

Abstract
Teenage binge drinking is a major health concern in the United States, with 21% of teenagers reporting binge-pattern drinking behavior in the last 30 days. Recently, our lab showed that alcohol-naïve offspring of rats exposed to alcohol during adolescence exhibited altered gene expression profiles in the hypothalamus, a brain region involved in stress regulation. We employed Enhanced Reduced Representation Bisulfite Sequencing as an unbiased approach to test the hypothesis that parental exposure to binge-pattern alcohol during adolescence alters DNA methylation profiles in their alcohol-naïve offspring. Wistar rats were administered a repeated binge-ethanol exposure paradigm during early (postnatal day (PND) 37-44) and late (PND 67-74) adolescent development. Animals were mated 24h after the last ethanol dose and subsequent offspring were produced. Analysis of male PND7 offspring revealed that offspring of alcohol-exposed parents exhibited differential DNA methylation patterns in the hypothalamus. The differentially methylated cytosines (DMCs) were distinct between offspring depending on which parent was exposed to ethanol. Moreover, novel DMCs were observed when both parents were exposed to ethanol and many DMCs from single parent ethanol exposure were not recapitulated with dual parent exposure. We also measured mRNA expression of several differentially methylated genes and some, but not all, showed correlative changes in expression. Importantly, methylation was not a direct predictor of expression levels, underscoring the complexity of transcriptional regulation. Overall, we demonstrate that adolescent binge ethanol exposure causes altered genome-wide DNA methylation patterns in the hypothalamus of alcohol-naïve offspring.
Keywords: Adolescence, alcohol, DNA methylation, epigenetics, inheritance
Introduction
Binge alcohol consumption among adolescents is a major health concern in the United States, with 21% of teenagers reporting binge-pattern drinking behavior in the last 30 days (White & Hingson, 2013). Americans under the age of 21 consume over 90% of alcohol in binge-like patterns, which is defined by the Centers for Disease Control as raising the blood alcohol concentration (BAC) above 0.08% within 2 hours (CDC 2014) (Miller, Naimi, Brewer, & Jones, 2007). This behavior is not only dangerous at the time, but can also lead to various health problems in adulthood such as increased risk for developing depression, mood disorders, alcohol dependence and neurodegenerative diseases (Allen, Rivier, & Lee, 2011; Coleman Jr, He, Lee, Styner, & Crews, 2011; Vargas, Bengston, Gilpin, Whitcomb, & Richardson, 2014).
Clinical studies have shown that children of alcoholics are at an increased risk for attention-deficit/hyperactivity disorder and have a greater propensity for abusing alcohol throughout life, but the root of these behaviors is confounded by child-rearing practices in homes of alcoholics (Hairston et al., 2016; Hill, Tessner, & McDermott, 2011; Sundquist, Sundquist, & Ji, 2014). However, experimental evidence from animal models suggests that molecular inheritance mechanisms could underlie these clinical findings (Finegersh, Rompala, Martin, & Homanics, 2015; Przybycien-Szymanska, Rao, Prins, & Pak, 2014). Data from our lab and others have demonstrated ethanol-induced long-term changes in gene expression in the hypothalamus as well as behavioral changes, such as increased preference for alcohol drinking and dysfunctional stress responsiveness in alcohol-naïve offspring (Finegersh & Homanics, 2014; Govorko, Berkdash, Zhang, & Sarkar, 2012; Przybycien-Szymanska, et al., 2014; Rompala, Finegersh, & Homanics, 2016). The hypothalamus has been investigated for its vulnerability to binge alcohol exposure as it has central importance in regulating the stress response (Przybycien-Szymanksa, Rao, & Pak, 2010). Together, these studies raise the possibility that epigenetic inheritance is one mechanism by which adolescent alcohol exposure can affect naïve offspring (Finegersh, et al., 2015; Shukla et al., 2008).
DNA methylation is a heritable epigenetic mark that is relatively stable but varies throughout development and can be influenced by environmental factors (Carone et al., 2010; Jones, 2012). Aberrant DNA methylation is implicated in many cognitive disorders such as schizophrenia, depression, and addiction (Gavin, Chase, & Sharma, 2013; Grayson & Guidotti, 2012; Manzardo & Butler, 2013). In the brain, DNA methylation is intimately involved in cellular differentiation as well as synaptic plasticity (Tognini, Napoli, & Pizzorusso, 2015). Therefore, proper patterning of the epigenetic landscape is necessary for neuronal function. Environmental factors are known to cause differential methylation of the brain during early development, which is a potential mechanism for lifetime adaptation. For example, early life stress through maternal deprivation can alter methylation of genes involved in mediating the physiological stress response and these methylation marks are persistent throughout adulthood (Chen et al., 2012). Exposure to adverse environmental factors and drugs of abuse during adolescence has also been demonstrated to have transgenerational consequences (Carone, et al., 2010; Minnes et al., 2014; Öst et al., 2014; Weyrich et al., 2016). For example, paternal cocaine exposure during puberty has been shown to alter DNA methylation and behavior in offspring (Killinger, Robinson, & Stanwood, 2012). Paternal preconception exposure to alcohol has also been associated with increased anxiety and depression in offspring (Liang et al., 2014). However, there have been very few studies to examine the effects of maternal preconception exposure to drugs of abuse on offspring (Vassoler, Byrnes, & Pierce, 2014).
Methylation occurs primarily at cytosine residues in the context of CpG dinucleotides, although other modified bases have recently been reported (Schübeler et al., 2011). CpG islands (CGIs) are GC-rich regions of the genome, an average of 1,000 bp in length, that are often close to transcription start sites (TSS) and tend to be unmethylated, which allows for active gene transcription (Jones, 2012; Schübeler, 2015). Methylated DNA found in promoter regions is thought to inhibit gene transcription by encouraging heterochromatin formation, therefore preventing binding of transcriptional activators, as well as recruiting repressive proteins to inhibit transcription of downstream genes (Jones, 2012; Schübeler, 2015; Smith & Meissner, 2013). Less is known about the role of methylation in other genic and intergenic regions, although it has been suggested that methylated DNA in coding regions of a gene can promote gene expression and/or alternative splicing (Maunakea, Chepelev, Cui, & Zhao, 2013).
We employed Enhanced Reduced Representation Bisulfite Sequencing (ERRBS) as an unbiased approach to test the hypothesis that preconception parental exposure to binge-pattern alcohol consumption during adolescence alters DNA methylation in the hypothalamus of alcohol-naïve male offspring. Our experimental design also allowed us to compare between the discrete maternal vs. paternal contributions to altered DNA methylation in offspring. This is the first study of this scope to analyze genome-wide changes in DNA methylation of offspring as a result of adolescent binge alcohol exposure of both parents.
Materials and Methods
Animals and Tissue Preparation
Male and female Wistar rats were purchased from Charles River Laboratory (Wilmington, MA) at post-natal day (PND) 23 and were allowed to acclimate for 7 days. Then, animals were handled by experimenters for 5 minutes once daily for 7 days to control for non-specific handling stress. Animals were pair-housed within the same treatment group. Food and water were available ad libitum and animals were kept on a 12:12 light/dark cycle, with lights on at 7:00 AM and handling/treatment began at 10:00 AM. Animal procedures were approved by the Loyola University Medical Center Institutional Animal Care and Use Committee (permit #2012021). All measures were taken to minimize pain and suffering.
