

Abstract
FOXO3a and FOXM1 are two forkhead transcription factors with antagonistic roles in cancer and DNA damage response. FOXO3a functions like a typical tumour suppressor, whereas FOXM1 is a potent oncogene aberrantly overexpressed in genotoxic resistant cancers. FOXO3a not only represses FOXM1 expression but also its transcriptional output. Recent research has provided novel insights into a central role for FOXO3a and FOXM1 in DNA damage response. The FOXO3a-FOXM1 axis plays a pivotal role in DNA damage repair and the accompanied cellular response through regulating the expression of genes essential for DNA damage sensing, mediating, signalling and repair as well as for senescence, cell cycle and cell death control. In this manner, the FOXO3a-FOXM1 axis also holds the key to cell fate decision in response to genotoxic therapeutic agents and controls the equilibrium between DNA repair and cell termination by cell death or senescence. As a consequence, inhibition of FOXM1 or reactivation of FOXO3a in cancer cells could enhance the efficacy of DNA damaging cancer therapies by decreasing the rate of DNA repair and cell survival while increasing senescence and cell death. Conceptually, targeting FOXO3a and FOXM1 may represent a promising molecular therapeutic option for improving the efficacy and selectivity of DNA damage agents, particularly in genotoxic agent resistant cancer. In addition, FOXO3a, FOXM1 and their downstream transcriptional targets may also be reliable diagnostic biomarkers for predicting outcome, for selecting therapeutic options, and for monitoring treatments in DNA-damaging agent therapy.
Keywords: Cancer, DNA damage, drug resistance, FOXM1, FOXO3a, genotoxic agents, transcriptional targets
Introduction
The genetic information of a living organism is encrypted within its DNA. Preserving this genetic information is essential for the correct functioning of the organism and the long-term survival of the species [1, 2]. Failure to conserve the genetic code can lead to instability of the genome, a prominent feature of cancer and degenerative diseases [3]. During the lifespan of an organism, its DNA is subject to constant assaults from endogenous and exogenous genotoxic stresses. The endogenous DNA damaging agents comprise of products of metabolism and reactive oxygen species (ROS), which cause oxidative damage to DNA [4], while the exogenous environmental agents include ultraviolet (UV) light, ionizing radiation, toxins, DNA damaging chemicals and genotoxic therapeutic agents [5]. In addition, DNA lesions can also arise through poorly regulated or faulty cellular metabolic processes, such as abortive topoisomerase activity and base mismatch during DNA replication [5, 6]. As a result of DNA damage, cells are unable to pair bases and DNA cannot be replicated properly [5]. This will lead to stalled DNA replication or the incorporation of erroneous genetic information. It is therefore imperative that the damaged DNA is repaired promptly and accurately.
DNA lesions come in various forms, which include nucleotide modifications, single stranded breaks (SSBs) and double stranded breaks (DSBs) [5]. DSBs occur when both of the complementary DNA strands are damaged simultaneously in close proximity [7]. DSBs are thought to be one of the most lethal forms of damage and, if left unrepaired, will increase the risk of chromosome breakages/rearrangement, mutagenesis and losing genetic information [8]. In response to DNA damage, cells trigger a complex molecular reaction mechanism called the DNA Damage Response (DDR). This detects DNA damage, arrests cell cycle proliferation for DNA repair to take place and signals for its repair or cell termination [9]. More specifically, it monitors chromatin integrity, and triggers a cascade of signals and reactions upon the detection of damaged DNA [10]. This mechanism also further propagates and amplifies the damage signals and ultimately coordinates DNA repair with cell cycle arrest and cell termination [9, 11]. To induce cell cycle arrest, the cells must activate cell cycle checkpoints in the G1-S or G2-M phases [12, 13]. The induction of cell cycle arrest prevents the transmission of faulty genomic information and allows more time for cells to repair the damage [1, 5].
Once DDR is activated, there are three potential outcomes. First, the repair mechanism is able to completely repair the damaged DNA and so cell proliferation can resume as normal [5]. Secondly, if the lesion is irreparable, the cell can either enter apoptosis, programmed cell death, mitotic catastrophe, or senescence, a state of permanent cell proliferative arrest [11]. In this way, the cell is removed from the population and so the passage of erroneous genetic information to the next generation is avoided. Finally, DDR can fail and the damaged DNA is not repaired. The accumulation of DNA lesions will result in genetic instability and can give rise to genetic disorders such as cancer, ataxias, and Huntington’s, etc [14, 15]. Cancer cells have a higher proliferation rate and a tendency to bypass cell cycle checkpoints and, therefore, an increased susceptibility to accumulating further DNA damage and mutations [1]. Of particular interest is the fact that some cancer cells are able to survive, and not undergo cell death or senescence, despite sustaining high levels of DNA damage. It is believed that an enhanced DNA damage repair and survival ability will allow some cancer cells to develop resistance to genotoxic agents and accumulate further mutations. In consequence, deregulated DDR will not only impact cancer initiation, but also cancer progression and genotoxic drug resistance. In this review, we explore the impact of the FOXO3a-FOXM1 forkhead transcription factor axis on the DNA damage response, focusing on the therapeutic potential of targeting FOXO3a and FOXM1 in overcoming genotoxic drug resistance.
FOXO3a
FOXO3a is a member of the class O subfamily of forkhead box (Fox) transcription factors, which share a common conserved ‘wing-helix’ DNA-binding domain [16, 17]. There is strong evidence that FOXO3a acts as a tumour suppressor and can inhibit cell growth by driving the transcription of genes, such as Bim, FasL, p27Kip1, p130 (RB2), essential for cell proliferative arrest, cell death and differentiation [18]. Consistently, inactivation of FOXO3a has been shown to be a crucial step for oncogenic transformation [18-21]. The activity, expression and subcellular localization of FOXO3a is regulated by a diverse range of post-translational modifications [22]. Phosphorylation by kinases, particularly Akt (also called PKB) [23], ERK [24], IKB kinase (IKK) [25] and serum and glucocorticoid-regulated kinase (SGK) [26] can promote FOXO3a nuclear to cytoplasmic shuttling [27], and it also provokes a conformational change in FOXO3a which facilitates recognition by Mdm2 (murine double minute 2) and Skp2 (S kinase phase protein 2) E3 ligases, leading to its nuclear exclusion, retention in the cytoplasm and subsequent proteasome degradation and inactivation [28]. Conversely, other kinases, such as p38 MAPK [29], stress activated c-Jun-NH2-kinase (JNK) [30], AMP-activated protein kinase (AMPK) [31] and Ste20-like protein kinase (MST1) [32], have been demonstrated to promote FOXO3a activity and expression. FOXO3a transcription factor can also be regulated by other post-translational modifications such as acetylation, methylation, ubiquitination and glycosylation [22]. Interestingly, FOXO3a overexpression can arrest cell cycle progression and prevent damage induced by genotoxic agents and oxidative stress [33-37].
Modulation of DNA Damage Response by FOXO3a
FOXO3a is a key factor in the control of the DNA damage response and that is mediated primarily through the regulation of a diverse range of genes involved in sensing DNA damage, propagating DNA damage response signals, cell cycle checkpoint arrest, and DNA repair [28]. It has been shown that DNA damaging agents, such as doxorubicin, can activate the p38 MAPK, which in turn will phosphorylate FOXO3a on Ser-7 to promote its nuclear localization and activation to mediate cell cycle arrest [29]. In this context, one of the direct transcriptional targets of FOXO3a is p27Kip1, a cyclin-dependent kinase (CDK) inhibitor that interacts with CDK-cyclin complexes to induce cell cycle arrest in G0/G1 as well as during S phase [38]. Interestingly, FOXO3a can induce cells to enter senescence, a permanent state of cell cycle arrest through promoting the expression of p27Kip1 [39]. In concordance, the cell cycle promoters cyclin D1 and -D2 are negatively regulated by FOXO3a [40]. FOXO3a overexpression also induces a decrease in cyclin D1/2 protein and mRNA levels followed by G1 arrest and the conditional activation of a FOXO3a mutant results in repression of cyclin D1 and D2 promoter activities, indicating that cyclin D1/2 is a transcriptional target of FOXO3a. Importantly, ectopic expression of cyclin D1 can prevent FOXO3a-mediated cell cycle arrest [40]. In addition to G1 arrest, G2/M phase-arrested cells also display an upregulation in FOXO3a expression along with an increase in FOXO3a binding to promoter regions of cyclin B1 and Polo-like kinase 1 (PLK1) and a decrease in their expression [41]. Nevertheless, the regulation of cyclins by FOXO3a does not always result in cell cycle arrest. Active forms of FOXO3a were shown to regulate cyclin G2 expression at the protein and mRNA levels and transactivate its promoter, which was associated with exit from cell cycle [42]. Unlike other cyclins, cyclin G2 does not promote cell growth, but might inhibit cell cycle progression and facilitate the maintenance of a quiescent state [43]. In addition, activation of FOXO3a also modulates the expression of the retinoblastoma family protein p130 (RB2). Although p130 levels are low in cycling cells, its expression is increased in quiescent and senescent cells [44]. In these cells, p130 is hypophosphorylated and can interact with the E2F-4 transcription factor to promote the repression of diverse genes required for reentry into cell cycle, thus contributing to the maintenance of the quiescence state [44]. In this context, FOXO3a has been described to upregulate the p130 gene and its protein levels, thus inducing cell cycle arrest, and eventually, cell quiescence [45-47], This further confirms that FOXO3a can contribute to the regulation of cell cycle checkpoint and exit. Recent evidence also suggests a role of FOXO3a in regulating microRNAs in response to DNA damage. FOXO3a expression in colorectal cells was shown to be required for the binding to miR-34c, which in turn downregulates Myc expression in response to etoposide treatment [48]. Consistently, conditional activation of FOXO3a resulted in a rapid accumulation of cells in the G1 phase, more pronounced in cells with Myc containing the 3-UTR sequence, therefore indicating that Myc downregulation is required for the FOXO3a-mediated cell cycle arrest.
