

Abstract
Arsenite has been shown to induce a variety of oxidative damage in mammalian cells. However, the mechanisms underlying cellular responses to its adverse effects remain unknown. We previously showed that the level of Nrf2, a nuclear transcription factor significantly increased in arsenite-treated human bronchial epithelial (HBE) cells suggesting that Nrf2 is involved in responding to arsenite-induced oxidative damage. To explore how Nrf2 can impact arsenite-induced oxidative damage, in this study, we examined Nrf2 activation and its regulation upon cellular arsenite exposure as well as its effects on arsenite-induced oxidative damage in HBE cells. We found that Nrf2 mRNA and protein levels were significantly increased by arsenite in a dose- and time-dependent manner. Furthermore, we showed that over-expression of Nrf2 significantly reduced the level of arsenite-induced oxidative damage in HBE cells including DNA damage, chromosomal breakage, lipid peroxidation and depletion of antioxidants. This indicates a protective role of Nrf2 against arsenite toxicity. This was further supported by the fact that activation of Nrf2 by its agonists, tertiary butylhydroquinone (t-BHQ) and sulforaphane (SFN) resulted in the same protective effects against arsenite toxicity. Moreover, we demonstrated that arsenite-induced activation of Nrf2 was mediated by the cyclic guanosine monophosphate (cGMP)-protein kinase G (PKG) signaling pathway. This is the first evidence showing that Nrf2 protects against arsenite-induced oxidative damage through the cGMP-PKG pathway. Our study suggests that activation of Nrf2 through the cGMP-PKG signaling pathway in HBE cells may be developed as a new strategy for prevention of arsenite toxicity.
Keywords: Arsenite, Nrf2, cGMP-PKG signaling pathway, Oxidative damage
Introduction
Arsenite is a ubiquitous toxic metalloid in the environment. It has been classified as one of the potent human carcinogens by International Agency for Research on Cancer (Hong et al. 2014; Hughes et al. 2011). Epidemiological studies have demonstrated that chronic exposure to arsenite can significantly increase the risk of a variety of diseases including diabetes (Kuo et al. 2015), cardiovascular diseases (Tsuji et al. 2014), neurodegenerative diseases (Gong and O'Bryant 2010) and cancer (Gentry et al. 2014). It has been shown that arsenite toxicity may result from oxidative stress, DNA damage, inhibition of DNA repair, chromosomal abnormalities, apoptosis, and disruption of cellular signal transduction (Hong et al. 2014; Hubaux et al. 2013; Hughes et al. 2011; Flora 2011; Jiang et al. 2015; Jomova et al. 2011). This is because arsenite can cause production of excessive amount of reactive oxygen species (ROS) by inhibiting energy metabolic enzymes as well as reduce the level of enzymatic and non-enzymatic antioxidants in cells. These result in damage of DNA, proteins and lipids, thereby disrupting the function of these biomacromolecules (Flora 2011). It has been shown that arsenite can induce the formation of superoxide anion (O2•−), hydroxyl radical (•OH), hydrogen peroxide (H2O2), singlet oxygen (1O2) and peroxyl radicals (Flora 2011) as well as the formation of oxidized lipid intermediates such as malondialdehyde (MDA) and 4-hydroxy-nonenal (HNE) that may in turn promote the production of several forms of ROS (Flora 2011; Jomova et al. 2011).
Nuclear factor erythroid 2-related factor 2 (Nrf2), a basic leucine zipper protein that can bind to antioxidant response element promoter sequence, is a redox-response transcription factor that activates the gene expression of antioxidants, detoxification enzymes or efflux pumps and confers cellular protection against oxidative damage in cells (Leinonen et al. 2014; Sinha et al. 2013). Moreover, Nrf2 plays an important role in attenuate inflammation. For instance, Nrf2 can effectively attenuate pro-inflammatory stimuli leading to decreased inflammation and inflammatory damage (So et al. 2008). Activation of Nrf2 exhibits chemoprotective effects during cancer treatment in animals and human patients. This appears result from its effects on suppressing carcinogenesis (Yates et al. 2009).
It has been shown that upregulation of Nrf2 gene expression is essential in protecting cells or animals from oxidative damage induced by environmental pollutants (Umemura et al. 2006). Previous studies indicate that Nrf2-mediated gene transcription is an important cellular mechanism in responding to arsenite exposure (Lau et al. 2013; Ray et al. 2015). Activation of Nrf2 in response to arsenite has been identified in multiple types of cells including hepatocytes (Liu et al. 2013b), keratinocytes (Zhao et al. 2012), lymphocytes (Morzadec et al. 2014) and dendritic cells (Macoch et al. 2015). Studies from our group and others have shown that Nrf2 gene expression was significantly increased by arsenite in a dose-dependent manner in human bronchial epithelial (HBE) (16-HBE, ATCC®CRL-2079™) cell line (Jiang et al. 2014; Viallet et al. 1994). It has been found that Nrf2 can also be activated to protect against arsenite-induced oxidative stress through other pathways. For example, Nrf2 can be activated through increased association between Keap1 and Cul3. This subsequently reduces E3 ubiquitin ligase activity preventing Nrf2 ubiquitination and degradation and increasing Nrf2 protein level (Eggler et al. 2009; Li et al. 2015). In addition, Nrf2 can cooperate with thioredoxin (Trx) to protect against the pathological changes of arsenic-related disease (Bai et al. 2016). On the other hand, Nrf2 can be downregulated by bach1 (BTB and CNC homology 1) through its interaction with the small Maf protein to form Nrf2/small Maf heterodimers. Bach1 can compete with Nrf2 to form a Bach1/small Maf heterodimer, thereby suppressing Nrf2 activation induced by arsenite (Liu et al. 2013a). However, it remains unknown how arsenite can activate Nrf2.
A recent report has indicated that release of Nrf2 proteins from cytoplasm, along with its translocation to the nucleus and its activation on antioxidant response elements may be promoted by the nitric oxide (NO)-cyclic guanosine monophosphate (cGMP)-protein kinase G (PKG) signaling pathway (Astort et al. 2014), which regulates cellular process including oxidative damage (Inserte et al. 2013). However, it remains unknown whether activation of Nrf2 by arsenite can be mediated through this pathway. We further hypothesize that Nrf2 protects against arsenite-induced oxidative damage through NO-cGMP-PKG signaling pathway. Since lung is the main target organ for arsenite toxicity, we tested this hypothesis in HBE cells. We showed that arsenite activated Nrf2 in HBE cells, and this significantly reduced arsenite-induced oxidative damage. However, we found that Nrf2 was activated by arsenite directly through the cGMP-PKG signaling pathway rather than the NO-dependent cGMP-PKG pathway. Moreover, activation of Nrf2 increased the levels of antioxidants, thereby protecting against arsenite-induced oxidative damage in HBE cells. Our discoveries provided new insight into the mechanisms underlying arsenite-induced oxidative damage. Our study will further help to identify Nrf2 and the cGMP-PKG pathway as a target for prevention of arsenite toxicity.