Beginning at PND37, which is defined as peri-puberty in the rat (Ketelslegers, Hetzel, Sherins, & Catt, 1978), animals were exposed to a repeated binge-pattern alcohol paradigm (Fig. 1). This 8-day paradigm has been used previously by our lab and others and is designed to mimic the reported drinking patterns of adolescents (Lauing, Himes, Rachwalski, Strotman, & Callaci, 2008; Przybycien-Szymanksa, et al., 2010; Przybycien-Szymanska, et al., 2014). This pattern of alcohol consumption raises the blood alcohol concentration (BAC) to 150–180 mg/dl in males and 210–240 mg/dl in females without altering body weight or normal growth patterns (Przybycien-Szymanksa, et al., 2010; Przybycien-Szymanska, et al., 2014). Animals were given food grade alcohol (Everclear, Luxco) diluted in tap water at a dose of 3g/kg body weight (20% v/v solution), or an equal volume of vehicle (water) via oral gavage. Treatment was given once per day for 3 days, followed by 2 days tap water and another 3 days alcohol (Fig. 1). Control groups received tap water for all 8 days. Animals were then left undisturbed until PND67, which is considered late puberty, when they underwent the same 8-day treatment.
Figure 1. Adolescent binge alcohol exposure paradigm.

An 8-day treatment paradigm was administered to male and female Wistar rats where animals received 3 g/kg body weight of ethanol (20% v/v in water) via oral gavage once per day. Starting at PND 37 (peri-puberty), ethanol-treated animals received 3 days ethanol, 2 days tap water, and 3 days ethanol, whereas control animals received tap water only for all 8 days. Animals were then left undisturbed until PND 67, when they underwent the same 8 day treatment. 24 hours after last ethanol dose, animals were mated (n=2–3 pairs/treatment) and offspring were born approximately 23 days later. Litters were culled to 10 pups per dam at PND 0 and animals were euthanized at PND 7.
For mating, pairs consisted of all combinations (maternal vehicle x paternal vehicle, maternal ethanol x paternal vehicle, maternal vehicle x paternal ethanol, maternal ethanol x paternal ethanol). Animals were paired for mating 24 hours following last gavage treatment, with 2–3 pairs of each treatment group. After 7 days, females were single-housed in order to properly nest and males were returned to pair-housing with previous cage-mate. There was no difference in litter size, pup weight, or sex ratio between treatment groups (Table 1). Maternal care was assessed based on gathering pups into the nest, crouching over pups to facilitate suckling, active licking/grooming, and growth rates of pups (indicative of nutritional status). Twice daily observations showed no apparent differences in maternal care between groups, although these observations were not scored for quantitative analysis. Within 1 hour of birth, litters were culled to 10 pups per dam (5 pups of each sex) and pups were raised by their biological mother until PND7. Pups were anesthetized on ice and euthanized by rapid decapitation. Brains were immediately removed and whole hypothalamus was microdissected on ice before flash freezing. Tissue was stored at −80°C until isolation of genomic DNA with phenol/chloroform extraction and RNA with TRIzol reagent (Life Technologies), according to manufacturer instructions.
Table 1.
Preconception binge alcohol exposure did not alter litter size, sex ratio or offspring growth.
| Litter | Total Pups | Sex Ratio | Average PND0 Weight (g) | Average PND7 Weight (g) |
|---|---|---|---|---|
| Control 1 | 15 | 8M:7F | 6.50 | 17.81 |
| Control 2 | 14 | 6M:8F | 6.11 | 14.54 |
| Control 3 | 13 | 5M:8F | 5.80 | 14.44 |
| Maternal Exposure 1 | 15 | 9M:6F | 6.09 | 14.86 |
| Maternal Exposure 2 | 11 | 4M:7F | 6.86 | 14.28 |
| Paternal Exposure 1 | 15 | 5M:10F | 6.84 | 17.37 |
| Paternal Exposure 2 | 8 | 5M:3F | 6.93 | 15.09 |
| Paternal Exposure 3 | 15 | 5M:10F | 6.09 | 17.59 |
| Both Exposed 1 | 13 | 5M:8F | 6.47 | 14.47 |
| Both Exposed 2 | 13 | 6M:7F | 5.73 | 13.13 |
Methylation Sequencing and Statistics
Enhanced Reduced Representation Bisulfite Sequencing (ERRBS) was performed at the University of Michigan Epigenomics Core, as described by Grimes et al. (2012). For ERRBS, genomic DNA from 3 male pups of each treatment group, with at least one from each mating pair, were used (12 total pups). Previous work using this paradigm has shown larger changes in gene expression of male offspring, therefore we chose to perform ERRBS analysis on only males (Przybycien-Szymanska, et al., 2014). Tissue from remaining male pups was used for mRNA expression analysis and female offspring were used for subsequent experiments.
We employed FASTQC (version 0.11.3) to assess the overall quality of each sequenced sample and identify specific reads and regions that may benefit from trimming (“http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/,”). TrimGalore (version 0.4.0) was used to trim low-quality bases (quality score lower than 20), adapter sequences (stringency 6) and end-repair bases from the 3′ end of reads (“http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/,”). For alignment and methylation calling we employed Bismark (version 0.14.3), an integrated alignment and methylation call program that performs unbiased alignment (by converting residual cytosines to thymines prior to alignment in both reads and reference) (Krueger & Andrews, 2011). Briefly, we aligned reads to the reference genome (UCSC rn6 from iGenomes, (“https://support.illumina.com/sequencing/sequencing_software/igenome.html,”) using Bowtie2 (Langmead & Salzberg, 2012) (version 2.2.1) with default parameters settings, except for maximum number of mismatches in seed alignment (N) set to 1, and length of seed substrings (L) set to 20. Methylation calls were reported for all nucleotides with a read depth of at least 10.
We used the methylSig R package (0.3.2) to assess the overall quality of methylation calls and coverage (Park, Figueroa, Rozek, & Sartor, 2014). We then employed it to identify differentially methylated positions by tiling the methylation data across windows of 25 bases. In addition, we used information from nearby CpG sites to improve variance estimates (local window size of 200 bases). For each pairwise comparison methylSig uses a beta-binomial approach to calculate differential methylation statistics, accounting for variation among replicates within each group. We adjusted the p-values for multiple testing using the FDR approach, and considered sites to be differentially methylated when they had a percent change in methylation of at least 20% and a q value < 0.05.
Finally, we annotated sites and regions using UCSC Genome Browser’s annotations for CpG islands, promoters and other genic regions (Rosenbloom et al., 2015). CpG shores were defined as the regions outside CpG islands but within 2,000 bp of a CpG island. CpG shelves were defined as the regions within 2,000 bp of a CpG shore. When regions overlapped, the priority was CpG island, followed by CpG shore. Gene promoters were defined as 1,000 bp upstream of reported transcription start sites.
All reported data passed QC analysis. In any given cell, any given cytosine is either methylated or not. Thus looking at a population of cells should yield a pattern where many C positions have high methylation and many C positions have low methylation. Percent methylation histograms should therefore have two peaks at either end, which we observed for all samples (Sup. Fig. 1). Histogram analysis of CpG coverage demonstrated the absence of a right-shift secondary peak, indicating that there was no PCR duplication bias in our samples (Sup. Fig 2). Differences in methylation at individual cytosine residues were analyzed, as this is a strength of the ERRBS technique. The base-pair resolution of ERRBS allows us to identify the exact residues which are susceptible to modification by parental binge alcohol exposure.