Reactive oxygen species (ROS) are generated as a by-product of normal aerobic activity, and, if not properly controlled, can cause substantial levels of DNA damage. As a result, the DDR is also activated in response to oxidative stress to protect against DNA damage. FOXO3a activation can also contribute to oxidative stress-resistance through direct transcriptional activation of the manganese superoxide dismutase (MnSOD, also called SOD2) gene [47]. Whereas upregulation of SOD2 by FOXO3a protects quiescent cells from apoptosis induced by ROS, Akt (PKB)-mediated phosphorylation and inactivation of FOXO3a culminates in reentry into the cell cycle and therefore, proliferation [46, 47]. Catalase, another scavenger of hydrogen peroxide, is also a direct transcriptional target of FOXO3a [49]. In agreement, the expression of FOXO3a and its targets, MnSOD and catalase, is reduced in caspase-2 deficient cells, which accumulate higher levels of oxidative stress and DNA damage following induction of ROS [50]. Caspase-2 knockout mice also develop early ageing symptoms in response to oxidative stress [51]. FOXO3a can induce DNA repair and oxidant scavenging by regulating Muc1, a protein highly expressed during oncogenic transformation [52]. Muc1 expression can attenuate the inhibition of FOXO3a by Akt and reduce the intracellular hydrogen peroxide levels, therefore preventing breast cancer cells from undergoing oxidative stress-mediated cell death [53]. FOXO3a-induced stress resistance can also be influenced by p53, which was described to inhibit its transcriptional activity in a SGK-dependent manner [54]. Upon treatment with UV radiation and etoposide, p53 expression was activated. This was followed by an increase in FOXO3a phosphorylation and its relocation to the cytoplasm, preventing transcriptional regulation of its downstream targets. Another negative regulator of FOXO3a expression is the latent membrane protein 1 (LMP1), an oncoviral protein crucial to EBV-mediated B-cell transformation, which is involved in genomic instability [55, 56]. LMP1 can suppress DNA repair, through the phosphorylation of Akt and FOXO3a, leading to FOXO3a nuclear exclusion in epithelial cells. These effects are associated with a decrease in FOXO3a’s ability to promote DNA repair; this effect can be completely reversed when these cells are transfected with a non-Akt-phosphorylatable FOXO3a [56]. This data suggests that LMP1 modulates the FOXO3a pathway to prevent DNA repair and proposes some mechanisms that may account for LMP1-mediated genomic instability.
In response to DNA damage, FOXO3a can not only regulate the expression of cell cycle regulator genes but also of those involved in DNA repair. One of the most studied transcriptional targets of FOXO3a in this context is Gadd45a [34, 57], a gene expressed in response to genotoxic stress [58]. Gadd45a is also relevant for inducing cell cycle arrest at the G2/M checkpoint upon DNA damage [59]. FOXO3a has been shown to promote DNA repair following exposure to UV irradiation, at least in part, through inducing Gadd45a expression [34]. Nevertheless, the key role of FOXO3a in DNA damage response is underscored by the revelation that FOXO3a can bind directly, through its carboxy-terminal region, to the FAT protein-binding domain of Ataxia-Telangiectasia mutated (ATM) protein and thus activate its DDR signalling function [13]. This suggests that FOXO3a may modulate the DNA-damage response through ATM. Consistently, FOXO3a overexpression promotes ATM-mediated signalling, the repair of the damaged DNA and the S-phase and G2-M cell-cycle checkpoints, while FOXO3a depletion leads to defects in these DDR functions [60]. Notably, the involvement of FOXO3a in the DNA damage response signalling goes beyond its interaction with ATM. FOXO3a has been shown to negatively regulate the expression and activity of FOXM1, a forkhead protein involved in the regulation of genes regulating several aspects of DDR and genotoxic agent resistance. Essentially, FOXM1 is one of the most important and relevant downstream transcriptional targets of FOXO3a, especially in terms of the regulation of the DNA damage response. In fact, FOXO3a not only represses FOXM1 transcription, it also competes for the binding to the same DNA motifs in target promoters (e.g. FOXM1 and VEGF) and produces opposing transcriptional outputs through the recruitment of HDACs to repress the transcription of FOXM1 target genes [61]. Thus, FOXO3a essentially antagonizes FOXM1-dependent transcription. In particular, FOXO3a activation will trigger the G1/S and G2/M cell cycle checkpoints as well as repressing DDR, cell proliferation and survival [62]. Collectively these findings suggest that FOXO3a plays a central part in sensing genotoxic stress, relaying the DDR signals and integrating them with the cell cycle checkpoints, anti-oxidative stress mechanisms, DNA damage repair pathways and the senescence and cell death machineries. Notably, while FOXO3a can function to antagonize ROS and promote DNA damage repair and cell survival, it can also impair the ability of FOXM1 to enhance DNA damage repair. These discrepancies might reflect the transformation status of the cells. Accumulating evidence suggests that the role of FOXO3a in inducing DNA repair and oxidant scavenging occurs in normal mammalian cells in order to maintain genome stability and integrity and suppress the emerging of cancer clones [42, 50, 63]. In addition, it also functions as a bona fide tumour suppressor to repress the oncogenic activity of FOXM1 [61, 62] and thereby, restricting cancer progression and the development of resistance to DNA damaging agents [60, 64].
Forkhead Box M1 (FOXM1) transcription factor regulates a broad spectrum of normal biological functions, including cell proliferation, cell cycle progression, cell renewal, cell differentiation, DNA damage repair, tissue homeostasis, cell migration, angiogenesis and cell survival [60]. While FOXO3a is a typical tumour suppressor, FOXM1 functions as a potent oncogene. Overexpression of FOXM1 is the hallmark of many malignancies, including cancers of the liver, prostate, brain, breast, lung, colon, pancreas, skin, cervix, ovary, mouth, blood and nervous system [65-79]. Furthermore, upregulation of FOXM1 expression has been proposed to be an early event during cancer development [75]. In agreement, genome-wide gene expression studies have independently identified FOXM1 as one of the most commonly overexpressed genes in different human cancers [80, 81]. Together, these findings suggest a central role for FOXM1 in cancer initiation. In addition, latest evidence reveals that FOXM1 also advances cancer progression by promoting cancer angiogenesis, invasion and metastasis as well as the development of genotoxic resistance [71, 82-86]. The role of FOXM1 in DNA repair is first defined by the finding that FOXM1-deficient cells accumulate high levels of damaged DNA, suggesting that FOXM1 has a DNA damage repair function. Subsequent studies have revealed that this is due, at least in part, to the ability of FOXM1 to regulate the transcriptional control of a network of DDR genes essential for DNA damage sensing, mediating, signalling and repair [83-85].
FOXM1 in the regulation of DNA damage repair pathways
Eukaryotic cells are constantly exposed to a broad range of exogenous and endogenous genotoxic stresses during their lifetime. To prevent the accumulation of damaged DNA, cells respond by activating a number of repair pathways, including nucleotide excision repair (NER), fanconi anaemia (FA)/BRCA pathway, mismatch repair (MMR), base excision repair (BER), homologous recombination (HR) and non-homologous end-joining (NHEJ) [87].
FOXM1 has been shown to be overexpressed in DNA-damaging cancer drug resistant cells and its expression can confer genotoxic agent resistance [64, 84, 85, 88]. For example, FOXM1 has been shown to regulate quiescence-associated radioresistance of human head and neck squamous carcinoma cells [89]. In agreement, a recent transcriptome meta-analysis across 11 microarray datasets have independently identified FOXM1 as one of the central transcription factors regulating radiation sensitivity [90]. A growing body of evidence has revealed that FOXM1 has a role in the regulation of almost every aspects of DNA damage repair. NER functions to fix distorting base lesions, such as pyrimidine dimers, generated through exposure to genotoxic agents, such as UV, ionizing irradiation, environmental mutagens, and cancer chemotherapeutic drugs (Fig. 1). Mutations in genes coding for NER factors can cause inherited disorders, such as xeroderma pigmentosum (XP), Cockayne syndrome, and trichothiodystrophy, as well as an increased in skin cancer risk [91]. Several components of the NER are downstream targets of FOXM1. Proteins key in recognizing these types of DNA lesions and in introducing incisions for NER repair are xeroderma pigmentosum group A (XPA), XPE, XPF, XPG, Cockayne syndrome A (CSA), CSB, the XPC-RAD23B complex, the transcription factor IIH (TFIIH) complex, the XPF-excision repair cross-complementation group 1 (ERCC1) complex and replication protein A (RPA). Following incision, a 24-32 base oligonucleotide containing the damaged DNA is excised and replaced with the correct DNA sequence through gap-filling and religation by replication factor C (RFC), proliferating cell nuclear antigen (PCNA), DNA polymerase (DNA pol) δror ε, DNA ligase I and RPA. Among these key NER factors, FOXM1 has been shown to transcriptionally regulate the expression of PolE2 and RFC4. The PolE gene encodes for DNA pol ε, while RFC4 is a subunit of RFC, which functions cooperatively with PCNA during the repair [92]. In addition, FOXM1 is further linked to NER through its transcriptional regulation of RAD23B, a cofactor of XPC, involved in the initiation of NER [93]. In quiescent cells, DNA pol δ and DNA ligase IIIαn along with their cofactor X-Ray Repair Cross-Complementing Protein 1 (XRCC1) are required for gap-filling and ligation for NER in an alternative mechanism. In this way, FOXM1 can also influence NER in quiescent cells via XRCC1, which is another downstream target of FOXM1 [93]. The (FA)/BRCA pathway is usually activated as a result of inter-strand DNA crosslinks caused by ionizing radiation [94, 95]. The (FA)/BRCA pathway often collaborates with NER to repair single strand DNA (ssDNA) damage by sharing common signalling components, and will in this way lend itself to the control by FOXM1. Furthermore, it is believed that the (FA)/BRCA pathway facilitates DNA repair by HR and cross-talks with DDR proteins, such as Nijmegen breakage syndrome 1 (NBS1), BRCA2 and RAD51, which are direct FOXM1 targets [84, 96, 97]. MMR mends errors from cellular metabolism, DNA replication and recombination that result in mispaired and unpaired bases. During MMR, the mispaired bases are detected by the MutS-MutL heterodimers (Fig. 1). FOXM1 is also linked to multiple repair pathways, including MMR, by its transcriptional target exonuclease 1 (Exo1) [92]. Upon mismatch detection, the MutS-MutL complexes direct exonuclease 1 (Exo1) to remove the segment containing the mismatched base. The importance of this regulatory FOXM1-Exo1 axis in DDR is highlighted by the findings that FOXM1 modulates the sensitivity to the DNA-damaging agents cisplatin and doxorubicin through regulating Exo1 in ovarian and breast cancer, respectively [88, 92]. The gap created by Exo1 is then filled with the correct base by DNA pol δ and εnand the remaining nick rejoined by DNA ligase. This repair process is again orchestrated by the RFC and PCNA that loads and clamps DNA pol, for DNA synthesis. As in NER, FOXM1 can also promote a number of SSB repair mechanisms, including MMR and BER, by transcriptionally activating the expression of genes, such as RFC4, Exo1, and PolE2 [92, 93]. During MMR, the co-operating RFC4, PCNA and DNA polmerases direct ssDNA to fill the gap left following the removal of the segment containing the mismatched base, whereas DNA pol ε is involved in sealing the ssDNA gap after processing by nucleases during BER, NER and MMR. Consistently, mutations to the POLE gene have recently been identified to be associated with familial colorectal adenomas and colorectal cancer (CRC) [98]. In addition, FOXM1 also directly regulates the transcription of BRCA1-interacting protein-terminal helicase 1 (BRIP1/BACH1/FRACJ). BRIP1 contributes to processing interstrand crosslinks (ICLs) during MMR. This is mediated, in a BRCA1 independent manner, through its interaction with the MutLα mismatch repair complex [99].