Materials and Methods
Chemical and reagents
Arsenite, purity≥99.0%, was obtained from Fluka Chemical Corp. (Buchs, Switzerland); The Alama blue cell viability assay kit was from Nanjing KeyGen Biotech (Nanjing, China). The lactate dehydrogenase (LDH)-release assay kit, the superoxide dismutase (SOD) activity assay kit, kits for measuring malondialdehyde (MDA) and NO levels, and kits for measuring total nitric oxide synthase (tNOS) and inducible NOS (iNOS) activity were all purchased from Nanjing Jiancheng Institute of Bioengineering (Jiangsu, China). The cGMP and PKG enzyme-linked immunosorbent assay (ELISA) kits were from Cell Signaling Technology (Beverly, MA, USA). Dulbecco’s modified Eagle’s medium (DMEM) was from Gibco Life Technologies (Grand Island, NY, USA). A ROS fluorescent probe 2’7’-dichlorofluorescin diacetate (DCFH-DA) was obtained from Applygen Technologies Inc., (Beijing, China). Low melting point agarose (LMPA), normal melting point agarose (LMPA), acridine orange and ethidium bromide were all from Amresco (Solon, OH, USA). N-acetylcysteine (NAC) and tertiary butylhydroquinone (t-BHQ) were purchased from Beijing Solarbio Science Technology Co., Ltd. (Beijing, China). Sulforaphane (SFN) was from Xian Haoxuan Biotechnology Co. (Xian, China). The Nuclear and cytoplasmic extraction kit and KT5823 were purchased from Beyotime Institute of Biotechnology (Jiangsu, China). Antibodies against Nrf2 and lamin B were obtained from Wuhan Boster Biological Technology Co. Ltd (Wuhan, China).
Cell culture
Human papillomavirus 16 E6/E7 transformed normal HBE cell line was generously provided by Stem Cells and Tissue Engineering Laboratory, State Key Laboratory of Biotherapy, Sichuan University, China (Chengdu, China). Cells were maintained in high glucose DMEM supplemented with 10% (v/v) fetal bovine serum, 100 units/mL penicillin and 100 μg/mL streptomycin. Cells were cultured at 37°C in a humidified atmosphere containing 5% CO2 and 95% air.
Alamar blue assay
Cell viability was determined by Alamar blue assay as described previously (Uzunoglu et al. 2010). Briefly, cells were seeded on a 96-well plate with 1 × 104 cells per well. After arsenite treatment, 10 μL Alama blue solution was added in each well and incubated with cells at 37 °C for 24 h. Absorbance at 570 nm was measured with a microplate spectrophotometer (Multiskan™ GO, Thermo Fisher Scientific, Inc., Waltham, MA, USA). Background absorbance at 600 nm was subtracted. The normalized index (Ro) and the reduction rate (AR570) were calculated according to the equation: Ro=Ao570/Ao600, AR570 (%)=[A570-(A600 × Ro)] ×100%.
Lactate dehydrogenase (LDH) release assay
LDH activity in the supernatant and total cell lysates were detected using LDH activity assay kit. In brief, supernatant resulting from treated cells was collected for measuring LDH activity. Triton X-100 (1%, v/v) was used to lyse cells for 10 min. LDH activity was then measured according to the manufacturer’s instruction. Absorbance at 440 nm was measured using a spectrophotometer (V-1100D, Mapada instruments Co., Ltd. Shanghai, China). LDH release rate was calculated according to the equation: LDH release rate (%)=LDH activity in supernatant/(LDH activity in supernatant + LDH activity in cells).
Measurement of ROS production, levels of GSH, MDA and SOD activity
Production of intracellular ROS was determined by an oxidation-sensitive fluorescent probe DCFH-DA as described previously (Gu et al. 2014). Briefly, cells treated with different concentrations of arsenite (5 μM-20 μM) were incubated with DCFH-DA at the final concentration of 2.5 μM for 1 h at 37°C in the dark. Cells were then collected, washed and resuspended in ice-cold PBS. The production of ROS was measured immediately by flow cytometry (Beckman Coulter, FC500, FL, USA). The concentration of GSH was measured as described previously (Chen et al. 2013a). Cellular levels of a lipid peroxidation product, MDA and SOD were measured by determining thiobarbituric acid reactivity, and the SOD activity for inhibiting the oxidation of hydroxylamine hydrochloride by superoxide, respectively as described previously (Jiang et al. 2014; Jiang et al. 2013). Absorbance at 420 nm (GSH concentration measurement), 550 nm (SOD activity measurement) and 532 nm (MDA concentration measurement), was measured by a spectrophotometer (V-1100D, Mapada instruments Co., Ltd. Shanghai, China).
Measurement of PKG and cGMP by enzyme linked immunosorbent assay (ELISA) assay
The concentrations of cGMP and activity of PKG were measured with ELISA. Cell extracts were made by lysing cells with freeze-thaw for three times. Fifty microgram proteins in total from each sample were analyzed. Absorbance at 450 nm was measured using a microplate spectrophotometer (Multiskan™ GO, Thermo Fisher Scientific Inc., Waltham, MA, USA). The concentration of cGMP and activity of PKG were calculated according to the standard curves prepared from standard solutions. All experiments were conducted in triplicate.
Measurement of the concentrations of NO and activities of total nitric oxide synthase (tNOS) and inducible NOS (iNOS)
After treatment with arsenite, cells were harvested by 0.2% EDTA (w/v), washed with cold PBS, and lysed. Cell extracts were made and used for subsequent experiments. The total NO level in cells was determined by the level of nitrite converted from nitrate by nitrate reductase. The NOS activity in HBE cells was determined by measuring the level of L-citrulline generated from L-arginine. The level of NO and activities of total NOS and iNOS were measured by spectrophotometer (V-1100D, Mapada instruments Co., Ltd. Shanghai, China) and normalized by protein concentrations.
Western blot analysis
Western blot was conducted according to the procedures as previously described (Chen et al. 2013b). The nuclear proteins were isolated by Nuclear and Cytoplasmic Extraction Kit according to the manufacturer’s instruction as described previously (Chen et al. 2013b). Proteins were denatured and subjected to SDS-PAGE and then transferred to polyvinylidencedifluoride membrane. The membrane was incubated with antibodies against Nrf2 (1:500) and lamin B (1:400) overnight at 4°C. The intensities of the protein were obtained and quantified using the software Quantity one (Bio-Rad, USA).
Comet assay
Alkaline comet assay was conducted according to the protocols described previously (Chen et al. 2013b). 1 × 105 cells were harvested. Cell viability was checked by trypan blue exclusion assay. Subsequently, cell suspensions were mixed with 80 μL 0.65% low melting agarose at 37°C and spread onto a microscope slide pre-coated with 100 μL 0.8% normal melting agarose. After gel solidification at 4°C for 10 min, slides were immersed in freshly prepared lysis solution in the dark at 4°C for 1 h, and then soaked in electrophoresis buffer for 30 min to allow DNA unwinding. Cells were then subjected to electrophoresis at 0.75 V/cm for 30 min. Slides were then stained with 20 μg/mL ethidium bromide and observed under a fluorescence microscopy (DMLB2, Leica, Wetzlar, Germany) at 200× magnification. Quantification of DNA damage was evaluated by measuring tail length, tail DNA and olive tail moment (OTM) of comet cells with Comet Assay Software Project (CASP, Free Software Foundation. Inc. Boston, MA, USA).