RT-qPCR
mRNA expression of differentially methylated target genes in offspring hypothalamus was measured using reverse transcription quantitative PCR (n=7–10/group). Total RNA (1.0 μg) was reverse transcribed using High Capacity Reverse Transcription Kit (Applied Biosystems) according to manufacturer instructions. FastStart SYBR Green Master Mix (Roche) was used for all RT-qPCR reactions, adding 2μl of cDNA and final primer concentrations of 0.25 μM for each gene (primer sequences listed in Table 2). RT-qPCR data were analyzed using the ΔΔCt method comparing to 18S RNA expression of each sample. Data were compared using one-way ANOVA to determine differences in expression between all treatment groups with Tukey’s Post-hoc analysis. P-value < 0.05 was considered significant.
Table 2.
RT-qPCR Primer Sequences
| Gene Name | Sequence (5′ to 3′) | |
|---|---|---|
| Begain | F | GATGGGCAGCGATCAGTCTTC |
| R | AGCTTCTCCAACTTGTGCGT | |
| Rtn4ip1 | F | GGACTCCCTGCTTGTTTGCT |
| R | ATTGCTACTGTGCTCCCCAC | |
| NPY | F | TACTCCGCTCTGCGACACTA |
| R | TGGGGGCATTTTCTGTGCTT | |
| Grm4 | F | ATGCGCGGTTGGTAGGAGTG |
| R | CAGCGCAGGTTCAGTAATGC | |
| FGFR1 | F | TCACAGCCACTCTCTGCACT |
| R | GTGATGCGTGTACGGTTGCT | |
| Acvr2a | F | CGGGGATTGTCATTTGTGCG |
| R | TCCAGGGTCCTGAGTAGGAA | |
| MC3r | F | TGCAACTCTGTCATCGACCC |
| R | CCATTGCAACCGCAGAGAAT | |
| Fzd10 | F | CTCCATGGACTTAGAGCGCC |
| R | TGGTGTTGTAGCCGATGTCC | |
| Esam | F | CCAGCTTACTGCGGGTTTTG |
| R | GATGAAGACTCCTCCCGTGC | |
| Arrdc1 | F | ACTACCCTTCCGAGCTATCCG |
| R | TGAAGTAGCTCTCCTCCACCA | |
| Gpank1 | F | TGAGGGACTTAGGTCGGGTGT |
| R | GGACATGGCTCAGGTTAGCG | |
| Ephb3 | F | AAGTTCGGGGGAGAACCCTA |
| R | TGAAGAGGTTTGGGGCACAC | |
| AVP | F | CGCAGTGCCCACCTATGCTC |
| R | AGGAAGCAGCCCAGCTCGTC | |
| 18S | F | CATTCGAACGTCTGCCCTAT |
| R | GTTTCTCAGGCTCCCTCTCC |
Results
Parental ethanol exposure induces differentially methylated cytosine residues in the hypothalamus of male ethanol-naïve offspring
We used a rat model of pubertal binge alcohol treatment previously established in our laboratory to determine intergenerational changes in DNA methylation patterns associated with parental adolescent exposure to binge alcohol (Fig. 1) (Przybycien-Szymanska, et al., 2014). Genome-wide DNA methylation in the hypothalamus was compared between male offspring of control x control mating pairs and all other combinations (maternal ethanol only, paternal ethanol only, both parents ethanol). Genomic DNA of three pups per treatment group, but from different litters, was analyzed with an average of 113,445,354 reads generated for each sample. Both pre-trimming and post-trimming QC reports indicated that the read data passed basic quality control, with exceptions following the expected patterns in ERRBS experiments. Alignment efficiencies and conversion rates were typical of ERRBS experiments and consistent across samples (Table 3). Raw methylation files will be deposited for public access upon publication.
Table 3.
Alignment efficiencies and conversion rates for ERRBS of all samples.
| Sample | Total Reads | Trimmed Reads | Unique Alignment | Alignment Efficiency | Conversion Rate |
|---|---|---|---|---|---|
| Control 1 | 101,048,410 | 100,855,039 | 62,220,409 | 61.70% | 99.40% |
| Control 2 | 127,821,721 | 127,215,674 | 78,361,090 | 61.60% | 99.00% |
| Control 3 | 102,862,686 | 102,170,841 | 58,944,438 | 57.70% | 99.10% |
| Maternal Ethanol 1 | 109,878,778 | 108,966,846 | 52,870,280 | 48.50% | 98.90% |
| Maternal Ethanol 2 | 124,275,848 | 123,559,339 | 74,581,912 | 60.40% | 99.20% |
| Maternal Ethanol 3 | 107,454,830 | 106,658,607 | 64,249,678 | 60.20% | 99.30% |
| Paternal Ethanol 1 | 114,893,371 | 113,982,695 | 69,455,351 | 60.90% | 99.30% |
| Paternal Ethanol 2 | 106,580,485 | 105,705,397 | 69,203,843 | 65.50% | 99.20% |
| Paternal Ethanol 3 | 99,166,858 | 98,773,966 | 61,786,238 | 62.60% | 99.20% |
| Maternal + Paternal 1 | 139,906,554 | 138,640,440 | 81,779,772 | 59.00% | 99.10% |
| Maternal + Paternal 2 | 121,250,474 | 120,463,927 | 69,505,491 | 57.70% | 99.10% |
| Maternal + Paternal 3 | 106,204,238 | 105,806,869 | 66,903,713 | 63.20% | 99.40% |
In general, we found more instances of hypermethylation in all groups compared to control and differentially methylated cytosines (DMCs) were distinct for all treatment groups with very little overlap; only 4 hypermethylated DMCs were common to all three groups (Fig. 2). The largest number of hypermethylated residues was found when both parents were exposed to ethanol (Fig. 2). Specifically, dual parent preconception ethanol exposure resulted in 168 hypermethylated DMCs. We also examined the differences in offspring DNA methylation when only one parent was exposed to binge ethanol preconception (the other parent received water), since both maternal and paternal gametes can affect offspring methylation patterns. We observed 95 hypermethylated DMCs in offspring where only the mother was exposed to ethanol and 54 hypermethylated DMCs when there was only paternal ethanol exposure (Fig. 2). Several genes were associated with more than one hypermethylated DMC, such as Rn5-8s, Bmp3 and Atg5 (Fig. 2; boldface type).
Figure 2. Annotated genes associated with hypermethylated residues vary between treatment groups.

Venn diagram describing bioinformatics results using UCSC Genome Browser for analysis. Statistically significant differentially methylated cytosine (DMC) residues between treatment groups were associated with the nearest downstream gene for hypermethylated cytosines. Genes are listed in alphabetical order for each treatment group. Gene names underlined in bold face type indicate that more than one cytosine was differentially methylated near that gene.
There were also a large number of hypomethylated DMCs among all groups with only 5 hypomethylated DMCs shared between all treatments (Fig. 3). In offspring where both parents were exposed to ethanol there were 105 hypomethylated DMCs compared to offspring from water-treated parents (Fig. 3). In maternal ethanol treated offspring, there were 79 hypomethylated residues compared to 47 hypomethylated residues when only the father was exposed to ethanol (Fig. 3). There were also multiple genes associated with more than one DMC, for example Exo5 was hypomethylated on multiple residues in both maternal ethanol and paternal ethanol offspring (Fig. 3; boldface type).
Figure 3. Annotated genes associated with hypomethylated residues vary between treatment groups.

Venn diagram describing bioinformatics results using UCSC Genome Browser for analysis. Statistically significant differentially methylated cytosine (DMC) residues between treatment groups were associated with the nearest downstream gene for hypomethylated cytosines. Genes are listed in alphabetical order for each treatment group. Gene names underlined in bold face type indicate that more than one cytosine was differentially methylated near that gene.