Fig. (1).
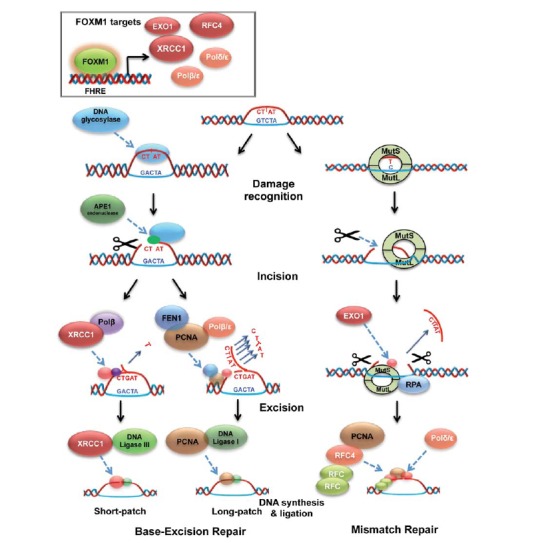
FOXM1 regulates proteins crucial for both base-excision repair and mismatch repair processes: schematic diagramme comparing proteins involved in short-patch and long patch base excision repair, as well as mismatch repair. All proteins high-lighted in red have shown to be under FOXM1 transcriptional regulation (XRCC1, Polβ/ε, BRIP1 and the RFC4 complex). For base excision repair, a process where an incorrect base has been inserted in a DNA template, DNA glycosylase is responsible for the detection of the erroneous base insertion. Alternatively, during mismatch repair, a process where two inserted base pairs are not corresponding, damage recognition is performed by a larger number of proteins: RFC4 (controlled by FOXM1) works in conjunction with a bigger complex of PCNA, the Mut and BRIP1 (also under FOXM1 transcriptional control). In this process, these proteins also perform the incision around the mismatching base, while incision is performed by APE1 endonuclease during base excision repair. The base excision repair process is then subdivided into short-patch and long-patch base excision repair: during short-patch, the single mismatched base is removed by XRCC1 (FOXM1 target) polymerase-β complex; In the latter, PCNA joins FEN1 and polymerase β/ε (regulated by FOXM1) to perform the excision. Alternatively, in mismatch repair, excision is performed by Mutsα, EXOI and RPA. Ligation again differs between the pathways: XRCC1 now binds to DNA ligase III for the short patch base excision repair; Long patch base excision repair ligation is perfomed by PCNA bound to DNA ligase I; Mis-match repair uses the RFC4 complex, coupled with PCNA, RPA and DNA polδ.
BER repairs damage to single bases caused by oxidation, alkylation, hydrolysis, or deamination throughout the cell cycle (Fig. 1). The damaged bases are recognized by DNA glycosylase enzymes, which also mediate base removal before the repair is completed by APE1 endonuclease, end processing enzymes (polynucleotide kinase-phosphatase), polymerases (Pol β and Pol λ for short-patch, and pol δ and pol ε for long-patch BER) and ligases (DNA ligase III along with its cofactor XRCC1 for short-patch, and DNA ligase III for long-patch BER). FOXM1 is also a transcriptional regulator of the BER factor X-ray cross-complementing group 1 (XRCC1). In a similar way as for NER and MMR, FOXM1 can also promote BER by driving the expression of ssDNA repair genes, such as RFC4, Exo1, and PolE2 [92, 93].
Double strand breaks (DSBs) are the most harmful species of DNA lesions and are predominantly repaired by HR and NHEJ [5, 100, 101]. HR is a relatively error-free DNA repair mechanism that uses the chromosome as a template to direct repair and therefore, only in S and G2 phases after DNA replication and before cell division [102] (Fig. 2). In HR, DSB response is initiated through the detection of DSBs by the MRN (Mre11-Rad50-NBS1) complex [100, 101], which helps to recruit and activate key DDR signalling kinases, including ATM at the sites of DNA damage.
Fig. (2).
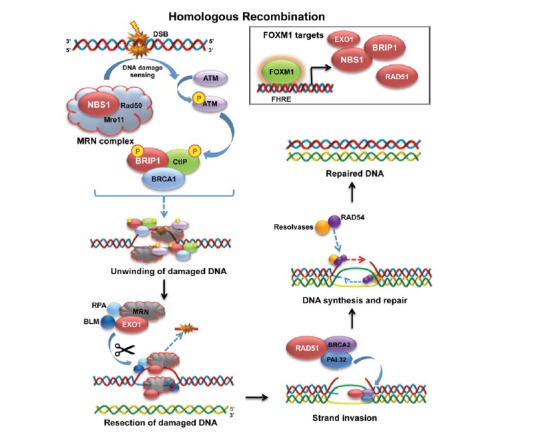
FOXM1 regulates crucial genes in homologous recombination: schematic diagramme representing the main steps in the homologous recombination repair of double stranded DNA damage. As shown in the top right corner, FOXM1 positively regulates the expression of NBS1, BRIP1, EXO1, BRAC2 and RAD51 (depicted in red throughout the diagramme). Following the instigation of DNA damage, FOXM1 promotes the transcription of NBS1, which forms part of the MRN complex. This participates in the detection of the DNA damage, as well as in the phosphorylation of ATM. Phosphorylated ATM in turn phosphorylates BRIP1 (also regulated by FOXM1), CltP and BRCA1. Together, these proteins form a complex which participates in the unwinding of the double stranded DNA, making it accessible for the RPA, BLM, EXO1 (regulated by FOXM1) complex, which cleaves and removes the damaged sections. RAD51, BRCA2 (both regulated by FOXM1) and PALB2 aid the strand invasion of a template double stranded DNA, upon which the new strand is modelled. Finally, resolvases coupled with RAD54 regulate the synthesis of the new strand, creating a complete double stranded DNA.
In turn, ATM phosphorylates H2AX, its downstream target histone, which ultimately culminates in the recruitment of DNA repair proteins to the damage sites [103]. FOXM1 has been found to regulate Nijmegen breakage syndrome 1 (NBS1) expression directly at the transcriptional level. In this way, FOXM1 can control the initiation of HR, as the assembly of the MRN complex is rate-limiting for the recruitment and activation of ATM [104]. In addition, there is evidence to suggest that this upregulation of the NBS1 expression by FOXM1 also indirectly enhances the stability of the other MRN subunits, including MRE11 and RAD50, and thereby further promotes the HR DNA damage repair response [84]. In turn, the activated ATM then phosphorylates its downstream substrates such as p53BP1, SMC1, BRCA1, NBS1 and CHK2 to trigger cell-cycle arrest, apoptosis, and DNA repair [100, 105-109]. FOXM1 can also promote HR repair indirectly through driving the transcription of S-phase kinase-associated protein 2 (Skp2) and cyclin-dependent kinases regulatory subunit 1 (Cks1) [110]. Skp2 and Cks1 are key components of the Skp2-SCF E3 ligase complex that mediates the K63-linked ubiquitination of NBS1. This process is critical for the activation of ATM and its recruitment to the DNA damage foci to initiate HR repair [111]. Appropriately, Skp2 deficient cells display HR repair defects and ionizing irradiation sensitivity [111].
The actual HR repair begins with nucleolytic resection of broken DNA ends facilitated by the CtBP-interacting protein (CtIP), BRCA1 and the MRN complex. This yields a 3’-ssDNA that is stabilized by association with RPA. During DNA resection, the MRN complex initiates a short 5'-end degradation, and the nucleases Exo1 and Dna2, together with the RecQ helicases degrade 5’-strands further exposing long 3’-strands [112]. During HR, the FOXM1 target Exo1 also unwinds duplex DNA and promotes DNA end resection. Next, the breast cancer susceptibility gene product 1 (BRCA1), BRCA2 and several RAD51-related proteins promote the displacement of RPA by the strand exchange protein RAD51, resulting in the formation of a RAD51 nucleoprotein filament. RAD51 then searches for homologous sequences and catalyzes an exchange strand between the broken duplex and the intact sister chromatid. Furthermore, FOXM1 has also been suggested to be an upstream transcriptional activator of BRCA2 [97], a vital HR regulator which binds the ssDNA and recruits the recombinase RAD51 to stimulate strand invasion during HR. Intriguingly, RAD51 itself is another direct transcriptional target of FOXM1. Induction of RAD51 by FOXM1 in glioblastomas has been shown to confer resistance to the genotoxic alkylating agent temozolomide [97]. In addition, the FOXM1 target BRIP1 also binds to and functions cooperatively with BRCA1 to promote HR repair. Bound BRIP1 unwinds damaged dsDNA to allow other repair proteins to access and process the damaged DNA [85, 113]. The importance of BRIP1 in HR is reflected by the fact that individuals with both copies of the BRIP1 gene being mutated are predisposed to the FA type J (FA-J) genetic disorder. These individuals are also prone to developing leukaemias and cancers of the head, neck, breast, stomach, ovary, cervix and skin [113-116].