Micronucleus assay
Micronucleus assay was performed according to the procedures described previously (Chen et al. 2014). 5× 105 cells per well were collected and resuspended in 0.075 M potassium chloride solutions for 5 min. Subsequently, cells were fixed in freshly prepared methanol-glacial acetic acid (3:1, v/v) solution for three times. After re-fixation with methanol-glacial acetic acid (99:1, v/v), cell suspension was dropped onto pre-cold glass slides and then stained with 40 μg/mL acridine orange. Cells were observed under a fluorescence microscopy (DMLB2, Leica, Wetzlar, Germany) at 400× magnification. For each treatment, quadruple slides were analyzed. For each slide, 1000 cells were randomly chosen for counting the number of micronucleated cells.
Immunofluorescence assay
5 × 105 cells were seeded onto a coverslip (24 mm × 24 mm). Cells were incubated with 4% paraformaldehyde for 10 min at room temperature and then rinsed with Tris-Buffer saline-Tween 20. Cells were subsequently blocked by 5% bovine serum albumin for 1 h, and then incubated with an antibody against Nrf2 (1:100) at 4°C overnight. Cy3-labeled secondary antibody was further incubated for additional 1 h. A coverslip was subsequently rinsed and incubated with antifade mounting medium. Cells were observed under a fluorescence microscopy. Fluorescent intensity was obtained and quantified by Image-Pro Plus software.
Quantitative PCR analysis
The level of Nrf2 mRNA in HBE cells was determined by quantitative PCR analysis. Total RNA was isolated by the Total RNA Extraction kit (KeyGen Biotech Co. Ltd. Nanjing, China) according to the manufacture’s protocol. Subsequently, the AccuPower® RocketScriptTM RT PreMix kit was used to synthesize cDNAs (BioNeer, Daejeon, South Korea), and quantitative PCR was carried out with AccuPower®2X GreenstarqPCR Master (BioNeer, Daejeon, South Korea). The primers of NFE2L2 (Nrf2) and GAPDH were synthesized by Bioneer Inc. (validated primers for amplifying Nrf2 sequence: No. P164742; the validated primers for amplifying GAPDH sequence: No. P267613)(Koo et al. 2013). PCR reactions were performed under the condition: 94 °C for 5 min, followed by 40 cycles of 95°C for 10 sec, 58°C for 25 sec and 72°C for 30 sec. The melting curve analysis of PCR product showed only one peak for each type of PCR product. mRNA relative abundance was calculated and normalized to the mean of GAPDH mRNA level.
Knockdown of Nrf2 gene with siRNA
Cells were cultured in a six-well dish to 30–50% con?uency for transfection. Nrf2-siRNAs (sense strand sequence: 5’-CAGCUAUGGAGACACACUA-3’; antisense strand sequence: 3’-UAGUGUGUCUCCAUAGCUG-5’) were purchased from BioNeer (Daejeon, South Korea). Transient transfection was performed with lipofectamine 2000 (Invitrogen, Carlsbad, CA). Nrf2-siRNA (40 pmol/per well) and negative control siRNA were mixed with lipofectamine 2000. The transfection reagent mixture was then added to a six-well culture dish with 0.1 mL free DMEM of FBS. Culture medium was replaced with DEAE with 10% FBS after 5 h. Cells were harvested for subsequent experiments.
Overexpression of Nrf2
Human NFE2L2 (Nrf2) expression plasmid was obtained by subcloning the PCR-amplified full-length Nrf2 cDNA into BamH1 and Not1-HF sites of the pCDH-CMV-MCS-EF1-copGFP (CD511B-1) expression vector. The sequence of the Nrf2 cDNA was confirmed by DNA sequence analysis. Subsequently, the lentiviral vectors and packaging vectors dR8.9 (pax-2) and VSVG were co-transfected into cells for packaging the lentivirus. GFP-positive cells were sorted and isolated by flow cytomerty for verifying Nrf2 expression. Overexpression of Nrf2 was confirmed by Western blot and quantitative PCR (Supplement Figure S1).
Statistical analysis
All experiments were conducted independently for at least three times. Results were reported as mean±standard deviation (S.D.). Significant differences among different groups were assessed by one-way analysis of variance (ANOVA) with the least significant difference (LSD) t-test. Non-parametric Kruskal-Wallis test was applied if original data had heterogeneity of variance. All statistical analysis was performed by Statistical Program for Social Sciences (SPSS) software, version 17.0 (SPSS Inc., Chicago, IL, USA). P<0.05 denotes a significant difference among different groups.
Results
Arsenite exhibits cytotoxicity in HBE cells through oxidative damage
Our previous work has shown that cell viability of HBE cells treated by arsenite was markedly reduced (Chen et al. 2015). In this study, we further examined arsenite cytotoxicity of HBE cells with a new sensitive and non-toxic assay, the Alamar blue assay. We found that HBE cell viability was significantly decreased by arsenite at a concentration of 10 μM or above with 24 h exposure (Figure 1a). To determine whether arsenite can cause cytotoxicity in HBE cells by directly promoting the release LDH, we examined this in HBE cells treated by various concentrations of arsenite ranging from 5 μM to 20 μM with 6 h, 12 h and 24 h exposure (Figure 1b). The results showed that arsenite significantly increased release of LDH in HBE cells in a dose- and time-dependent manner with 25% of LDH release resulting from cellular exposure to 20 μM arsenite (Figure 1b). The results indicated that arsenite exhibited toxicity in HBE cells.
Figure 1. Arsenite reduced cell viability and induced oxidative damage in HBE cells.







(a) Cells were treated with 1 μM-60 μM arsenite for 24 h and then subjected to Alama Blue assay for obtaining AR570 value. (b) Cells were treated with 0, 5 μM, 10 μM and 20 μM arsenite for 6, 12 and 24 h, respectively, and LDH release rate was measured as described in the Materials and Methods. (c) Cells were treated with 5 μM, 10 μM, 20 μM arsenite for 24 h or pretreated with 10 mM NAC for 2 h and subsequently treated with 10 μM arsenite for 24 h. The level of ROS was detected with DCFH-DA probe through flow cytometry. The Gm value was obtained from Windows Multiple Document Interface for Flow Cytometry (Version 2.8). Representative images of ROS determination were showed in (d). The levels of MDA (e), SOD (f) and GSH (g) in untreated HBE cells and cells treated with various concentrations of arsenite were determined as described in Materials and Methods. “*” denotes a significant difference (P<0.05) was detected between treated cells and untreated cells, whereas “**” denotes a significant difference (P<0.05) was detected between cells pretreated with NAC and cells treated with 10 μM arsenite alone.
Because the concentration of arsenite in the environment ranges from 20 to 2000 ppb, which is equivalent to 0.3 μM to 27 μM (Gentry et al. 2014; Schoen et al. 2004; Snow et al. 2005), we examined oxidative damage and antioxidant response of HBE cells under treatment with 5 μM to 20 μM arsenite for 24 h by measuring cellular level of ROS, a lipid peroxidation product, malondialdehyde (MDA) as well as the level and activity of antioxidants, glutathione (GSH) and superoxide dismutase (SOD). The results showed that 10 μM and 20 μM arsenite significantly increased ROS level (Figure 1c and 1d) and the level of MDA (Figure 1e) in cells. In contrast, the same concentrations of arsenite significantly reduced the activities of SOD in a dose-dependent manner (from 10 to 20 μM) (Figure 1f). Surprisingly, we found that low concentration of arsenite at 5 μM significantly increased the level of GSH in cells (Figure 1g). However, arsenite ranging from 10 μM to 20 μM significantly decreased the level of GSH in cells (Figure 1g). Employing a potent antioxidant, N-acetylcysteine (NAC) that can inhibit oxidative damage, we showed that pretreatment of HBE cells with 10 mM NAC for 2 h significantly attenuated arsenite-induced oxidative damage by decreasing the levels of ROS and MDA and increasing the levels of GSH and SOD in cells (Figure 1c-1g). This further demonstrated that arsenite induced oxidative damage.