Differentially methylated cytosines were distributed across all chromosomes and the extent of hypo-versus hypermethylation was dependent on parental ethanol exposure
Overall, there were discrete yet robust changes in DNA methylation across the genome and we did not observe clustering of DMCs on a particular chromosome or region of the genome (Fig. 4). In addition, there were no ethanol-induced global changes in DNA methylation and ethanol exposure to both parents did not have an additive effect on DMCs for individual chromosomes (Fig. 4). For example, maternal ethanol exposure caused hypomethylation of cytosine residues on chromosome 11, whereas paternal ethanol exposure had no effect (Fig. 4A, B). By contrast, ethanol induced a combination of both hypo- and hypermethylated sites on chromosome 11 when both parents were exposed (Fig. 4C). As another example, the X chromosome was hypomethylated in the offspring of both maternal-only ethanol and paternal-only ethanol exposed animals (Fig. 4A, B), but the X chromosome was hypermethylated when both parents were treated (Fig. 4C). Similarly, the Y chromosome had hypermethylated DMCs only when the father was ethanol exposed (Fig. 4B). These examples underscore the lack of an additive effect from dual parental exposure and the complexity of offspring DNA methylation.
Figure 4. Distribution of differentially methylated cytosines across chromosomes.

Histogram analysis of differentially methylated cytosines (DMCs) on each chromosome in (A) offspring from maternal ethanol-exposed, (B) offspring from paternal ethanol-exposed, and (C) offspring from both maternal and paternal ethanol-exposed. Blue region indicates number of hypomethylated DMCs and red region indicates number of hypermethylated DMCs on each chromosome.
In addition to genome-wide changes in methylation, we examined the percentage differences in methylation at each residue. Residues with the greatest percent difference in methylation were ranked for hyper- and hypomethylation and the top 5 hypermethylated and 5 hypomethylated DMCs for each group are listed in Table 2. The same cytosine on chromosome 2, upstream of Hmox-ps1, was the most hypermethylated residue in maternal ethanol and paternal ethanol offspring, but was not changed in offspring of maternal and paternal ethanol exposure (Table 4).
Table 4. Top 5 Hyper and 5 Hypomethylated DMCs as ranked by greatest percentage difference for each treatment group.
Residues with significant hypermethylation compared to control are in white rows, with significant hypomethylated residues in gray rows.
| Treatment | Location | Position | P value | Q value | Methylation Difference (%) | Nearest Gene | Distance to gene body (bp) | Region |
|---|---|---|---|---|---|---|---|---|
| Maternal Ethanol | chr2 | 215352989 | 1.38E-07 | 3.89E-03 | 92.44 | Hmox2-ps1 | 17205 | Inter CGI |
| chr2 | 177162515 | 1.59E-07 | 4.12E-03 | 90.63 | Rapgef2 | 673686 | Inter CGI | |
| chr12 | 8190387 | 5.32E-06 | 4.17E-02 | 86.25 | Slc7a1 | 81414 | Inter CGI | |
| chr3 | 33440426 | 5.97E-06 | 4.40E-02 | 85.81 | Orc4 | 364739 | Inter CGI | |
| chr15 | 2798203 | 7.63E-11 | 3.02E-05 | 85.10 | Dupd1 | 0 | Coding | |
| chr9 | 45676004 | 2.21E-07 | 5.29E-03 | −84.74 | Chst10 | 116862 | Coding | |
| chr16 | 11212749 | 6.04E-08 | 2.39E-03 | −85.30 | Opn4 | 260198 | Coding | |
| chr20 | 6421481 | 1.18E-07 | 3.66E-03 | −87.00 | Cdkn1a | 62616 | Exon | |
| chr15 | 4643007 | 9.53E-11 | 3.02E-05 | −89.17 | Kcnk5 | 46006 | Inter CGI | |
| chr1 | 215031715 | 7.29E-08 | 2.64E-03 | −95.36 | Dusp8 | 0 | Exon | |
| Paternal Ethanol | chr2 | 215352989 | 6.01E-10 | 2.68E-04 | 95.30 | Hmox2-ps1 | 17205 | Inter CGI |
| chr5 | 71464665 | 6.25E-07 | 1.74E-02 | 89.32 | Klf4 | 818645 | Inter CGI | |
| chr9 | 39855580 | 2.37E-06 | 3.27E-02 | 80.00 | Khdrbs2 | 0 | Coding | |
| chr14 | 42190320 | 1.63E-06 | 2.59E-02 | 78.80 | LOC360933 | 756401 | Inter CGI | |
| chr6 | 26853900 | 3.29E-07 | 1.16E-02 | 77.01 | Agbl5 | 0 | Exon | |
| chr10 | 103823050 | 2.81E-06 | 3.55E-02 | −67.20 | Tmem104 | 29835 | Coding | |
| chr1 | 114898684 | 1.12E-07 | 6.84E-03 | −68.63 | LOC100365773 | 0 | Coding | |
| chr16 | 11212749 | 2.23E-06 | 3.21E-02 | −78.36 | Opn4 | 260198 | Coding | |
| chr9 | 86252196 | 3.07E-06 | 3.76E-02 | −80.77 | Cul3 | 149036 | Inter CGI | |
| chr1 | 215031715 | 6.91E-07 | 1.89E-02 | −91.00 | Dusp8 | 0 | Exon | |
| Maternal + Paternal Ethanol | chr2 | 186682537 | 6.68E-08 | 2.78E-03 | 93.39 | Fcrl2 | 77443 | Inter CGI |
| chr3 | 2137673 | 1.06E-05 | 4.32E-02 | 92.26 | Arrdc1 | 736 | Promoter | |
| chr12 | 48646837 | 4.00E-08 | 2.13E-03 | 91.05 | Ficd | 12829 | Coding | |
| chr2 | 39211484 | 1.31E-09 | 2.18E-04 | 90.32 | Kif2a | 1002717 | Inter CGI | |
| chr7 | 131964758 | 2.75E-06 | 2.34E-02 | 89.12 | Alg10 | 628574 | Inter CGI | |
| chr17 | 78631069 | 7.02E-07 | 1.12E-02 | −81.86 | Fam107b | 69454 | Inter CGI | |
| chr18 | 73900477 | 3.46E-06 | 2.63E-02 | −82.27 | Rnf165 | 27232 | Inter CGI | |
| chr2 | 42671444 | 1.20E-07 | 3.68E-03 | −82.33 | Gpbp1 | 330599 | Inter CGI | |
| chr12 | 45206285 | 9.70E-07 | 1.31E-02 | −85.42 | Pebp1 | 175136 | Inter CGI | |
| chr2 | 185450313 | 4.10E-11 | 2.73E-05 | −86.61 | Rps3a | 5465 | Inter CGI |
DMCs were primarily observed outside of CpG islands and in intergenic regions
The distribution of DMCs throughout the genome can have important functional implications for their role in gene expression.15 Therefore, we identified the relationship between DMC location and the defined functional genomic region for each treatment group. Fig. 5 (A–C) shows the percentage of differentially methylated residues that fall within the defined classes of CpG rich regions. Interrogation was similar between CpG islands and InterCGI regions in all samples (48% and 40%, respectively), but the majority of DMCs were found outside of CpG islands. The priority analysis for functional overlapping elements was gene promoter, coding DNA sequence (CDS), noncoding region, 5′UTR, and then 3′UTR. Fig. 5 (D–F) shows the distribution of differentially methylated residues that fall within defined genic elements according to RefGene. The majority of DMCs were found at nucleotides outside genic regions (intergenic), as well as in the introns of coding genes, regardless of parental ethanol exposure (percent DMCs in intergenic + introns = maternal 86%; paternal 88%; dual parent 89%; Fig. 5D–F). The functional role of methylation in intergenic regions is not completely understood, but these might mediate the activity of distant enhancer elements and non-coding RNAs, or modulate overall chromatin structure (Schübeler, 2015). The other chromosomal regions had very low incidence of ethanol-induced DMCs and were similar between treatment groups (Fig. 5).