NHEJ repairs double-strand DNA breaks by directly ligating together the broken DNA ends (Fig. 3), without the need of a homologous template [117]. NHEJ can accurately join compatible breaks with no damaged nucleotides, but can also introduce mutations when joining mismatched termini or termini that harbour damaged nucleotides. In NHEJ, DSBs are recognized by the Ku heterodimer complex (Ku70-Ku80), which binds to and activates the catalytic subunit of DNA-PK (DNA-PKcs). This catalytic subunit recruits and activates end-processing enzymes (e.g. Artemis), polymerases (e.g. pol μ and λ), DNA ligase IV and its cofactor XRCC4. The Artemis-DNA-PKcs complex is thought to have 5’ and 3’ nuclease activity that can cleave the damaged DNA that overhangs to form blunt ends in order to prepare them for ligation by DNA ligase IV and its cofactor XRCC4, with the help of Cernunnos/XLF. The MRN complex is also involved in NHEJ repair (Fig. 3), particularly in response to etoposide-induced DSBs [118]. Cells deficient in Mre11 or NBS1, but not ATM, exhibit a major NHEJ repair defect, suggesting that the function of the MRN in NHEJ repair is independent of ATM. This role of MRN also helps to link FOXM1 to the NHEJ pathway.
Fig. (3).
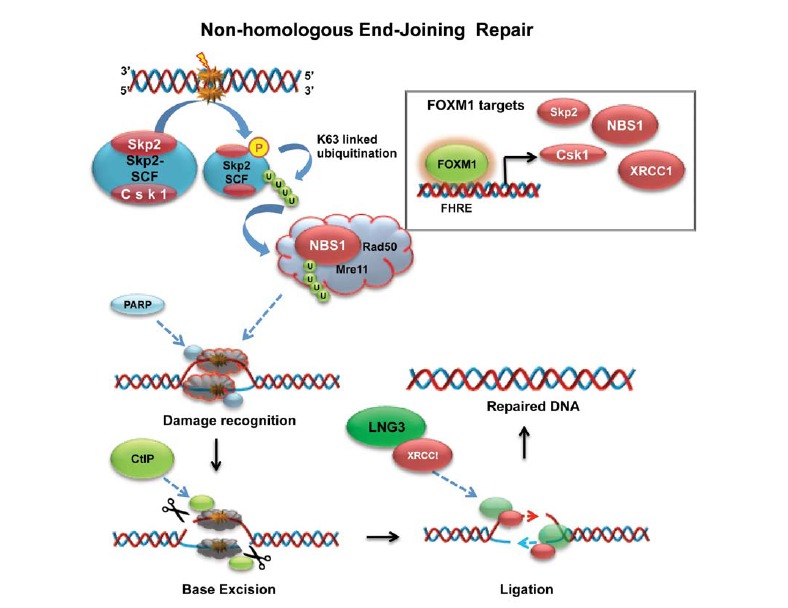
FOXM1 controls initial steps of non-homologus end joining repair mechanism: schematic diagramme describing the main stages of non-homologous end-joining repair (NHEJ) of double stranded DNA damage. FOXM1 exerts transcriptional control over the proteins highlighted in red, Skp2 and Csk1 as part of the Skp2-SCF complex and NBS1, which are also shown in the top right corner. Upon insurgence of a double-stranded DNA break, the FOXM1 promotes the transcription of Skp2 and Csk1 proteins, necessary for the formation of the Skp2-SCF complex. This will be phosphorylated, making it eligible for the K63-linked ubiquitination. The poly-ubiquitin chain (depicted in green) is then transferred to NBS1 (also a FOXM1 target), activating the MRN complex for an ATM independent repair process. Damage recognition is performed by MRN coupled with PARP. Ctlp (CtPB-interacting protein) then cleaves individual DNA strand, to permit base excision. Finally, LNG3 and XRCC1 replace the cleaved damaged DNA strand by promoting the synthesis of the correct template, leaving a repaired double stranded DNA helix.
Recent evidence also shows that the chromatin structure can also influence DNA repair, and that the repair of damaged DNA located proximal to compact chromatin is less effective than that in comparatively open chromatin, probably because compact chromatin can be a barrier for the access of repair proteins to the damaged DNA [119-121]. In this regard, FOXM1 can facilitate DNA repair through modulating chromatin structure. For example, FOXM1 has been shown to regulate the expression of the DNA methyltransferase DNMT1 through the chromatin remodelling factor HELLS [122]. Intriguingly, DNMT1 has a methyltransferase-independent role in promoting DNA damage repair through decondensing chromatin local to sites of DNA damage [123]. Therefore, FOXM1 can play an indirect role in DNA repair through promoting the expression of gene products that can modulate chromatin remodelling at sites of DNA damage to enhance repair. Collectively, these findings provide strong indications that FOXM1 plays an integral part in DNA damage response through driving the transcription of genes encoding for DNA damage sensors, mediators, signal transducers and effectors.
Role of FOXM1 in genotoxic agent-induced cell cycle checkpoints and cell fate decisions
FOXM1 is a principal promoter of cell cycle progression and its overexpression has been shown to confer proliferative advantages to cancer cells. On the other hand, FOXM1 is downregulated in response to genotoxic agents to evoke multiple cell cycle checkpoints, in particular those at G1/S, G2/M and M phases [124]. Compelling evidence has demonstrated that FOXM1 is a cellular target of genotoxic agents and the expression and transcriptional activity of FOXM1 is substantially downregulated in response to genotoxic stress through transcriptional and post-translational mechanisms. Upon treatment with genotoxic drugs such as epirubicin, p53 has been shown to repress FOXM1 transcription through an E2F-element on its promoter in breast cancer cells [125]. However, in the absence of functional p53, genotoxic stress will induce FOXM1 expression through ATM and E2F1 to promote DNA repair and survival. Furthermore, epirubicin has been shown to induce FOXM1 transcription via E2F1 through activating the p38 MAPK-MK2 signalling axis [126]. Apart from transcription, the activity of FOXM1 is also controlled by genotoxic stress via post-translational modifications. Previous studies have shown that treatment with DNA-damaging agents, such as γ-irradiation, etoposide and UV, promotes CHK2-induced phosphorylation of FOXM1. Such phosphorylation results in the stabilization of FOXM1 and transcriptional activation of downstream DNA repair and survival genes [97]. Recent evidence also suggests that DNA damaging agents can also modulate the stability of the FOXM1 protein through SUMOylation [86].
The downregulation of FOXM1 expression through transcriptional and post transcriptional mechanisms in response to genotoxic stress is critical for the DNA damage signals to execute the cell cycle checkpoints at G1/S, S, G2/M and M phases. These cell cycle checkpoints are mediated by FOXM1 through the downregulation of cell cycle regulatory genes, such as Cyclin D1, Cyclin A2, CDC25B, PLK1, Aurora B kinase, Cyclin B1, PLK1, MYC, BUB1B and CENPF, which are known transcriptional targets of FOXM1 [127]. The inhibition of FOXM1 also results in the downregulation of its targets Csk1 and Skp2 [110], which are key components of the Skp2-SCF(Skp1-Cullin1-F-box protein) E3 ligase complex that mediates the degradation of the cyclin-dependent kinase inhibitors (CKIs) p21Cip1 and p27Kip1 [128, 129]. In this context, downregulation of FOXM1 in response to genotoxic treatments will lead to stabilization of p21Cip1 and p27Kip1 and thereby, the inhibition of the cyclin-CDK1/2 kinases and cell cycle arrest at the G1/S, S and G2/M checkpoints [130]. Notably, FOXM1 also cooperates with other cell cycle regulatory oncogenic transcription factors, such as nuclear factor kB (NF-kB), E2F1, and B-myb to extend its influence to a greater network of cell cycle genes [131-134]. It is believed that upon genotoxic stress, cells will undergo cell death or permanent cell cycle arrest-senescence, if the damaged DNA is so extensive that it is irreparable or that it cannot be rectified in time. FOXM1 also contributes to the modulation of cell fate decisions in response to DNA damage, through controlling the transcriptional activity of anti-apoptotic and anti-senescence genes, including Bcl-2, Survivin (BIRC5), and Bmi-1, respectively [131, 135, 136]. There is now clear evidence that in response to DNA damage cells are also eliminated through mitotic catastrophe, a form of non-apoptotic cell death also mediated by FOXM1 [124]. Accordingly, depletion of FOXM1 can lead to centrosome amplification and mitotic catastrophe [124]. In agreement, MEIS2 a protein involved in preventing mitotic catastrophe has been found to be a direct transcription activator of FOXM1 as well as a promoter of the MuvB-BMYB-FOXM1 cell cycle gene regulatory complex [137]. In addition, the role of FOXM1 in evading mitotic catastrophe has been shown to involve the expression of genes, required for faithful chromosome segregation and mitosis, including Nek2, KIF20A, CENP-A and BUB1B (BUBR1) [124, 138]. For example, BUB1B depletion has been shown to result in chromosome missegregation and mitotic catastrophe in neuroblastoma cells [139]. Hence, downregulation of FOXM1 by genotoxic agents can also trigger suppression in the expression of essential component of the mitotic checkpoints. Collectively, these findings corroborate the idea that FOXM1, in addition to modulating DNA damage signalling and repair pathways, also governs the downstream cellular responses, involving apoptosis, mitotic catastrophe and senescence, in DNA damage response.