Employing comet assay and micronucleus assay, we found that 10 μM and 20 μM arsenite increased the tail length, the percentage of tail DNA and Olive tail moment of comet cells (Figure 2a-2d) indicating that arsenite at these doses induced single-strand DNA breaks in HBE cells. In consistency with this, we found that increasing concentrations of arsenite significantly increased the frequency of micronucleated cells (Figure 2e) demonstrating the genotoxic effects of arsenite in HBE cells. To determine whether arsenite-induced genotoxicity resulted from oxidative damage, HBE cells were pretreated with 10 mM NAC for 2 h followed by treatment of 10 μM arsenite. The results showed that NAC significantly reduced DNA damage and chromosomal breakage induced by arsenite (Figure 2a-2e). Taken together, our results indicated that arsenite-induced DNA damage and chromosomal breakages through oxidative damage.
Figure 2. Arsenite induced DNA damage and chromosomal breakage in HBE cells.





Cells were treated with 5 μM, 10 μM, 20 μM arsenite for 24 h or pretreated with 10 mM NAC for 2 h and subsequently treated with 10μM arsenite. Arsenite-induced DNA damage and chromosomal breakage were determined by comet assay (a)-(d) and micronucleus assay (e), respectively. Representative images in comet assay were showed (200×) and illustrated in (a). The effects of arsenite on tail DNA (%), tail length and OTM were illustrated in (b)-(d). “*” denotes a significant difference (P<0.05) detected between treated cells and untreated cells, whereas “**” denotes a significant difference (P<0.05) detected between cells pretreated with NAC and cells treated with 10 μM arsenite.
Arsenite activated Nrf2 signaling pathway by stimulating Nrf2 gene expression in HBE cells
Nrf2-mediated signaling pathway is a major mechanism to protect against oxidative damage in cells (Leinonen et al. 2014). Because Nrf2 is expressed constitutively in the cytoplasm and translocates to the nucleus in responding to oxidative damage (Kaspar et al. 2009), we initially determined the level of Nrf2 protein in the nucleus of HBE cells. We found that 10 μM arsenite increased the level of Nrf2 protein in the nucleus within 12 h (Figure 3a-3c). This effect reached the highest level within 24 h (Figure 3a-3c). Increased concentrations of arsenite ranging from 5 μM to 20 μM also significantly increased Nrf2 protein level in the nucleus (Figure 3a-3c). This was further confirmed by the results from immunofluorescence staining of Nrf2 showing that 5-20 μM arsenite increased Nrf2 protein level in the nucleus by 2-2.5-fold (Figure 3d-3e). Consistent with this, we also found that 5-20 μM arsenite significantly increased the level of Nrf2 mRNA with increasing doses and exposure time (Figure 3f-3g). The results indicated that Nrf2 gene expression was significantly stimulated by arsenite in an exposure time- and dose-dependent manner. Thus, our results demonstrated that arsenite activated Nrf2 suggesting that the Nrf2 signaling pathway protected cells from arsenite-induced oxidative damage.
Figure 3. Arsenite activated Nrf2 in HBE cells.
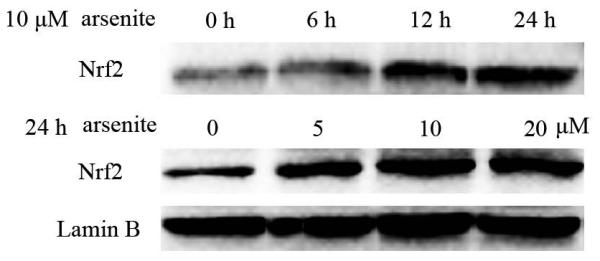






Cells were treated with 5 μM, 10 μM and 20 μM arsenite for 24 h or 10 μM arsenite for 6 h, 12 h and 24 h, respectively. The effects of arsenite on Nrf2 protein level at different time points and doses were shown in (a)-(c). The level of Nrf2 in the nucleus of HBE cells treated with arsenite was determined by immunofluorescence (d) and (e). Representative images (400×) of immunofluorescence results were showed in (d). Fluorescence intensity was obtained from Image-Pro Plus software. The mRNA level of Nrf2 in HBE cells was illustrated in (f) and (g). "*" denotes a significant difference (P<0.05) detected between treated and untreated cells.
Nrf2 combats arsenite-induced oxidative damage in HBE cells
To further explore if Nrf2 may reduce arsenite-induced oxidative damage in HBE cells, we examined arsenite-induced oxidative damage under a low and high level of Nrf2 by knocking down and overexpressing Nrf2 in HBE cells. We found that with treatment of 10 μM arsenite for 24 h, more comet cells occurred in HBE cells with Nrf2 gene knockdown than normal cells (Figure 4a, compare the panel in the middle with the one on the left). Accordingly, in Nrf2 knockdown cells, arsenite significantly increased tail length of comet cells, OTM and tail DNA (Figure 4b). In contrast, under the same treatment, Nrf2 overexpression HBE cells exhibited significantly lower number of comet cells and lower level of OTM and tailed DNA than normal cells (Figure 4a, compare the panel on the left with that on the right)(Figure 4b). The results indicated that a low level of Nrf2 promoted accumulation of arsenite-induced oxidative DNA damage, whereas a high level of Nrf2 reduced arsenite-induced oxidative DNA damage. Moreover, we found that knockdown of Nrf2 significantly increased the frequency of micronucleated cells from HBE cells treated with 10 μM arsenite compared with normal cells (Figure 4c), whereas overexpression of Nrf2 significantly decreased the frequency of micronucleated cells (Figure 4c). This indicated that Nrf2 reduced arsenite-induced oxidative DNA damage and chromosomal breakage. Based on the results, we then hypothesize that Nrf2 reduces oxidative DNA damage by increasing the level of antioxidants in HBE cells. To test this, we examined the level of GSH, SOD and LDH release in the normal, Nrf2 knockdown and Nrf2 overexpression HBE cells treated by 10 μM arsenite. We found that Nrf2 gene knockdown significantly reduced cellular level of antioxidants, GSH and SOD (Figure 4d), while it increased the level of MDA in HBE cells (Figure 4d). Overexpression of Nrf2 increased the levels of GSH and SOD in HBE cells by about 1.5-fold compared with normal cells (Figure 4d), while it also significantly reduced the level of MDA induced by arsenite (Figure 4d). In addition, we found that knockdown of Nrf2 gene increased cellular LDH release, whereas overexpression of the gene reduced arsenite-induced LDH release by about 2-fold compared with normal cells (Figure 4e). In consistency with these results, we found that pretreatment of cells with Nrf2 agonist, t-BHQ (20 μM) and SFN (10 μM) also led to the protective effects against arsenite-induced oxidative damage by significantly reducing the elevated levels of ROS (Figure 5a and 5b), oxidative DNA damage (Figure 5c and 5d), chromosomal breakage (Figure 5e) and MDA (Figure 5f) induced by arsenite. Moreover, it attenuated the reduction of GSH and SOD in HBE cells treated by arsenite (Figure 5f). In summary, our results indicated that activation of Nrf2 protected HBE cells against arsenite-induced oxidative damage by inhibiting cellular production of ROS, MDA and LDH leakage and increasing the levels of GSH and SOD.