Figure 5. Location of differentially methylated cytosines within CpG and functional regions of the genome.
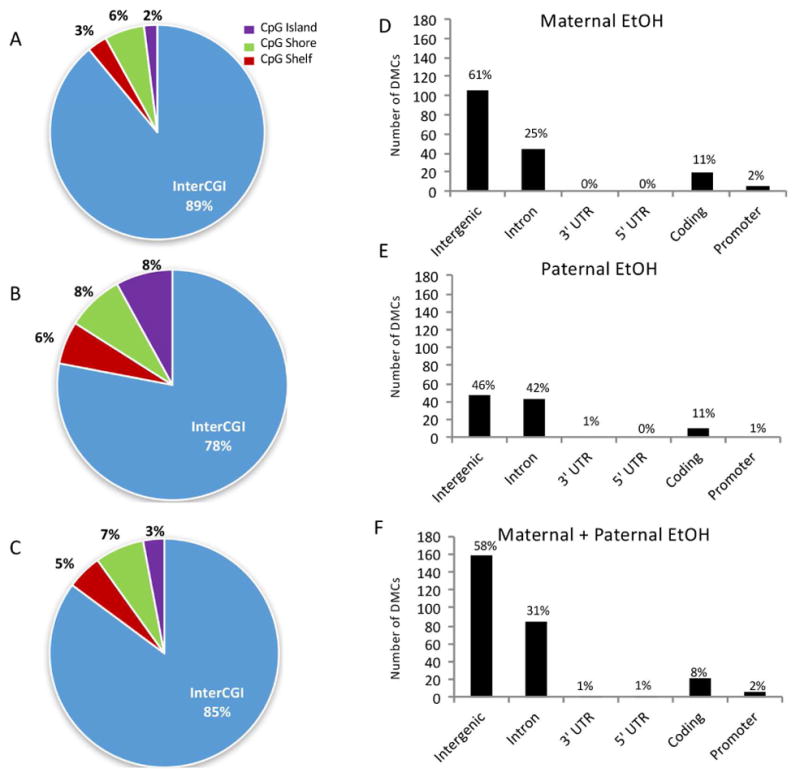
Pie charts represent the percentage of DMCs found in each region as defined by CpG status in pups from (A) maternal ethanol, (B) paternal ethanol, and (C) dual parent exposure. Bar graphs representing the number of DMCs detected in each defined genic regions in (D) offspring from maternal ethanol-exposed, (E) offspring from paternal ethanol-exposed, and (F) offspring from both maternal and paternal ethanol-exposed. UTR = untranslated region; CDS = coding DNA sequence
Methylation of gene promoter regions is considered to have the greatest potential impact on transcriptional gene activity (Jones, 2012). Our data revealed that 35–45% of all annotated promoters were interrogated with sufficient depth in each sample to detect reliable differences. Of those interrogated, only 10 genes had DMCs in their promoter regions (Fig. 5, Table 5). Residues found in the promoters of Fam110a and Esam were hypermethylated and Olr286 was hypomethylated in offspring of mating pairs with only maternal exposure to ethanol. The same hypermethylated cytosine was found in the Esam promoter with only paternal exposure, and this was the only instance of differential promoter methylation in these offspring (Table 5). Unexpectedly, the Esam promoter was not differentially methylated in offspring where both parents were exposed. Instead, adolescent ethanol exposure of both parents induced hypermethylation of cytosine residues in the offspring gene promoters of Arrdc1, Ephb3, and miR6216. Conversely, this same treatment induced hypomethylation in the gene promoters for Golt1b, Gpank1, and Sparcl1 (Table 5).
Table 5. Genes associated with differential promoter methylation for each treatment group.
Those with significant hypermethylation compared to control are in red rows, with significant hypomethylation in blue rows.
| Maternal Ethanol | Paternal Ethanol | Maternal + Paternal Ethanol |
|---|---|---|
| Esam | Esam | Arrdc1 |
| Fam110a | Ephb3 | |
| Olr286 | Mir6216 | |
| Gpank1 | ||
| Golt1b | ||
| Sparcl1 |
mRNA expression of differentially methylated genes is altered in the hypothalamus
Next, we measured the hypothalamic mRNA expression of several genes associated with DMCs that are known to have functions in the nervous system in order to determine if the gene methylation status correlated with gene expression. Genes harboring differential promoter methylation were expected to show a reduction in gene expression, however four genes with differential promoter methylation were excluded because they have not been well characterized (Fam110a, Olr286, Mir6216, Golt1b). Several genes associated with coding or intergenic differential methylation were also measured as they are related to neuro-development or function, with uncertainty as to the effect of methylation at these positions on their mRNA expression.
Hypermethylation of arrestin domain containing protein 1 (Arrdc1), a gene involved in vesicle formation, correlated with a 50% reduction in mRNA expression in offspring where both parents were exposed to alcohol (Table 6). The modified methylated cytosine residue was located within the promoter region (736 bp upstream of the TSS), suggesting that promoter hypermethylation of Arrdc1 reduced downstream gene transcription. However, there was little correlation between gene expression and DMCs for the other genes we tested, suggesting that the relationship between DNA methylation and gene expression may be more complex (Table 6). For example, Ephrin-type B receptor (Ephb3), a receptor tyrosine kinase thought to inhibit synaptic stability, had a hypermethylated residue in the promoter region, 863 bp upstream of the TSS, in offspring in which both parents were exposed to alcohol, yet there were no changes in Ephb3 gene expression (Table 6).
Table 6.
Summary data for mRNA expression of DMC-associated genes.