Targeting FOXO3a and FOXM1 in DNA damage-response and cancer drug resistance
The DNA damage response plays a crucial role in cancer initiation, progression and drug resistance. Whereas a deficiency in DNA damage repair contributes to tumorigenesis and increased risks of disease progression, aberrant activation of DNA damage repair also plays a key role in resistance to genotoxic anticancer drugs. Convincing evidence illustrates that the FOXO3a and FOXM1 transcription factors are regulated by cytotoxic and targeted-therapeutic agents and mediate their effects through modulating the transcription of their targets involved in apoptosis, cell cycle progression, senescence and DNA damage repair. As a consequence, abrogating the DDR pathways through targeting the FOXO3a-FOXM1 axis may represent an effective strategy for enhancing the therapeutic index of genotoxic agents (Fig. 4). Besides genotoxic drugs, such as doxorubicin and cisplatin [29, 140], other cancer therapeutic agents, including paclitaxel [37], lapatinib [141], gefitinib [142, 143] and imatinib [144], have also been shown to exert their cytotoxic and cytostatic effects through FOXO3a. These findings highlight that FOXO3a is common cancer drug target and that combining genotoxic treatments with agents that target FOXO3a may have enhanced therapeutic effects. Indeed, the use of drug combinations to treat cancer and to overcome cancer drug resistance is a well-established principle of cancer therapy. In addition to cancer therapeutics, FOXO3a can also be activated by agents targeting its upstream regulatory PI3K-Akt pathway. For instance, the Akt inhibitor, OSU-03012, has been shown to induce FOXO3a dephosphorylation and nuclear relocation in breast cancer cells [145]. A similar study has also demonstrated that another AKT inhibitor MK-2206 can cause FOXO3a dephosphorylation and activation, and is able to synergize with conventional genotoxic drug, such as doxorubicin, in liver cancer treatment. NVP-BEZ235, a dual PI3K/mTOR inhibitor, has also been reported to sensitize coloncarcinoma cells to genotoxic drugs through targeting FOXO3a [146]. Another effective strategy to enhance the activity of DNA damaging agents and to overcome genotoxic resistance is through targeting the Sirtuin family of class III histone deacetylases (SIRT1-7), which are crucial regulators of FOXO3a activity [147, 148]. The regulation of FOXO3a by SIRT1 was first demonstrated in cells under oxidative stress (e.g. heat shock or hydrogen peroxide-exposed) where expression of SIRT1 resulted in FOXO3a deacetylation, increased expression of the DNA repair Gadd45a gene but decreased expression of pro-apoptotic targets such as FasL and Bim. This suggests that SIRT1 modulates the balance between FOXO3a-mediated stress resistance and cell termination [147]. Recently, it has also been reported that the FOXO3a-mediated induction of Gadd45a can be negatively regulated by nicotinamide-phosphoribosyltransferase (NAMPT), a stress-induced protein, and SIRT1 [149]. In agreement, chemical inhibition of NAMPT or SIRT1 knockdown can result in increased FOXO3a acetylation and Gadd45a upregulation. Another Sirtuin protein, SIRT6, has also been shown to modulate FOXO3a expression [148]. In the context of DNA damage repair, SIRT6 overexpression can promote epirubicin resistance, whereas cells lacking SIRT6 accumulate significantly more DNA damage in response to genotoxic agents, including epirubicin and γ-irradiation. In the same way, depletion of SIRT6 in breast cancer cells also results in inefficient repair of double-strand breaks and thus, accumulation of damaged DNA in response to DNA-damaging agents. Intriguingly, these effects of SIRT6 on DNA repair and drug resistance appear to be mediated, at least in part, through the regulation of FOXO3a acetylation [148]. Inhibitors against individual or multiple SIRTs have been developed and some of them have been shown to be able to reactivate tumour suppressors, like p53 and FOXO3a [150, 151]. Consistently, small molecule SIRT-inhibitors, such as Sirtinol, Salermide, and EX527, have been shown to have anti-proliferative activity and be able to combine with DNA damaging agents, such as doxorubicin, to eliminate breast cancer cells [152]. Collectively, these findings suggest targeting FOXO3a is conceptually an effective therapeutic approach for cancer treatment. This is because not only FOXO3a transcriptionally regulates genes involved in cell cycle control and DDR but also it negatively regulates the transcriptional output of FOXM1, a key regulator of DNA repair and cell survival. Considering the critical role played by FOXM1 in DDR, attention has also focused on the generation of therapeutic strategies that will specifically inhibit FOXM1 expression and function. It is interesting to note that FOXM1 inhibition can sensitize breast cancer cells to genotoxic agent-induced senescence, but FOXM1 depletion alone is enough to cause genotoxic drug resistant cells to enter senescence [84]. This provides further proof that FOXM1 is a key determinant of genotoxic drug resistance, and is likely to be due to the fact that drug-resistant cancer cells have become overreliance on high levels of FOXM1 to protect them from genotoxic stress. This observation also suggests that the dependency on FOXM1 overexpression in cancer and in genotoxic resistance can be exploited for therapeutic benefits. Siomycin A and thiostrepton are potent thiazole antibiotics that have been shown to suppress the FOXM1 mRNA and protein expression [153, 154]. These thiazoles function by binding specifically to the DNA binding domain [155] and can kill cancer cells with tolerable toxicity to untransformed cells [153, 154]. Another natural compound, 2-deprenyl-rheediaxanthone B, isolated from the fern Metaxya rostrata, has also been reported to activate cell death in colorectal tumor cells through targeting FOXM1 [156]. Approaches based on small peptides have also been developed to target FOXM1 directly. Small peptides which mimic the tumour suppressor p19ARF and can disrupt the interaction between FOXM1-p19ARF have been generated. These ARF peptides have been shown to inhibit the expression and transcriptional activity of FOXM1 in hepatocellular carcinoma cells and suppress cancer cell growth [157]. Together, these studies provide proof-of-concept evidence that the FOXO3a-FOXM1 axis can be targeted to specifically eliminate cancer cells and drug resistant clones and pave the way for future FOXO3a-FOXM1 targeting therapeutics development.
Fig. (4).
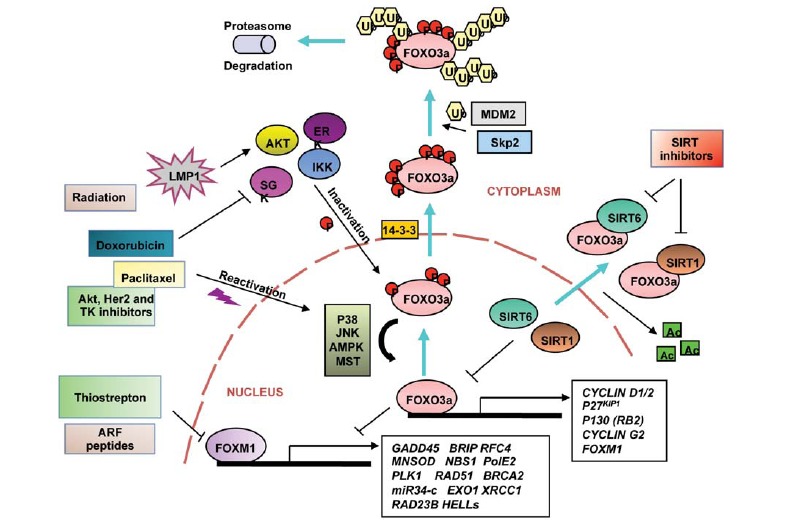
Targeting FOXO3a and FOXM1 in DNA damage response. Schematic diagramme representing upstream and downstream FOXO3a networks involved in modulation of its function and regulation of crucial transcriptional targets. FOXO3a is phosphorylated by several key oncogenic kinases such as Akt (also called PKB), IKB kinase (IKK) and serum and glucocorticoid-regulated kinase (SGK) and ERK, which facilitates recognition by Mdm2 (murine double minute 2) and Skp2 (S kinase phase protein 2) E3 ligases, leading to its nuclear exclusion, proteasome degradation and thus, inactivation of its function. The latent membrane protein 1 (LMP1), an oncoviral protein, can modulate FOXO3a expression in an Akt-dependent way to prevent DNA repair. Conversely, FOXO3a can be reactivated through phosphorylation by p38 MAPK, stress activated c-Jun-NH2-kinase (JNK), AMP-activated protein kinase (AMPK) and Ste20-like protein kinase (MST1), which are stimulated upon drug treatment or genotoxic stress such as exposure to doxorubicin, paclitaxel, UV radiation and Akt, Her2 and tyrosine kinase (TK) inhibitors. FOXO3a can also be regulated by acetylation, and SIRT 1 and SIRT6 histone deacetylase proteins play a crucial role in suppressing FOXO3a function. This phenomenon can be rescued by treatment with SIRT inhibitors, which can prevent the FOXO3a deacetylation and thus, inactivation. Cyclin D1/2 and G2, p27Kip1, p130 (RB2), GADD45, MnSOD, Polo-like kinase 1 (PLK1), Atm and the microRNA34-c are regulated by FOXO3a at the transcriptional level, modulating the processes of DNA damage response, resistance to oxidative stress, cell cycle checkpoints and senescent/quiescent state. Additionaly, FOXO3a role in regulation of the aforementioned biological processes relies, at least in part, on its ability to suppress the expression of the FOXM1 transcription factor, an important regulator of DNA repair and DNA damage response.
Evidence from these studies also argues strongly that FOXO3a, FOXM1 and their downstream gene signatures can be reliable diagnostic markers for cancer progression as well as genotoxic drug resistance.