Figure 4. The effects of Nrf2 on arsenite-induced oxidative damage in HBE cells.





The effects of Nrf2 on DNA damage induced by arsenite were examined under Nrf2 gene downregulation and overexpression (a) and (b) with treatment of 10 μM arsenite for 24 h. Representative images of comet assay were shown in (a) (200×). The effect of Nrf2 on arsenite-induced chromosomal breakage was determined under the same condition (c), whereas the effects of the protein on the level of GSH, SOD, MDA (d) as well as LDH release (e) were also determined. "*" denotes a significant difference (P<0.05), detected between HBE cells with Nrf2 gene knockdown or Nrf2 overexpression and normal HBE cells.
Figure 5. The effects of agonists of Nrf2 on arsenite-induced oxidative damage in HBE cells.






The effects of agonists of Nrf2, t-BHQ and SFN on arsenite-induced oxidative damage were determined as described in Materials and Methods. The effects of t-BHQ and SFN on arsenite-induced production of ROS were illustrated in (a) and (b). The effects of t-BHQ and SFN on arsenite-induced DNA damage determined by comet assay were shown in (c) and (d). The effects of t-BHQ and SFN on arsenite-induced chromosomal breakage was determined by micronucleus assay (e), whereas their effects on the level of GSH and MDA as well as the activity of SOD in HBE cells treated by 10 μM arsenite were illustrated in (f). "*" denotes a significant difference (P<0.05) detected between HBE cells treated with arsenite alone and cells treated with arsenite along with t-BHQ or SFN.
Arsenite activates Nrf2 signaling pathway through activating cGMP-protein kinase G (PKG) signaling pathway directly rather than through a nitric oxide (NO)-dependent cGMP-PKG pathway in HBE cells
Activation of NO-cGMP-PKG signaling pathway has been shown to be critical for regulating cellular physiological function (Inserte et al. 2013). It is also involved in some pathophysiological conditions including oxidative damage. It has been shown that NO can bind to guanylate cyclase, increasing cGMP production and activating PKG (Gonzalez et al. 2008). This can then induce Nrf2 translation to the nucleus, thereby activating antioxidant-related genes (Astort et al. 2014). To explore a possibility that arsenite may activate Nrf2 through a NO-dependent cGMP-PKG pathway, we initially examined the effects of arsenite on the levels of NO, NOS, tNOS and iNOS, cGMP and PKG in HBE cells treated with increasing concentrations of arsenite (5 μM-20 μM) for 12, 16 and 24 h. Surprisingly, we found that 10 μM-20 μM arsenite significantly decreased the levels of NO, tNOS and iNOS (Figure 6a), indicating that arsenite inhibited the synthesis of NO by inhibiting the activities of tNOS and iNOS. More interestingly, we found that exposure to 20 μM arsenite for 12 h still led to increased cellular levels of cGMP and PKG (Figure 6b and 6c). This indicated that arsenite activated the cGMP-PKG pathway independent of NO. To further determine whether cGMP-PKG pathway is necessary in mediating arsenite-induced activation of Nrf2, we examined the level of Nrf2 in HBE cells pretreated with 0.25 μM KT5823, a PKG inhibitor for 2 h. The results showed that inhibition of PKG by KT5823 significantly reduced the increased of Nrf2 mRNA (Figure 7a) and protein level (Figure 7b-7c) induced by arsenite as well as inhibited Nrf2 nuclear translocation (Figure 7d-7e). The results indicated that arsenite-induced Nrf2 activation was directly mediated by cGMP-PKG pathway. In summary, our results indicated that arsenite activated Nrf2 by directly activating cGMP-PKG signaling pathway rather than through NO in HBE cells.
Figure 6. Arsenite increased the levels of PKG and cGMP in HBE cells independent of NO.



The effects on arsenite on the levels of NO, tNOS and iNOS in HBE cells were determined as described in Materials and Methods. (a). Cells were treated with 5 μM, 10 μM and 20 μM arsenite for 24 h, respectively. "*" denotes a significant difference (P<0.05) detected in untreated HBE cells and cells treated with various concentrations of arsenite. The levels of PKG and cGMP in HBE cells treated with various concentrations of arsenite (5 μM-20 μM) for 6 h-24 h were determined as described in Materials and Methods. The effect of arsenite on PKG level in HBE cells was illustrated in (b), whereas its effect on cellular cGMP level was shown in (c). "*" denotes a significant difference detected between untreated cells and cells treated with various concentrations of arsenite for 6 h-24 h.
Figure 7. Inhibition of cGMP-PKG pathway suppressed arsenite-induced Nrf2 activation in HBE cells.

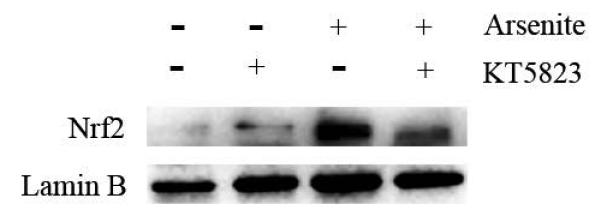



Cells were treated with 10 μM arsenite for 24 h or pretreated with KT5823 for 2 h followed by treatment of 10 μM arsenite for additional 24 h. The effect of KT5823 on the mRNA level of Nrf2 was determined by quantitative real-time PCR (a). The effect of KT5823 on Nrf2 protein level was determined with Western blot (b) and (c). The effect of KT5823 on the protein level of Nrf2 in nucleus was determined by immunofluorescence (d) and (e). The immunofluorescence intensity was obtained from Image-Pro Plus software. “*” denotes a significant difference (P<0.05) detected between cells treated with arsenite alone and cells treated with arsenite along with KT5823.