| Gene | Expression Fold Change ± SEM | P value | DMC state | Location of DMC | ||
|---|---|---|---|---|---|---|
| Maternal Ethanol | Paternal Ethanol | Maternal + Paternal Ethanol | ||||
| Sparcl1 | 0.61 ± 0.15 | 0.71 ± 0.12 | 0.38 ± 0.10 | 0.047* | Both Exposed (2): −53%, −54% | Promoter, CpG shore |
| Arrdc1 | 0.90 ± 0.16 | 1.15 ± 0.04 | 0.53 ± 0.13 | 0.005* | Both Exposed: +92% | Promoter |
| Esam | 1.04 ± 0.14 | 1.25 ± 0.15 | 0.51 ± 0.10 | 0.005* | Maternal: +28%, Paternal: +26% | Promoter |
| Gpank1 | 0.99 ± 0.06 | 1.25 ± 0.08 | 0.89 ± 0.10 | 0.009* | Both Exposed: −35% | Promoter, exon |
| Ephb3 | 1.35 ± 0.10 | 1.30 ± 0.11 | 1.19 ± 0.12 | 0.223 | Both Exposed: +85% | Promoter |
| Grm4 | 1.10 ± 0.12 | 1.25 ± 0.10 | 1.28 ± 0.16 | 0.454 | Maternal (3): +48%, +60%, −82% | Promoter (3) |
| AVP | 3.52 ± 1.30 | 3.00 ± 0.52 | 2.22 ± 0.61 | 0.122 | Maternal: −36%, Paternal: −37% | Coding |
| Acvr2a | 0.91 ± 0.07 | 0.93 ± 0.07 | 0.98 ± 0.11 | 0.943 | Maternal: +67% | Intergenic |
| Begain | 1.55 ± 0.24 | 1.20 ± 0.14 | 1.41 ± 0.27 | 0.351 | Both Exposed: +75% | Intergenic |
| FGFR1 | 1.19 ± 0.13 | 1.06 ± 0.17 | 0.998 ± 0.10 | 0.756 | Both Exposed: −62% | Intergenic |
| Fzd10 | 1.21 ± 0.32 | 1.65 ± 0.36 | 1.58 ± 0.28 | 0.462 | Paternal +37%, Both Exposed +26% | Intergenic |
| MC3r | 1.29 ± 0.19 | 1.05 ± 0.13 | 1.26 ± 0.24 | 0.668 | Paternal: +53% | Intergenic |
| NPY | 1.23 ± 0.11 | 0.93 ± 0.17 | 0.84 ± 0.21 | 0.335 | Paternal: −43% | Intergenic |
| Rtn4ip1 | 1.16 ± 0.09 | 1.17 ± 0.9 | 1.08 ± 0.08 | 0.650 | Both Exposed: −34% | Intergenic |
One-way ANOVA (n=7–10 per group) with p <0.05 considered significant. When there was a significant difference, pairwise comparisons were performed with Tukey’s post hoc analysis. Values in boldface type represent statistically significant differences in those offspring as compared to offspring of control mating pairs.
Discussion
The results from this study revealed three novel findings and highlight the potential for both maternal and paternal preconception binge-like alcohol abuse during adolescence to alter the epigenetic landscape of first-generation offspring. First, there was a lack of global DNA methylation changes in offspring as a result of parental preconception exposure to binge ethanol treatment, suggesting that ethanol mobilizes distinct molecular machinery that confers specificity to DNA methylation sites within the genome. This observation would also support the conclusion that intergenerational ethanol effects are not due to broad ethanol-induced dysfunction in the gametes. Second, the modes of epigenetic inheritance are more complex than that of classical genetic inheritance, and do not necessarily reflect equal contributions of both parents. Unexpectedly, genes that were differentially methylated with either maternal or paternal ethanol exposure (i.e. Esam, See Table 5), were not differentially methylated when both parents were exposed. These results suggest that recombination events during early conception may mask or redefine individual parental epigenetic marks. Third, there was a high prevalence of intergenic, non-promoter methylation in the genome and, methylation of gene promoter regions did not always correspond to changes in gene expression. The stringent analysis parameters used along with our mild animal paradigm underscore the remarkable nature of our results, showing preconception exposure of either parent to just a few episodes of binge-pattern alcohol consumption can cause differential methylation in the hypothalamus of offspring.
To our knowledge, this is the first report of a genome-wide approach examining DNA methylation patterns in ethanol-naïve offspring of parents exposed to binge ethanol treatment during puberty. Previous work examining adult brain tissue reported that repeated adolescent ethanol treatment reduces the function of enzymes involved in epigenetic patterning, including DNA methyltransferases and histone deacetylases (Sakharkar et al., 2014). Therefore, it could be predicted that epigenetic marks as a whole would be reduced in all tissues following ethanol exposure. Instead, our results showed that adolescent binge ethanol exposure had specific consequences at particular residues within the genome for first generation offspring. The lack of global changes in DNA methylation of the hypothalamus in these animals suggests that alcohol does not cause a deficiency in the epigenetic machinery as a whole, especially in the gametes of these exposed animals. Rather, our results suggest that there might be a wide range of nucleotide residues in gametes that are susceptible to alcohol-induced modifications, but the underlying molecular basis for vulnerability at these cytosine residues requires further research. Additionally, further research into the epigenetic changes in specific nuclei of the hypothalamus, such as the paraventricular nucleus, may reveal more noticeable changes in methylation patterns as each nucleus has distinct gene expression patterns and functional outputs.
Our experimental design allowed us to differentiate between the maternal and paternal contributions to offspring methylation patterns, providing some insight into sex-specific mechanism(s) by which epigenetic marks are transmitted to offspring. DNA methylation was altered in the hypothalamus of alcohol-naïve offspring regardless of which parent was exposed. However, very few of the detected DMCs were common to all treatment groups and there were very few DMCs that could be attributed to maternal and paternal exposure, separately, that were then combined in offspring when both parents were treated. Additionally, the differentially methylated residues fall outside regions of known parental imprinting. These results highlight the complexity of epigenetic inheritance and also allow us to speculate about the epigenetic vulnerabilities of gametes to binge alcohol exposure. One possibility is that offspring methylation is reflective of changes in parental gamete methylation that are simply passed on to offspring. Alternatively, it is possible that the gametes of both parents transmit dysfunctional epigenetic machinery to the offspring, preventing proper epigenetic patterning. For example, hypermethylated residues that we observed in alcohol naïve offspring might have escaped demethylation during normal embryogenesis, while hypomethylated residues were skipped during remethylation processes. Another possibility is that post-natal treatment of offspring by the mother is changed when either parent is exposed to alcohol, as has been a suggested mechanism in other preconception treatment experiments (Mashoodh, Franks, Curley, & Champagne, 2012). Our experimental design precludes determining which of these possibilities represents the mechanistic basis for sex-specific contributions of offspring methylation patterns following preconception alcohol use. Future studies that include maternal cross-fostering and quantification of gamete methylation prior to conception will further refine our understanding.
Based on published literature, we can speculate that site specific changes to methylation patterns, such as the ones observed in this study, would require protein or nucleic acid “guides” that would direct these epigenetic events to specific cytosine residues in the genome. Putative molecular candidates for this process include non-coding RNAs, which can be transmitted via gametes to the embryo and are known to be critical for embryonic development (A. B. Rodgers, Morgan, Bronson, Revello, & Bale, 2013; Ali B. Rodgers, Morgan, Leu, & Bale, 2015). Recently, mechanisms demonstrating that non-coding RNAs can mediate DNA methylation were described and this process has been hypothesized to effect transgenerational epigenetic inheritance (Holoch & Moazed, 2015; Matzke & Mosher, 2014; Peschansky & Wahlestedt, 2013; Yan, 2014). Similarly, previous work in our lab demonstrated that adolescent binge ethanol treatment can alter the long-term expression of microRNAs in the hypothalamus, and these small non-coding RNAs could also dictate changes in offspring gene expression (Prins, Przybycien-Szymanska, Rao, & Pak, 2014). Taken together, the emerging evidence supports the hypothesis that adolescent exposure to binge alcohol alters the expression of non-coding RNA in both the sperm and egg and those RNAs can direct a different epigenetic landscape in multiple organ systems in the offspring.
This study revealed that a large percentage of discrete changes in DNA methylation were located in different functional regions of the genome, with the highest prevalence in intergenic regions as well as in introns and coding regions. One possible conclusion is that the intergenic methylation sites correspond to enhancer regions, which may influence gene expression of proximal or more distal genes. The current analysis only examined the relationship of DMCs to the nearest downstream gene and further work needs to be done to test the possibility of their interaction with distant elements. Differential methylation within the coding region of a gene has been previously shown to have case-dependent impacts on gene expression. Some reports have shown that gene body methylation can increase transcription, while others have shown that it might inhibit transcription (Jones, 2012; Watson et al., 2015). Still others have shown that intron methylation may cause alternative splicing of the transcript (Maunakea, et al., 2013). In this study we did not measure a direct relationship between the methylation status and expression pattern for all of the select genes we investigated, however, it is important to carefully interpret the causal relationship between methylation and gene expression (Birney, Smith, & Greally, 2016).