ACKNOWLEDGEMENTS
This work is funded by grants from Breast Cancer Campaign, and Cancer Research UK. Gabriela Nestal de Moraes is supported in part by an internship from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Brazil. Stefania Zona is a post-doctoral fellow supported by Breast Cancer Campaign UK, and Laura Bella is supported by a studentship also from Breast Cancer Campaign. Matthew J. Burton is a recipient of the Wellcome Trust Biomedical Vacation Scholarship 2014.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
REFERENCES
- 1.Cheung-Ong K., Giaever G., Nislow C. DNA-Damaging Agents in Cancer Chemotherapy: Serendipity and Chemical Biology. Chem. Biol. 2013;20:648–659. doi: 10.1016/j.chembiol.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Stratton M.R., Campbell P.J., Futreal P.A. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Motoyama N., Naka K. DNA damage tumor suppressor genes and genomic instability. Curr. Opin. Genet. Dev. 2004;14:11–16. doi: 10.1016/j.gde.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Storr S.J., Woolston C.M., Zhang Y., Martin S.G. Redox environment, free radical, and oxidative DNA damage. Antioxid. Redox Signal. 2013;18:2399–2408. doi: 10.1089/ars.2012.4920. [DOI] [PubMed] [Google Scholar]
- 5.Jackson S.P., Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiricny J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006;7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 7.Podhorecka M, Skladanowski A, Bozko P. H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. . J Nucleic Acids . 2010. [DOI] [PMC free article] [PubMed]
- 8.Ridgway R.A., Serrels B., Mason S., et al. Focal adhesion kinase is required for beta-catenin induced mobilisation of epidermal stem cells. Carcinogenesis. 2012;33(12):2369–2376. doi: 10.1093/carcin/bgs284. [DOI] [PubMed] [Google Scholar]
- 9.Harper J.W., Elledge S.J. The DNA Damage Response: Ten Years After. Mol. Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 10.Gospodinov A., Herceg Z. Chromatin structure in double strand break repair. DNA Repair (Amst.) 2013;12:800–810. doi: 10.1016/j.dnarep.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 11.d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat. Rev. Cancer. 2008;8:512–522. doi: 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- 12.Gorgoulis V.G., Vassiliou L-V., Karakaidos P., et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 13.Tsai W.B., Chung Y.M., Takahashi Y., Xu Z., Hu M.C. Functional interaction between FOXO3a and ATM regulates DNA damage response. Nat. Cell Biol. 2008;10:460–467. doi: 10.1038/ncb1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rass U., Ahel I., West S.C. Defective DNA Repair and Neurodegenerative Disease. Cell. 2007;130:991–1004. doi: 10.1016/j.cell.2007.08.043. [DOI] [PubMed] [Google Scholar]
- 15.Kulkarni A., Wilson Iii D.M. The Involvement of DNA-Damage and -Repair Defects in Neurological Dysfunction. Am. J. Hum. Genet. 2008;82:539–566. doi: 10.1016/j.ajhg.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaestner K.H., Knochel W., Martinez D.E. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 2000;14:142–146. [PubMed] [Google Scholar]
- 17.Myatt S.S., Lam E.W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer. 2007;7:847–859. doi: 10.1038/nrc2223. [DOI] [PubMed] [Google Scholar]
- 18.Yang J.Y., Hung M.C. A new fork for clinical application: targeting forkhead transcription factors in cancer. Clin. Cancer Res. 2009;15:752–757. doi: 10.1158/1078-0432.CCR-08-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arden K.C. Multiple roles of FOXO transcription factors in mammalian cells point to multiple roles in cancer. Exp. Gerontol. 2006;41:709–717. doi: 10.1016/j.exger.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 20.Myatt S.S., Lam E.W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer. 2007;7:847–859. doi: 10.1038/nrc2223. [DOI] [PubMed] [Google Scholar]
- 21.Yang J.Y., Hung M.C. Deciphering the role of forkhead transcription factors in cancer therapy. Curr. Drug Targets. 2011;12:1284–1290. doi: 10.2174/138945011796150299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Y., Wang Y., Zhu W.G. Applications of post-translational modifications of FoxO family proteins in biological functions. J. Mol. Cell Biol. 2011;3:276–282. doi: 10.1093/jmcb/mjr013. [DOI] [PubMed] [Google Scholar]
- 23.Brunet A., Bonni A., Zigmond M.J., et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 24.Yang J.Y., Zong C.S., Xia W., et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008;10:138–148. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu M.C., Lee D-F., Xia W., et al. IκB Kinase Promotes Tumorigenesis through Inhibition of Forkhead FOXO3a. Cell. 2004;117:225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- 26.Brunet A., Park J., Tran H., Hu L.S., Hemmings B.A., Greenberg M.E. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell. Biol. 2001;21:952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Der Heide L.P., Hoekman M.F., Smidt M.P. The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem. J. 2004;380:297–309. doi: 10.1042/BJ20040167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang H, Tindall DJ. Regulation of FOXO protein stability via ubiquitination and proteasome degradation. . Biochim Biophys Acta. 2011. [DOI] [PMC free article] [PubMed]
- 29.Ho K.K., McGuire V.A., Koo C.Y., et al. Phosphorylation of FOXO3a on Ser-7 by p38 promotes its nuclear localization in response to doxorubicin. J. Biol. Chem. 2012;287:1545–1555. doi: 10.1074/jbc.M111.284224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sunters A., Madureira P.A., Pomeranz K.M., et al. Paclitaxel-induced nuclear translocation of FOXO3a in breast cancer cells is mediated by c-Jun NH2-terminal kinase and Akt. Cancer Res. 2006;66:212–220. doi: 10.1158/0008-5472.CAN-05-1997. [DOI] [PubMed] [Google Scholar]
- 31.Greer E.L., Oskoui P.R., Banko M.R., et al. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 2007;282:30107–30119. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 32.Lehtinen M.K., Yuan Z., Boag P.R., et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 33.Burgering B.M., Kops G.J. Cell cycle and death control: long live Forkheads. Trends Biochem. Sci. 2002;27:352–360. doi: 10.1016/s0968-0004(02)02113-8. [DOI] [PubMed] [Google Scholar]
- 34.Tran H., Brunet A., Grenier J.M., et al. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–534. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- 35.Lei H., Quelle F.W. FOXO transcription factors enforce cell cycle checkpoints and promote survival of hematopoietic cells after DNA damage. Mol. Cancer Res. 2009;7:1294–1303. doi: 10.1158/1541-7786.MCR-08-0531. [DOI] [PubMed] [Google Scholar]
- 36.Blake D.C., Jr, Mikse O.R., Freeman W.M., Herzog C.R. FOXO3a elicits a pro-apoptotic transcription program and cellular response to human lung carcinogen nicotine-derived nitrosaminoketone (NNK). Lung Cancer. 2010;67:37–47. doi: 10.1016/j.lungcan.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 37.Sunters A., Fernandez de Mattos S., et al. FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J. Biol. Chem. 2003;278:49795–49805. doi: 10.1074/jbc.M309523200. [DOI] [PubMed] [Google Scholar]
- 38.Toyoshima H., Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994;78:67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- 39.Collado M., Medema R.H., Garcia-Cao I., et al. Inhibition of the phosphoinositide 3-kinase pathway induces a senescence-like arrest mediated by p27Kip1. J. Biol. Chem. 2000;275:21960–21968. doi: 10.1074/jbc.M000759200. [DOI] [PubMed] [Google Scholar]
- 40.Schmidt M., Fernandez de Mattos S., van der Horst A., et al. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol. Cell. Biol. 2002;22:7842–7852. doi: 10.1128/MCB.22.22.7842-7852.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alvarez B., Martinez A.C., Burgering B.M., Carrera A.C. Forkhead transcription factors contribute to execution of the mitotic programme in mammals. Nature. 2001;413:744–747. doi: 10.1038/35099574. [DOI] [PubMed] [Google Scholar]
- 42.Martinez-Gac L., Marques M., Garcia Z., Campanero M.R., Carrera A.C. Control of cyclin G2 mRNA expression by forkhead transcription factors: novel mechanism for cell cycle control by phosphoinositide 3-kinase and forkhead. Mol. Cell. Biol. 2004;24:2181–2189. doi: 10.1128/MCB.24.5.2181-2189.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bennin D.A., Don A.S., Brake T., et al. Cyclin G2 associates with protein phosphatase 2A catalytic and regulatory B' subunits in active complexes and induces nuclear aberrations and a G1/S phase cell cycle arrest. J. Biol. Chem. 2002;277:27449–27467. doi: 10.1074/jbc.M111693200. [DOI] [PubMed] [Google Scholar]
- 44.Grana X., Garriga J., Mayol X. Role of the retinoblastoma protein family, pRB, p107 and p130 in the negative control of cell growth. Oncogene. 1998;17:3365–3383. doi: 10.1038/sj.onc.1202575. [DOI] [PubMed] [Google Scholar]
- 45.Smith E.J., Leone G., DeGregori J., Jakoi L., Nevins J.R. The accumulation of an E2F-p130 transcriptional repressor distinguishes a G0 cell state from a G1 cell state. Mol. Cell. Biol. 1996;16:6965–6976. doi: 10.1128/mcb.16.12.6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kops G.J., Dansen T.B., Polderman P.E., et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 47.Kops G.J., Medema R.H., Glassford J., et al. Control of cell cycle exit and entry by protein kinase B-regulated forkhead transcription factors. Mol. Cell. Biol. 2002;22:2025–2036. doi: 10.1128/MCB.22.7.2025-2036.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kress T.R., Cannell I.G., Brenkman A.B., et al. The MK5/PRAK Kinase and Myc Form a Negative Feedback Loop that Is Disrupted during Colorectal Tumorigenesis. Mol. Cell. 2011;41:445–457. doi: 10.1016/j.molcel.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 49.Tan W.Q., Wang K., Lv D.Y., Li P.F. Foxo3a inhibits cardiomyocyte hypertrophy through transactivating catalase. J. Biol. Chem. 2008;283:29730–29739. doi: 10.1074/jbc.M805514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shalini S., Dorstyn L., Wilson C., Puccini J., Ho L., Kumar S. Impaired antioxidant defence and accumulation of oxidative stress in caspase-2-deficient mice. Cell Death Differ. 2012;19:1370–1380. doi: 10.1038/cdd.2012.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y., Padalecki S.S., Chaudhuri A.R., et al. Caspase-2 deficiency enhances aging-related traits in mice. Mech. Ageing Dev. 2007;128:213–221. doi: 10.1016/j.mad.2006.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yin L., Huang L., Kufe D. MUC1 oncoprotein activates the FOXO3a transcription factor in a survival response to oxidative stress. J. Biol. Chem. 2004;279:45721–45727. doi: 10.1074/jbc.M408027200. [DOI] [PubMed] [Google Scholar]