Discussion
Oxidative damage caused by overproduction of ROS and depletion of cellular antioxidants has been shown as one of the mechanisms underlying arsenite toxicity (Flora 2011; Gupta et al. 2013; Jomova et al. 2011). Arsenite-induced overproduction of ROS can directly oxidize DNA bases and proteins, lipids, resulting in inactivation of enzyme activities, lipid peroxidation, oxidative DNA damage and chromosomal breakage (Flora 2011). On the other hand, arsenite-induced ROS can deplete enzymatic and non-enzymatic antioxidants, such as SOD and GSH in cells. This in turn can reduce the ability for cells to scavenge ROS (Flora 2011). In this study, we showed that arsenite induced cytotoxicity and oxidative damage in HBE cells (Figure 1a) by increasing the levels of LDH release (Figure 1b), ROS (Figure 1c-1d) and MDA (Figure 1e) and decreasing the level of enzymatic and non-enzymatic antioxidants in HBE cells (Figure 1f-1g). The results also indicated that arsenite-induced ROS overproduction and oxidative damage resulted in DNA damage and chromosomal breakage (Figure 2) through depletion of cellular antioxidants. This was supported by the results showing that supply of an antioxidant, NAC to cells treated by arsenite, significantly relieved arsenite-induced oxidative damage (Figure 1 and Figure 2). We further demonstrated that increasing concentrations of arsenite under various exposure time points significantly increased the level of Nrf2 protein and its nuclear translocation in HBE cells (Figure 3). Moreover, we showed that knockdown of Nrf2 gene in HBE cells promoted arsenite-induced oxidative damage (Figure 4), whereas overexpression of Nrf2 protein in cells significantly reduced arsenite-induced oxidative damage (Figure 4). This indicated that Nrf2 activation protected against arsenite-induced oxidative damage. This was further supported by the fact that HBE cells treated with Nrf2 agonists, t-BHQ and SFN along with arsenite, exhibited reduced oxidative damage compared with cells treated with arsenite along (Figure 5). We further demonstrated that arsenite-induced Nrf2 activation was not mediated by a NO-dependent cGMP-PKG signaling pathway (Figure 6a). Instead its activation was accomplished directly by activation of cGMP-PKG signaling pathway (Figure 6b and 6c). This was also supported by the results showing that inhibition of PKG attenuated arsenite-induced Nrf2 activation in HBE cells (Figure 7). Our results allowed us to propose a model for cellular response to arsenite-induced oxidative damage through activation of Nrf2, in which arsenite induces oxidative damage while it activates the cGMP-PKG signaling pathway that in turn activates Nrf2 in cells. This subsequently increases the levels of antioxidants such as GSH and the activity of SOD, thereby relieving aresnite-induced overproduction of ROS and its-resulted oxidative damage on DNA and lipids (Figure 8).
Figure 8. A model for arsenite-induced Nrf2 activation to protect against oxidative damage via the cGMP-PKG signaling pathway in HBE cells.
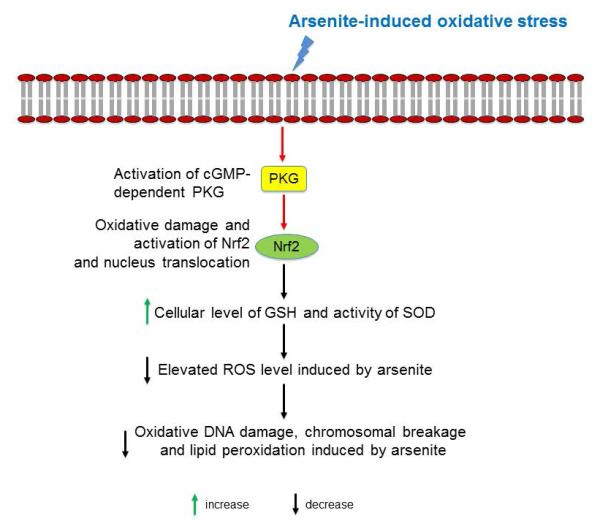
Arsenite induces oxidative damage while it activates cGMP-PKG signaling pathway by increasing cellular level of cGMP and PKG in HBE cells. This in turn increases the level of Nrf2 which subsequently increases cellular level of GSH and SOD activity, thereby reducing the elevated level of ROS induced by arsenite. This then significantly reduces oxidative DNA damage, chromosomal breakage and lipid peroxidation induced by arsenite.
Arsenite has been shown as to induce oxidative stress through modulating cellular Nrf2 signaling pathway (Ma 2013). Yet, it remains to be identified which genes mediate arsenite-induced activation of Nrf2. In this study, for the first time, we revealed that arsenite-induced oxidative damage and activation of Nrf2 were mediated by the cGMP-PKG signaling pathway. We showed that inhibition of this pathway by a specific PKG inhibitor, KT5823, significantly decreased the elevated Nrf2 expression in HBE cells induced by arsenite. This subsequently abolished arsenite-induced oxidative damage. Thus, our results indicate that inhibition of the cGMP-PKG pathway can alleviate oxidative damage induced by arsenite in HBE cells further suggesting that inhibition of PKG with its inhibitors can be potentially developed as a remedy for relieving arsenite toxicity.
It should be noted that arsenite markedly enhanced the level of GSH at a low concentration, 5 μM (Figure 1g), but decreased GSH level at high doses ranging from 10 to 20 μM (Figure 1g). The results were consistent with our previous findings (Jiang et al. 2014). Maiti et al., also revealed that a low dose of arsenite significantly increased GSH level in hepatocytes in mice (Maiti and Chatterjee 2001). The effect was proposed as a low dose adaptive response by arsenite, which is also named as hormesis of arsenite. Previous studies have demonstrated that arsenite can facilitate cell proliferation at the doses below 2.5 μM, whereas it inhibits cell growth at high doses (>5 μM). The phenomenon of hormesis has been also observed in cells treated by heavy metals, antibiotics and pro-oxidants (Mattson 2008). This further indicates that a low dose of arsenite increased the levels of cellular antioxidant that were sufficient to combat a relatively small amount of ROS. However, with increasing doses of arsenite in cells, antioxidants in cells were ultimately depleted, thereby leading to failure of protecting cells from oxidative damage by cellular antioxidants.
Nrf2 has been shown as the master regulator of a cellular defense mechanism against toxic insults (Lau et al. 2013; Sinha et al. 2013). The protein is retained in the cytoplasm and maintained at a low level by its negative regulator, Kelch-like ECH associated protein 1 (Keap1). Upon oxidative damage in cells, Nrf2 gene expression is activated while Nrf2 protein is translocated to the nucleus. Subsequently it binds to the antioxidant response element in the promoter region of cytoprotective genes for detoxification and elimination of toxicants such as arsenite (Lau et al. 2013). Our study showed that arsenite significantly increased Nrf2 gene expression (Figure 3a-3c and 3f-3g) and nuclear translocation of Nrf2 protein in HBE cells (Figure 3d-3e). This in turn reduced arsenite-induced oxidative damage in cells. It is noteworthy that our results showed a simultaneous increase in both oxidative stress and Nrf2 in HBE cells that were treated by 10 μM and 20 μM arsenite. The results further indicate that HBE cells effectively increased the cellular level of Nrf2 to combat the oxidative stress induced by the arsenite exposure at these doses. It is conceivable that arsenite exposure at high doses and/or long period of time would result in massive oxidative stress that in turn deplete Nrf2 ultimately leading to a significant decrease in cellular Nrf2 level.
In this study, we showed that NO, an important cell signaling molecule that regulates multiple physiological processes, was not involved in arsenite-induced oxidative damage. It has been found that NO plays an active role in mediating oxidative damage (Drechsel et al. 2012; Förstermann and Sessa 2012) and stimulation of cellular guanylate cyclase activity and production of cGMP that in turn activates protein kinase PKG (Takahashi et al. 2008). However, we found that the activities of tNOS, iNOS were all decreased upon cellular exposure to arsenite (Figure 6a). Since endogenous synthesis of NO is catalyzed by NOS, it is possible that decreased NO in HBE cells may result from inhibition of NO synthesis. In supporting this notion, we found that arsenite at above 5 μM significantly reduced the NOS activity and NO level in HBE cells (Figure 6a). Interestingly, we found that arsenite still increased cellular levels of cGMP and PKG (Figure 6b and 6c) with decreased levels of NO and NOS indicating that arsenite directly activated cGMP-PKG signaling pathway that in turn activated Nrf2.