Many biological systems have shown that differential methylation at an individual residue can impact gene transcription, mainly through altering interactions of transcription factors with the genome (Wyatt et al., 2013). However, recent studies have demonstrated that the relationship between hypermethylation of promoters and gene expression is both gene- and tissue-specific (Birney, et al., 2016; Jones, 2012). Therefore, the reported methylation marks in the young offspring could lead to altered hypothalamic development and/or predispose them for adverse responses to alcohol or other stressors later in life. The DNA may be “poised” for further environmental influence, which could manifest as more pronounced phenotypic differences in adulthood. Alternatively, these methylation marks could represent evolutionary adaptation to environmental toxins and will confer resilience in the offspring. For example, a recent study in Wild guinea pigs found that exposure of fathers to high heat causes adaptive responses in offspring via DNA methylation and differential expression of a key thermoregulation gene, Stat3 (Weyrich, et al., 2016).
Preconception use of other common drugs such as nicotine, opioids and marijuana has been a focus of several previous studies, but this study is the first of our knowledge to examine the effects of maternal preconception exposure to alcohol on offspring (Vassoler, et al., 2014). Our results are consistent with a recent report on the use of marijuana and the intergenerational effects of its active ingredient tetrahydrocannabinol (THC) (Watson, et al., 2015). In that study, DNA methylation profiles from the nucleus accumbens brain region of drug-naïve offspring revealed discrete, yet genome-wide, changes with some correlating with altered gene expression when both parents were treated with THC throughout pubertal development (Watson, et al., 2015). Taken together these results provide evidence that there is epigenetic vulnerability to drugs of abuse that extend beyond the exposed individual.
In conclusion, this study provides the first genome-wide interrogation of the intergenerational effects of adolescent binge-pattern alcohol consumption in rats. Remarkably, we demonstrated that there were altered DNA methylation patterns in alcohol-naïve male offspring, regardless of which parent was exposed to alcohol. These changes were at discrete residues throughout the genome and differed between maternal and paternal ethanol exposure, underscoring the complexity of epigenetic inheritance. Additionally, DMCs were mostly found in intergenic, intronic and coding functional regions and did not directly correlate with gene mRNA expression. These results provide insight into the mechanism of intergenerational epigenetics and the potential vulnerability of offspring to both maternal and paternal preconception binge-pattern alcohol consumption.
Supplementary Material
Highlights.
Adolescent binge alcohol abuse impacts male offspring DNA methylation
Naïve male offspring harbor genome-wide, yet discrete changes in DNA methylation
Maternal and paternal preconception alcohol cause distinct methylation marks
Differential methylation is found in intergenic and coding regions of DNA
DNA methylation did not predict gene expression of all downstream genes
Acknowledgments
The authors would like to thank the Epigenomics Core at the University of Michigan for their technical and bioinformatics work. This research was supported by the National Institutes of Health, National Institute on Alcohol Abuse and Alcoholism (NIH RO1 AA021517).
List of abbreviations
- Arrdc1
Arrestin domain containing protein 1
- BAC
blood alcohol concentration
- bp
base pair
- C
cytosine
- CDS
coding DNA sequence
- CGI
CpG Island
- DMC
Differentially methylated cytosine
- Ephb3
Ephrin type B receptor 3
- ERRBS
Enhanced Reduced Representation Bisulfite Sequencing
- Esam
Endothelial cell adhesion molecule
- PND
Post-natal day
- THC
tetrahydrocannabinol
- TSS
transcription start site
- UTR
untranslated region
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen CD, Rivier CL, Lee SY. Adolescent alcohol exposure alters the central brain circuits known to regulate the stress response. Neuroscience. 2011;182:162–168. doi: 10.1016/j.neuroscience.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Smith GD, Greally JM. Epigenome-wide association studies and the interpretation of disease -omics. PLoS Genet. 2016;12(6):e1006105. doi: 10.1371/journal.pgen.1006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carone BR, Fauquier L, Habib N, Shea JM, Hart CE, Li R, et al. Paternally Induced Transgenerational Environmental Reprogramming of Metabolic Gene Expression in Mammals. Cell. 2010;143(7):1084–1096. doi: 10.1016/j.cell.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Evans AN, Liu Y, Honda M, Saavedra JM, Aguilera G. Maternal Deprivation in Rats is Associated with Corticotrophin-Releasing Hormone (CRH) Promoter Hypomethylation and Enhances CRH Transcriptional Responses to Stress in Adulthood. Journal of Neuroendocrinology. 2012;24(7):1055–1064. doi: 10.1111/j.1365-2826.2012.02306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman LG, Jr, He J, Lee J, Styner M, Crews FT. Adolescent binge drinking alters adult brain neurotransmitter gene expression, behavior, brain regional volumes, and neurochemistry in mice. Alcohol Clin Exp Res. 2011;35(4):671–688. doi: 10.1111/j.1530-0277.2010.01385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finegersh A, Homanics GE. Paternal alcohol exposure reduces alcohol drinking and increases behavioral sensitivity to alcohol selectively in male offspring. PLoS ONE. 2014;9(6):e99078. doi: 10.1371/journal.pone.0099078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finegersh A, Rompala GR, Martin DIK, Homanics GE. Drinking beyond a lifetime: new and emerging insights into paternal alcohol exposure on subsequent generations. Alcohol. 2015;49(5):461–470. doi: 10.1016/j.alcohol.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin DP, Chase KA, Sharma RP. Active DNA demethylation in post-mitotic neurons: a reason for optimism. Neuropharmacology. 2013;75:233–245. doi: 10.1016/j.neuropharm.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govorko D, Berkdash RA, Zhang C, Sarkar DK. Male germline transmits fetal alcohol adverse effect on hypothalamic proopiomelanocortin gene across generations. Biol Psychiat. 2012;72:378–388. doi: 10.1016/j.biopsych.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson DR, Guidotti A. The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacol. 2012;38(1):138–166. doi: 10.1038/npp.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hairston IS, Conroy DA, Heitzeg MM, Akbar NZ, Brower KJ, Zucker RA. Sleep mediates the link between resiliency and behavioural problems in children at high and low risk for alcoholism. J Sleep Res. 2016;25:341–349. doi: 10.1111/jsr.12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SY, Tessner KD, McDermott MD. Psychopathology in offspring from families of alcohol dependent female probands: a prospective study. J Psychiat Res. 2011;45:285–294. doi: 10.1016/j.jpsychires.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holoch D, Moazed D. RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet. 2015;16:71–84. doi: 10.1038/nrg3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/.
- http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/.
- https://support.illumina.com/sequencing/sequencing_software/igenome.html.