- 53.Yin L., Li Y., Ren J., Kuwahara H., Kufe D. Human MUC1 carcinoma antigen regulates intracellular oxidant levels and the apoptotic response to oxidative stress. J. Biol. Chem. 2003;278:35458–35464. doi: 10.1074/jbc.M301987200. [DOI] [PubMed] [Google Scholar]
- 54.You H., Jang Y., You-Ten A.I., et al. p53-dependent inhibition of FKHRL1 in response to DNA damage through protein kinase SGK1. Proc. Natl. Acad. Sci. USA. 2004;101:14057–14062. doi: 10.1073/pnas.0406286101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu M.T., Chen Y.R., Chen S.C., et al. Epstein-Barr virus latent membrane protein 1 induces micronucleus formation, represses DNA repair and enhances sensitivity to DNA-damaging agents in human epithelial cells. Oncogene. 2004;23:2531–2539. doi: 10.1038/sj.onc.1207375. [DOI] [PubMed] [Google Scholar]
- 56.Chen Y.R., Liu M.T., Chang Y.T., Wu C.C., Hu C.Y., Chen J.Y. Epstein-Barr virus latent membrane protein 1 represses DNA repair through the PI3K/Akt/FOXO3a pathway in human epithelial cells. J. Virol. 2008;82:8124–8137. doi: 10.1128/JVI.00430-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Furukawa-Hibi Y., Yoshida-Araki K., Ohta T., Ikeda K., Motoyama N. FOXO forkhead transcription factors induce G(2)-M checkpoint in response to oxidative stress. J. Biol. Chem. 2002;277:26729–26732. doi: 10.1074/jbc.C200256200. [DOI] [PubMed] [Google Scholar]
- 58.Takekawa M., Saito H. A Family of Stress-Inducible GADD45-like Proteins Mediate Activation of the Stress-Responsive MTK1/MEKK4 MAPKKK. Cell. 1998;95:521–530. doi: 10.1016/s0092-8674(00)81619-0. [DOI] [PubMed] [Google Scholar]
- 59.Smith C.C., Aylott M.C., Fisher K.J., Lynch A.M., Gooderham N.J. DNA damage responses after exposure to DNA-based products. J. Gene Med. 2006;8:175–185. doi: 10.1002/jgm.827. [DOI] [PubMed] [Google Scholar]
- 60.Lam E.W., Brosens J.J., Gomes A.R., Koo C.Y. Forkhead box proteins: tuning forks for transcriptional harmony. Nat. Rev. Cancer. 2013;13:482–495. doi: 10.1038/nrc3539. [DOI] [PubMed] [Google Scholar]
- 61.Karadedou C.T., Gomes A.R., Chen J., et al. FOXO3a represses VEGF expression through FOXM1-dependent and -independent mechanisms in breast cancer. Oncogene. 2012;31:1845–1858. doi: 10.1038/onc.2011.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koo CY, Muir KW, Lam EW. FOXM1: From cancer initiation to progression and treatment. Biochim Biophys Acta . [DOI] [PubMed]
- 63.Smith M.L., Ford J.M., Hollander M.C., et al. p53-mediated DNA repair responses to UV radiation: studies of mouse cells lacking p53, p21, and/or gadd45 genes. Mol. Cell. Biol. 2000;20:3705–3714. doi: 10.1128/mcb.20.10.3705-3714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zona S, Bella L, Burton MJ, Nestal de Moraes G, Lam EW. FOXM1: An emerging master regulator of DNA damage response and genotoxic agent resistance. . Biochim Biophys Acta. 2014. [DOI] [PMC free article] [PubMed]
- 65.Kalinina O.A., Kalinin S.A., Polack E.W., et al. Sustained hepatic expression of FoxM1B in transgenic mice has minimal effects on hepatocellular carcinoma development but increases cell proliferation rates in preneoplastic and early neoplastic lesions. Oncogene. 2003;22:6266–6276. doi: 10.1038/sj.onc.1206640. [DOI] [PubMed] [Google Scholar]
- 66.Kalin T.V., Wang I.C., Ackerson T.J., et al. Increased levels of the FoxM1 transcription factor accelerate development and progression of prostate carcinomas in both TRAMP and LADY transgenic mice. Cancer Res. 2006;66:1712–1720. doi: 10.1158/0008-5472.CAN-05-3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim I.M., Ackerson T., Ramakrishna S., et al. The Forkhead Box m1 transcription factor stimulates the proliferation of tumor cells during development of lung cancer. Cancer Res. 2006;66:2153–2161. doi: 10.1158/0008-5472.CAN-05-3003. [DOI] [PubMed] [Google Scholar]
- 68.Liu M., Dai B., Kang S.H., et al. FoxM1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Cancer Res. 2006;66:3593–3602. doi: 10.1158/0008-5472.CAN-05-2912. [DOI] [PubMed] [Google Scholar]
- 69.Madureira P.A., Varshochi R., Constantinidou D., et al. The Forkhead box M1 protein regulates the transcription of the estrogen receptor alpha in breast cancer cells. J. Biol. Chem. 2006;281:25167–25176. doi: 10.1074/jbc.M603906200. [DOI] [PubMed] [Google Scholar]
- 70.Ma R.Y., Tong T.H., Cheung A.M., Tsang A.C., Leung W.Y., Yao K.M. Raf/MEK/MAPK signaling stimulates the nuclear translocation and transactivating activity of FOXM1c. J. Cell Sci. 2005;118:795–806. doi: 10.1242/jcs.01657. [DOI] [PubMed] [Google Scholar]
- 71.Wang Z., Banerjee S., Kong D., Li Y., Sarkar F.H. Down-regulation of Forkhead Box M1 transcription factor leads to the inhibition of invasion and angiogenesis of pancreatic cancer cells. Cancer Res. 2007;67:8293–8300. doi: 10.1158/0008-5472.CAN-07-1265. [DOI] [PubMed] [Google Scholar]
- 72.Yoshida Y., Wang I.C., Yoder H.M., Davidson N.O., Costa R.H. The forkhead box M1 transcription factor contributes to the development and growth of mouse colorectal cancer. Gastroenterology. 2007;132:1420–1431. doi: 10.1053/j.gastro.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 73.Teh M.T., Wong S.T., Neill G.W., Ghali L.R., Philpott M.P., Quinn A.G. FOXM1 is a downstream target of Gli1 in basal cell carcinomas. Cancer Res. 2002;62:4773–4780. [PubMed] [Google Scholar]
- 74.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gemenetzidis E., Bose A., Riaz A.M., et al. FOXM1 upregulation is an early event in human squamous cell carcinoma and it is enhanced by nicotine during malignant transformation. PLoS One. 2009;4:e4849. doi: 10.1371/journal.pone.0004849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Teh M.T., Gemenetzidis E., Chaplin T., Young B.D., Philpott M.P. Upregulation of FOXM1 induces genomic instability in human epidermal keratinocytes. Mol. Cancer. 2010;9:45. doi: 10.1186/1476-4598-9-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huynh K.M., Soh J.W., Dash R., Sarkar D., Fisher P.B., Kang D. FOXM1 expression mediates growth suppression during terminal differentiation of HO-1 human metastatic melanoma cells. J. Cell. Physiol. 2011;226:194–204. doi: 10.1002/jcp.22326. [DOI] [PubMed] [Google Scholar]
- 78.Janus J.R., Laborde R.R., Greenberg A.J., et al. Linking expression of FOXM1, CEP55 and HELLS to tumorigenesis in oropharyngeal squamous cell carcinoma. Laryngoscope. 2011;121:2598–2603. doi: 10.1002/lary.22379. [DOI] [PubMed] [Google Scholar]
- 79.Nakamura S., Hirano I., Okinaka K., et al. The FOXM1 transcriptional factor promotes the proliferation of leukemia cells through modulation of cell cycle progression in acute myeloid leukemia. Carcinogenesis. 2010;31:2012–2021. doi: 10.1093/carcin/bgq185. [DOI] [PubMed] [Google Scholar]
- 80.Pilarsky C., Wenzig M., Specht T., Saeger H.D., Grutzmann R. Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia. 2004;6:744–750. doi: 10.1593/neo.04277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Curtis C., Shah S.P., Chin S.F., et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bella L., Zona S., Nestal de Moraes G., Lam E.W. FOXM1: A key oncofoetal transcription factor in health and disease. Semin. Cancer Biol. 2014;29:32–39. doi: 10.1016/j.semcancer.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 83.Wang I.C., Chen Y.J., Hughes D.E., et al. FoxM1 regulates transcription of JNK1 to promote the G1/S transition and tumor cell invasiveness. J. Biol. Chem. 2008;283:20770–20778. doi: 10.1074/jbc.M709892200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Khongkow P., Karunarathna U., Khongkow M., Gong C., Gomes A.R., Yague E., et al. FOXM1 targets NBS1 to regulate DNA damage-induced senescence and epirubicin resistance. Oncogene. 2014;33:4144–4155. doi: 10.1038/onc.2013.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Monteiro L.J., Khongkow P., Kongsema M., et al. The Forkhead Box M1 protein regulates BRIP1 expression and DNA damage repair in epirubicin treatment. Oncogene. 2013;32:4634–4645. doi: 10.1038/onc.2012.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Myatt S.S., Kongsema M., Man C.W., et al. SUMOylation inhibits FOXM1 activity and delays mitotic transition. Oncogene. 2014;33:4316–4329. doi: 10.1038/onc.2013.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wood R.D., Shivji M.K. Which DNA polymerases are used for DNA-repair in eukaryotes? Carcinogenesis. 1997;18:605–610. doi: 10.1093/carcin/18.4.605. [DOI] [PubMed] [Google Scholar]
- 88.Park Y.Y., Jung S.Y., Jennings N., et al. FOXM1 mediates Dox resistance in breast cancer by enhancing DNA repair. Carcinogenesis. 2012 doi: 10.1093/carcin/bgs1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eckers J.C., Kalen A.L., Sarsour E.H., et al. Forkhead box m1 regulates quiescence-associated radioresistance of human head and neck squamous carcinoma cells. Radiat. Res. 2014;182:420–429. doi: 10.1667/RR13726.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lu T.P., Hsu Y.Y., Lai L.C., Tsai M.H., Chuang E.Y. Identification of gene expression biomarkers for predicting radiation exposure. Sci. Rep. 2014;4:6293. doi: 10.1038/srep06293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Andressoo J.O., Hoeijmakers J.H. Transcription-coupled repair and premature ageing. Mutat. Res. 2005;577:179–194. doi: 10.1016/j.mrfmmm.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 92.Zhou J., Wang Y., Wang Y., et al. FOXM1 Modulates Cisplatin Sensitivity by Regulating EXO1 in Ovarian Cancer. PLoS One. 2014;9:e96989. doi: 10.1371/journal.pone.0096989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ye H., Holterman A.X., Yoo K.W., Franks R.R., Costa R.H. Premature expression of the winged helix transcription factor HFH-11B in regenerating mouse liver accelerates hepatocyte entry into S phase. Mol. Cell. Biol. 1999;19:8570–8580. doi: 10.1128/mcb.19.12.8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.D'Andrea A.D., Grompe M. The Fanconi anaemia/BRCA pathway. Nat. Rev. Cancer. 2003;3:23–34. doi: 10.1038/nrc970. [DOI] [PubMed] [Google Scholar]