In summary, in this study, we demonstrated that Nrf2 was activated during arsenite-induced oxidative damage through the cGMP-PKG signaling pathway in HBE cells. Activation of Nrf2 effectively reduced arsenite-induced production of ROS and oxidative damage by increasing cellular level of GSH and activity of SOD, thereby combating arsenite-induced depletion of antioxidants in cells. We suggest that PKG inhibitors can be developed as a strategy for relieving arsenite toxicity.
Supplementary Material
Acknowledgment
This study was supported by grants from National Science Foundation of China (No.81372945) to Z. Zhang and National Institutes of Health (R01ES023569) to Y. Liu.
Footnotes
Conflict of Interests
The authors declare that they have no conflict of interests.
Reference
- Astort F, Mercau M, Giordanino E, Degese MS, Caldareri L, Coso O, Cymeryng CB. Nitric oxide sets off an antioxidant response in adrenal cells: involvement of sGC and Nrf2 in HO-1 induction. Nitric Oxide. 2014;37:1–10. doi: 10.1016/j.niox.2013.12.006. [DOI] [PubMed] [Google Scholar]
- Bai J, Yao X, Jiang L, Qiu T, Liu S, Qi B, Zheng Y, Kong Y, Yang G, Chen M. Taurine protects against As2O3-induced autophagy in pancreas of rat offsprings through Nrf2/Trx pathway. Biochimie. 2016;123:1–6. doi: 10.1016/j.biochi.2016.01.002. others. [DOI] [PubMed] [Google Scholar]
- Chen C, Gu S, Jiang X, Zhang Z. Nuclear translocation of nuclear factor kappa B is regulated by G protein signaling pathway in arsenite-induced apoptosis in HBE cell line. Environ Toxicol. 2015 doi: 10.1002/tox.22183. doi: 10.1002/tox.22183. [DOI] [PubMed] [Google Scholar]
- Chen C, Jiang X, Hu Y, Zhang Z. The protective role of resveratrol in the sodium arsenite-induced oxidative damage via modulation of intracellular GSH homeostasis. Biol Trace Elem Res. 2013a;155:119–131. doi: 10.1007/s12011-013-9757-x. [DOI] [PubMed] [Google Scholar]
- Chen C, Jiang X, Lai Y, Liu Y, Zhang Z. Resveratrol protects against arsenic trioxide-induced oxidative damage through maintenance of glutathione homeostasis and inhibition of apoptotic progression. Environ Mol Mutagen. 2014;56:333–346. doi: 10.1002/em.21919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Jiang X, Zhao W, Zhang Z. Dual role of resveratrol in modulation of genotoxicity induced by sodium arsenite via oxidative stress and apoptosis. Food Chem Toxicol. 2013b;59C:8–17. doi: 10.1016/j.fct.2013.05.030. [DOI] [PubMed] [Google Scholar]
- Drechsel DA, Estévez AG, Barbeito L, Beckman JS. Nitric oxide-mediated oxidative damage and the progressive demise of motor neurons in ALS. Neurotoxic Res. 2012;22:251–264. doi: 10.1007/s12640-012-9322-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggler AL, Small E, Hannink M, Mesecar AD. Cul3-mediated Nrf2 ubiquitination and antioxidant response element (ARE) activation are dependent on the partial molar volume at position 151 of Keap1. Biochem J. 2009;422:171–180. doi: 10.1042/BJ20090471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Euro Heart J. 2012;33:829–837. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flora SJS. Arsenic-induced oxidative stress and its reversibility. Free Radic Biol Med. 2011;51:257–281. doi: 10.1016/j.freeradbiomed.2011.04.008. [DOI] [PubMed] [Google Scholar]
- Gentry PR, Clewell HJ, 3rd, Greene TB, Franzen AC, Yager JW. The impact of recent advances in research on arsenic cancer risk assessment. Regul Toxicol Pharmacol. 2014;69:91–104. doi: 10.1016/j.yrtph.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Gong G, O'Bryant SE. The arsenic exposure hypothesis for Alzheimer disease. Alzheimer Dis Assoc Disord. 2010;24:311–316. doi: 10.1097/WAD.0b013e3181d71bc7. [DOI] [PubMed] [Google Scholar]
- Gonzalez DR, Fernandez IC, Ordenes PP, Treuer AV, Eller G, Boric MP. Differential role of S-nitrosylation and the NO-cGMP-PKG pathway in cardiac contractility. Nitric Oxide. 2008;18:157–167. doi: 10.1016/j.niox.2007.09.086. [DOI] [PubMed] [Google Scholar]
- Gu S, Chen C, Jiang X, Zhang Z. Resveratrol synergistically triggers apoptotic cell death with arsenic trioxide via oxidative stress in human lung adenocarcinoma A549 cells. Biol Trace Elem Res. 2014;163:112–123. doi: 10.1007/s12011-014-0186-2. [DOI] [PubMed] [Google Scholar]
- Gupta D, Inouhe M, Rodríguez-Serrano M, Romero-Puertas M, Sandalio L. Oxidative stress and arsenic toxicity: role of NADPH oxidases. Chemosphere. 2013;90:1987–1996. doi: 10.1016/j.chemosphere.2012.10.066. [DOI] [PubMed] [Google Scholar]
- Hong YS, Song KH, Chung JY. Health effects of chronic arsenic exposure. J Prev Med Public Health. 2014;47:245–252. doi: 10.3961/jpmph.14.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubaux R, Becker-Santos DD, Enfield KS, Rowbotham D, Lam S, Lam WL, Martinez VD. Molecular features in arsenic-induced lung tumors. Mol Cancer. 2013;12:20. doi: 10.1186/1476-4598-12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes MF, Beck BD, Chen Y, Lewis AS, Thomas DJ. Arsenic exposure and toxicology: a historical perspective. Toxicol Sci. 2011;123:305–332. doi: 10.1093/toxsci/kfr184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inserte J, Hernando V, Vilardosa U, Abad E, Poncelas-Nozal M, Garcia-Dorado D. Activation of cGMP/protein kinase G pathway in postconditioned myocardium depends on reduced oxidative stress and preserved endothelial nitric oxide synthase coupling. J Am Heart Assoc. 2013;2:e005975. doi: 10.1161/JAHA.112.005975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R, Xu L, Liu X, Chen JD, Jonas JB, Wang YX. Optic nerve head changes after short-term intraocular pressure elevation in acute primary angle-closure suspects. Ophthalmology. 2015;122:730–737. doi: 10.1016/j.ophtha.2014.11.008. [DOI] [PubMed] [Google Scholar]
- Jiang X, Chen C, Liu Y, Zhang P, Zhang Z. Critical role of cellular glutathione homeostasis for trivalent inorganic arsenite-induced oxidative damage in human bronchial epithelial cells. Mutat Res Genet Toxicol Environ Mutagen. 2014;770:35–45. doi: 10.1016/j.mrgentox.2014.04.016. [DOI] [PubMed] [Google Scholar]
- Jiang X, Chen C, Zhao W, Zhang Z. Sodium arsenite and arsenic trioxide differently affect the oxidative stress, genotoxicity and apoptosis in A549 cells: An implication for the paradoxical mechanism. Environ Toxicol Pharmacol. 2013;36:891–902. doi: 10.1016/j.etap.2013.08.002. [DOI] [PubMed] [Google Scholar]