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- Ketelslegers J, Hetzel W, Sherins R, Catt K. Developmental changes in testicular gonadotropin receptors: plasma gonadotropins and plasma testosterone in the rat. Endocrinology. 1978;103(1):212–222. doi: 10.1210/endo-103-1-212. [DOI] [PubMed] [Google Scholar]
- Killinger CE, Robinson S, Stanwood GD. Subtle biobehavioral effects produced by paternal cocaine exposure. Synapse. 2012;66(10):902–908. doi: 10.1002/syn.21582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews S. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27(11):1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg S. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauing K, Himes R, Rachwalski M, Strotman P, Callaci JJ. Binge alcohol treatment of adolescent rats followed by alcohol abstinence is associated with site-specific differences in bone loss and incomplete recovery of bone mass and strength. Alcohol. 2008;42:649–656. doi: 10.1016/j.alcohol.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F, Dia L, Liu J, Jiang N, Zhang J, Want H, et al. Paternal ethanol exposure and behavioral abnormities in offspring: associated alterations in imprinted gene methylation. Neuropharmacology. 2014;81:126–133. doi: 10.1016/j.neuropharm.2014.01.025. [DOI] [PubMed] [Google Scholar]
- Manzardo AM, Butler MG. Introduction to genetics: DNA methylation, histone modification and gene regulation. iConcept Press; 2013. The Status of Epigenetics and DNA Methylation in Alcoholism. [Google Scholar]
- Mashoodh R, Franks B, Curley JP, Champagne FA. Paternal social enrichment effects on maternal behavior and offspring growth. PNAS. 2012;109:17232–17238. doi: 10.1073/pnas.1121083109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzke MA, Mosher RA. RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat Rev Genet. 2014;15(6):394–408. doi: 10.1038/nrg3683. [DOI] [PubMed] [Google Scholar]
- Maunakea AK, Chepelev I, Cui K, Zhao K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013;23(11):1256–1269. doi: 10.1038/cr.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J, Naimi T, Brewer R, Jones S. Binge drinking and associated health risk behaviors among high school students. Pediatrics. 2007;119(1):76–85. doi: 10.1542/peds.2006-1517. [DOI] [PubMed] [Google Scholar]
- Minnes S, Singer L, Min MO, Wu M, Lang A, Yoon S. Effects of prenatal cocaine/polydrug exposure on substance use by age 15. Drug Alcohol Depen. 2014;134:201–210. doi: 10.1016/j.drugalcdep.2013.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öst A, Lempradl A, Casas E, Weigert M, Tiko T, Deniz M, et al. Paternal diet defines offspring chromatin state and intergenerational obesity. Cell. 2014;159(6):1352–1364. doi: 10.1016/j.cell.2014.11.005. [DOI] [PubMed] [Google Scholar]
- Park Y, Figueroa M, Rozek L, Sartor M. MethylSig: a whole genome DNA methylation analysis pipeline. Bioinformatics. 2014;30(17):2414–2422. doi: 10.1093/bioinformatics/btu339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschansky VJ, Wahlestedt C. Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics. 2013;9(1):3–12. doi: 10.4161/epi.27473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins SA, Przybycien-Szymanska MM, Rao YS, Pak TR. Long-Term Effects of Peripubertal Binge EtOH Exposure on Hippocampal microRNA Expression in the Rat. PLoS ONE. 2014;9(1):e83166. doi: 10.1371/journal.pone.0083166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybycien-Szymanksa MM, Rao YS, Pak TR. Binge-pattern alcohol exposure during puberty induces sexually dimorphic changes in genes regulating the HPA axis. Am J Physiol Endocrinol Metab. 2010;298:E320–E328. doi: 10.1152/ajpendo.00615.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybycien-Szymanska MM, Rao YS, Prins SA, Pak TR. Parental binge alcohol abuse alters F1 generation hypothalamic gene expression in the absence of direct fetal alcohol exposure. PLoS ONE. 2014;9(2):e89320. doi: 10.1371/journal.pone.0089320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers AB, Morgan CP, Bronson SL, Revello S, Bale TL. Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J Neurosci. 2013;33(21):9003–9012. doi: 10.1523/JNEUROSCI.0914-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers AB, Morgan CP, Leu NA, Bale TL. Transgenerational epigenetic programming via sperm microRNA recapitulates effects of paternal stress. PNAS. 2015;112(44):13699–13704. doi: 10.1073/pnas.1508347112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rompala GR, Finegersh A, Homanics GE. Paternal preconception ethanol exposure blunts hypothalamic-pituitary-adrenal axis responsivity and stress-induced excessive fluid intake in male mice. Alcohol. 2016;53:19–25. doi: 10.1016/j.alcohol.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom K, Armstrong J, Barber G, Casper J, Clawson H, Diekhans M, et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 2015;43:D670–681. doi: 10.1093/nar/gku1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakharkar AJ, Tang L, Zhang H, Chen Y, Grayson DR, Pandey SC. Effects of acute ethanol exposure on anxiety measures and epigenetic modifiers in the extended amygdala of adolescent rats. The International Journal of Neuropsychopharmacology. 2014;17(12):2057–2067. doi: 10.1017/S1461145714001047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schübeler D. Function and information content of DNA methylation. Nature. 2015;517(7534):321–326. doi: 10.1038/nature14192. [DOI] [PubMed] [Google Scholar]
- Schübeler D, Ziller MJ, Müller F, Liao J, Zhang Y, Gu H, et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011;7(12):e1002389. doi: 10.1371/journal.pgen.1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla SD, Velazquez J, French SW, Lu SC, Ticku MK, Zakhari S. Emerging role of epigenetics in the actions of alcohol. Alcohol Clin Exp Res. 2008;32(9):1525–1534. doi: 10.1111/j.1530-0277.2008.00729.x. [DOI] [PubMed] [Google Scholar]
- Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14(3):204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- Sundquist J, Sundquist K, Ji J. Autism and attention-deficit/hyperactivity disorder among individuals with a family history of alcohol use disorders. eLife. 2014 doi: 10.7554/eLife.02917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognini P, Napoli D, Pizzorusso T. Dynamic DNA methylation in the brain: a new epigenetic mark for experience-dependent plasticity. Front Cell Neurosci. 2015;9:1–11. doi: 10.3389/fncel.2015.00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas WM, Bengston L, Gilpin NW, Whitcomb BW, Richardson HN. Alcohol binge drinking during adolescence or dependence during adulthood reduces prefrontal myelin in male rats. J Neurosci. 2014;34(44):14777–14782. doi: 10.1523/JNEUROSCI.3189-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassoler F, Byrnes E, Pierce R. The impact of exposure to addictive drugs on future generations: physiological and behavioral effects. Neuropharmacology. 2014;76 doi: 10.1016/j.neuropharm.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson CT, Szutorisz H, Garg P, Martin Q, Landry JA, Sharp AJ, et al. Genome-wide DNA methylation profiling reveals epigenetic changes in the rat nucleus accumbens associated with cross-generational effects of adolescent THC exposure. Neuropsychopharmacol. 2015;40(13):2993–3005. doi: 10.1038/npp.2015.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyrich A, Benz S, Karl S, Jeschek M, Jewgenow K, Fickel J. Paternal heat exposure causes DNA methylation and gene expression changes of Stat3 in Wild guinea pig sons. Ecology and Evolution. 2016;6(9):2657–2666. doi: 10.1002/ece3.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A, Hingson R. The burden of alcohol use: excessive alcohol consumption and related consequences among college students. Alcohol Res. 2013 doi: 10.35946/arcr.v35.2.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt AK, Zavodna M, Viljoen JL, Stanton JL, Gemmell NJ, Jasoni CL. Changes in methylation patterns of Kiss1 and Kiss1r gene promoters across puberty. Genet Epigenet. 2013;5:51–62. doi: 10.4137/GEG.S12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W. Potential roles of noncoding RNAs in environmental epigenetic transgenerational inheritance. [Review] Mol Cell Endocrinol. 2014;398:24–30. doi: 10.1016/j.mce.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.