol Pharmacol. 2009;75:331–341. doi: 10.1124/mol.108.050963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Drubbisch V, Lameh L, Philip M, Sharma YK, Sadee W. Pharm Res. 1992;9:1644–1647. doi: 10.1023/a:1015885029612. [DOI] [PubMed] [Google Scholar]
- 105.Gentry P, Kokubo M, Bridges T, Kett N, Harp J, Cho H, Smith E, Chase P, Hodder P, Niswender C, Daniels J, Conn P, Wood M, Lindsley C. J Med Chem. 2013;22:9351–5. doi: 10.1021/jm4013246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Berizzi A, Gentry PR, Rueda P, den Hoedt S, Sexton PM, Langmead CJ, Christopoulos A. Mol Pharmacol. 2016 doi: 10.1124/mol.116.104182. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.