- Jomova K, Jenisova Z, Feszterova M, Baros S, Liska J, Hudecova D, Rhodes C, Valko M. Arsenic: toxicity, oxidative stress and human disease. J Applied Toxicol. 2011;31(2):95–107. doi: 10.1002/jat.1649. [DOI] [PubMed] [Google Scholar]
- Kaspar JW, Niture SK, Jaiswal AK. Nrf2: INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47:1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo K, Kim H, Bae Y, Kim K, Park B, Lee C, Kim Y. Salinomycin induces cell death via inactivation of Stat3 and downregulation of Skp2. Cell Death Dis. 2013;4:e693. doi: 10.1038/cddis.2013.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CC, Howard BV, Umans JG, Gribble MO, Best LG, Francesconi KA, Goessler W, Lee E, Guallar E, Navas-Acien A. Arsenic exposure, arsenic metabolism, and incident diabetes in the strong heart study. Diabetes Care. 2015;38:620–627. doi: 10.2337/dc14-1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A, Whitman SA, Jaramillo MC, Zhang DD. Arsenic-mediated activation of the Nrf2-Keap1 antioxidant pathway. J Biochem Mol Toxicol. 2013;27:99–105. doi: 10.1002/jbt.21463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen HM, Kansanen E, Polonen P, Heinaniemi M, Levonen AL. Role of the Keap1-Nrf2 pathway in cancer. Adv Cancer Res. 2014;122:281–320. doi: 10.1016/B978-0-12-420117-0.00008-6. [DOI] [PubMed] [Google Scholar]
- Li J, Duan X, Dong D, Zhang Y, Li W, Zhao L, Nie H, Sun G, Li B. Hepatic and nephric NRF2 pathway up-regulation, an early antioxidant response, in acute arsenic-exposed mice. Int J Environ Res Public Health. 2015;12:12628–12642. doi: 10.3390/ijerph121012628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Duan X, Dong D, Bai C, Li X, Sun G, Li B. Activation of the Nrf2 pathway by inorganic arsenic in human hepatocytes and the role of transcriptional repressor Bach1. Oxid Med Cell Longev. 2013;2013:984546. doi: 10.1155/2013/984546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macoch M, Morzadec C, Genard R, Pallardy M, Kerdine-Romer S, Fardel O, Vernhet L. Nrf2-dependent repression of interleukin-12 expression in human dendritic cells exposed to inorganic arsenic. Free Radic Biol Med. 2015;88:381–390. doi: 10.1016/j.freeradbiomed.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Maiti S, Chatterjee AK. Effects on levels of glutathione and some related enzymes in tissues after an acute arsenic exposure in rats and their relationship to dietary protein deficiency. Arch Toxicol. 2001;75:531–537. doi: 10.1007/s002040100240. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Hormesis defined. Ageing Res Rev. 2008;7(1):1–7. doi: 10.1016/j.arr.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morzadec C, Macoch M, Sparfel L, Kerdine-Romer S, Fardel O, Vernhet L. Nrf2 expression and activity in human T lymphocytes: stimulation by T cell receptor activation and priming by inorganic arsenic and tert-butylhydroquinone. Free Radic Biol Med. 2014;71:133–145. doi: 10.1016/j.freeradbiomed.2014.03.006. [DOI] [PubMed] [Google Scholar]
- Ray PD, Huang BW, Tsuji Y. Coordinated regulation of Nrf2 and histone H3 serine 10 phosphorylation in arsenite-activated transcription of the human heme oxygenase-1 gene. Biochim Biophys Acta. 2015;1849:1277–1288. doi: 10.1016/j.bbagrm.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoen A, Beck B, Sharma R, Dube E. Arsenic toxicity at low doses: epidemiological and mode of action considerations. Toxicol Appl Pharmacol. 2004;198:253–267. doi: 10.1016/j.taap.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Sinha D, Biswas J, Bishayee A. Nrf2-mediated redox signaling in arsenic carcinogenesis: a review. Arch Toxicol. 2013;87:383–396. doi: 10.1007/s00204-012-0920-5. [DOI] [PubMed] [Google Scholar]
- Snow ET, Sykora P, Durham TR, Klein CB. Arsenic, mode of action at biologically plausible low doses: what are the implications for low dose cancer risk? Toxicol Appl Pharmacol. 2005;207:557–564. doi: 10.1016/j.taap.2005.01.048. [DOI] [PubMed] [Google Scholar]
- So H, Kim H, Kim Y, Kim E, Pae HO, Chung HT, Kim HJ, Kwon KB, Lee KM, Lee HY. Evidence that cisplatin-induced auditory damage is attenuated by downregulation of pro-inflammatory cytokines via Nrf2/HO-1. J Assoc Res Otolaryngol. 2008;9:290–306. doi: 10.1007/s10162-008-0126-y. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Lin H, Geshi N, Mori Y, Kawarabayashi Y, Takami N, Mori MX, Honda A, Inoue R. Nitric oxide–cGMP–protein kinase G pathway negatively regulates vascular transient receptor potential channel TRPC6. J Physiol. 2008;586:4209–4223. doi: 10.1113/jphysiol.2008.156083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji JS, Perez V, Garry MR, Alexander DD. Association of low-level arsenic exposure in drinking water with cardiovascular disease: a systematic review and risk assessment. Toxicology. 2014;323:78–94. doi: 10.1016/j.tox.2014.06.008. [DOI] [PubMed] [Google Scholar]
- Umemura T, Kuroiwa Y, Kitamura Y, Ishii Y, Kanki K, Kodama Y, Itoh K, Yamamoto M, Nishikawa A, Hirose M. A crucial role of Nrf2 in in vivo defense against oxidative damage by an environmental pollutant, pentachlorophenol. Toxicol Sci. 2006;90:111–119. doi: 10.1093/toxsci/kfj076. [DOI] [PubMed] [Google Scholar]
- Uzunoglu S, Karaca B, Atmaca H, Kisim A, Sezgin C, Karabulut B, Uslu R. Comparison of XTT and Alamar blue assays in the assessment of the viability of various human cancer cell lines by AT-101 (−/− gossypol) Toxicol Mech Methods. 2010;20:482–486. doi: 10.3109/15376516.2010.508080. [DOI] [PubMed] [Google Scholar]
- Viallet J, Liu C, Emond J, Tsao MS. Characterization of human bronchial epithelial cells immortalized by the E6 and E7 genes of human papillomavirus type 16. Exp Cell Res. 1994;212:36–41. doi: 10.1006/excr.1994.1115. [DOI] [PubMed] [Google Scholar]
- Yates MS, Tran QT, Dolan PM, Osburn WO, Shin S, McCulloch CC, Silkworth JB, Taguchi K, Yamamoto M, Williams CR. Genetic versus chemoprotective activation of Nrf2 signaling: overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis. 2009;30:1024–1031. doi: 10.1093/carcin/bgp100. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Hou Y, Zhang Q, Woods CG, Xue P, Fu J, Yarborough K, Guan D, Andersen ME, Pi J. Cross-regulations among NRFs and KEAP1 and effects of their silencing on arsenic-induced antioxidant response and cytotoxicity in human keratinocytes. Environ Health Perspect. 2012;120:583–589. doi: 10.1289/ehp.1104580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.