- 95.Kim H., D'Andrea A.D. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26:1393–1408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang N., Wu X., Yang L., et al. FoxM1 Inhibition Sensitizes Resistant Glioblastoma Cells to Temozolomide by Downregulating the Expression of DNA-Repair Gene Rad51. Clin. Cancer Res. 2012;18:5961–5971. doi: 10.1158/1078-0432.CCR-12-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tan Y., Raychaudhuri P., Costa R.H. Chk2 mediates stabilization of the FoxM1 transcription factor to stimulate expression of DNA repair genes. Mol. Cell. Biol. 2007;27:1007–1016. doi: 10.1128/MCB.01068-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Valle L., Hernandez-Illan E., Bellido F., et al. New insights into POLE and POLD1 germline mutations in familial colorectal cancer and polyposis. Hum. Mol. Genet. 2014;23(13):3506–3512. doi: 10.1093/hmg/ddu058. [DOI] [PubMed] [Google Scholar]
- 99.Peng M., Litman R., Xie J., Sharma S., Brosh R.M., Jr, Cantor S.B. The FANCJ/MutLalpha interaction is required for correction of the cross-link response in FA-J cells. EMBO J. 2007;26:3238–3249. doi: 10.1038/sj.emboj.7601754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lee J.H., Paull T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 101.Paull T.T., Lee J.H. The Mre11/Rad50/Nbs1 complex and its role as a DNA double-strand break sensor for ATM. Cell Cycle. 2005;4:737–740. doi: 10.4161/cc.4.6.1715. [DOI] [PubMed] [Google Scholar]
- 102.San Filippo J., Sung P., Klein H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 103.So S., Davis A.J., Chen D.J. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J. Cell Biol. 2009;187:977–990. doi: 10.1083/jcb.200906064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dupre A., Boyer-Chatenet L., Gautier J. Two-step activation of ATM by DNA and the Mre11-Rad50-Nbs1 complex. Nat. Struct. Mol. Biol. 2006;13:451–457. doi: 10.1038/nsmb1090. [DOI] [PubMed] [Google Scholar]
- 105.Riches L.C., Lynch A.M., Gooderham N.J. Early events in the mammalian response to DNA double-strand breaks. Mutagenesis. 2008;23:331–339. doi: 10.1093/mutage/gen039. [DOI] [PubMed] [Google Scholar]
- 106.Kitagawa R., Bakkenist C.J., McKinnon P.J., Kastan M.B. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 2004;18:1423–1438. doi: 10.1101/gad.1200304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Matsuoka S., Huang M., Elledge S.J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- 108.Carney J.P., Maser R.S., Olivares H., et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–486. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- 109.Kobayashi J., Antoccia A., Tauchi H., Matsuura S., Komatsu K. NBS1 and its functional role in the DNA damage response. DNA Repair (Amst.) 2004;3:855–861. doi: 10.1016/j.dnarep.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 110.Wang I.C., Chen Y.J., Hughes D., et al. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol. Cell. Biol. 2005;25:10875–10894. doi: 10.1128/MCB.25.24.10875-10894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wu J., Zhang X., Zhang L., et al. Skp2 E3 ligase integrates ATM activation and homologous recombination repair by ubiquitinating NBS1. Mol. Cell. 2012;46:351–361. doi: 10.1016/j.molcel.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mohaghegh P., Karow J.K., Brosh R.M., Jr, Bohr V.A., Hickson I.D. The Bloom's and Werner's syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001;29:2843–2849. doi: 10.1093/nar/29.13.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hiom K. FANCJ: solving problems in DNA replication. DNA Repair (Amst.) 2010;9:250–256. doi: 10.1016/j.dnarep.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 114.Cantor S.B., Guillemette S. Hereditary breast cancer and the BRCA1-associated FANCJ/BACH1/BRIP1. Future Oncol. 2011;7:253–261. doi: 10.2217/fon.10.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ma L., Wang H., Yao H., Zhu L., Liu W., Zhou Z. Bmi1 expression in oral lichen planus and the risk of progression to oral squamous cell carcinoma. Ann. Diagn. Pathol. 2013;17:327–330. doi: 10.1016/j.anndiagpath.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 116.Mori R., Yoshida K., Tanahashi T., Yawata K., Kato J., Okumura N., et al. Decreased FANCJ caused by 5FU contributes to the increased sensitivity to oxaliplatin in gastric cancer cells. Gastric Cancer. 2013;16:345–354. doi: 10.1007/s10120-012-0191-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lieber M.R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- 118.Quennet V., Beucher A., Barton O., Takeda S., Lobrich M. CtIP and MRN promote non-homologous end-joining of etoposide-induced DNA double-strand breaks in G1. Nucleic Acids Res. 2011;39:2144–2152. doi: 10.1093/nar/gkq1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Goodarzi A.A., Noon A.T., Deckbar D., et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 120.Green C.M., Almouzni G. When repair meets chromatin. First in series on chromatin dynamics. EMBO Rep. 2002;3:28–33. doi: 10.1093/embo-reports/kvf005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Green C.M., Almouzni G. Local action of the chromatin assembly factor CAF-1 at sites of nucleotide excision repair in vivo. EMBO J. 2003;22:5163–5174. doi: 10.1093/emboj/cdg478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Teh M.T., Gemenetzidis E., Patel D., Tariq R., Nadir A., Bahta A.W., et al. FOXM1 induces a global methylation signature that mimics the cancer epigenome in head and neck squamous cell carcinom. PLoS One. 2012;7:e34329. doi: 10.1371/journal.pone.0034329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jin B., Robertson K.D. DNA methyltransferases, DNA damage repair, and cancer. Adv. Exp. Med. Biol. 2013;754:3–29. doi: 10.1007/978-1-4419-9967-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wonsey D.R., Follettie M.T. Loss of the forkhead transcription factor FoxM1 causes centrosome amplification and mitotic catastrophe. Cancer Res. 2005;65:5181–5189. doi: 10.1158/0008-5472.CAN-04-4059. [DOI] [PubMed] [Google Scholar]
- 125.Millour J., de Olano N., Horimoto Y., et al. ATM and p53 regulate FOXM1 expression via E2F in breast cancer epirubicin treatment and resistance. Mol. Cancer Ther. 2011;10:1046–1058. doi: 10.1158/1535-7163.MCT-11-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.de Olano N., Koo C.Y., Monteiro L.J., et al. The p38 MAPK-MK2 axis regulates E2F1 and FOXM1 expression after epirubicin treatment. Mol. Cancer Res. 2012;10:1189–1202. doi: 10.1158/1541-7786.MCR-11-0559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang X., Quail E., Hung N.J., Tan Y., Ye H., Costa R.H. Increased levels of forkhead box M1B transcription factor in transgenic mouse hepatocytes prevent age-related proliferation defects in regenerating liver. Proc. Natl. Acad. Sci. USA. 2001;98:11468–11473. doi: 10.1073/pnas.201360898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bornstein G., Bloom J., Sitry-Shevah D., Nakayama K., Pagano M., Hershko A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J. Biol. Chem. 2003;278:25752–25757. doi: 10.1074/jbc.M301774200. [DOI] [PubMed] [Google Scholar]
- 129.Tsvetkov L.M., Yeh K.H., Lee S.J., Sun H., Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr. Biol. 1999;9:661–664. doi: 10.1016/s0960-9822(99)80290-5. [DOI] [PubMed] [Google Scholar]
- 130.Wang X., Kiyokawa H., Dennewitz M.B., Costa R.H. The Forkhead Box m1b transcription factor is essential for hepatocyte DNA replication and mitosis during mouse liver regeneration. Proc. Natl. Acad. Sci. USA. 2002;99:16881–16886. doi: 10.1073/pnas.252570299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Down CF, Millour J, Lam EW, Watson RJ. Binding of FoxM1 to G2/M gene promoters is dependent upon B-Myb. . Biochim Biophys Acta . 1819. [DOI] [PubMed]
- 132.Sadasivam S., Duan S., DeCaprio J.A. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev. 2012;26:474–489. doi: 10.1101/gad.181933.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Grant G.D., Brooks L., III, Zhang X., et al. Identification of cell cycle-regulated genes periodically expressed in U2OS cells and their regulation by FOXM1 and E2F transcription factors. Mol. Biol. Cell. 2013;24:3634–3650. doi: 10.1091/mbc.E13-05-0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhao B., Barrera L.A., Ersing I., et al. The NF-kappaB Genomic Landscape in Lymphoblastoid B Cells. Cell Reports. 2014;8:1595–1606. doi: 10.1016/j.celrep.2014.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tian T., Li J., Li B., et al. Genistein exhibits anti-cancer effects via down-regulating FoxM1 in H446 small-cell lung cancer cells. Tumour Biol. 2014;35:4137–4145. doi: 10.1007/s13277-013-1542-0. [DOI] [PubMed] [Google Scholar]
- 136.Li S.K., Smith D.K., Leung W.Y., et al. FoxM1c counteracts oxidative stress-induced senescence and stimulates Bmi-1 expression. J. Biol. Chem. 2008;283:16545–16553. doi: 10.1074/jbc.M709604200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zha Y., Xia Y., Ding J., et al. MEIS2 is essential for neuroblastoma cell survival and proliferation by transcriptional control of M-phase progression. Cell Death Dis. 2014;5:e1417. doi: 10.1038/cddis.2014.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wan X., Choh Y., Kim S.Y., et al. Identification of the FoxM1/Bub1b signaling pathway as a required component for growth and survival of rhabdomyosarcoma. Cancer Res. 2012;72(22):5889–5899. doi: 10.1158/0008-5472.CAN-12-1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Al-Ejeh F., Kumar R., Wiegmans A., Lakhani S.R., Brown M.P., Khanna K.K. Harnessing the complexity of DNA-damage response pathways to improve cancer treatment outcomes. Oncogene. 2010;29:6085–6098. doi: 10.1038/onc.2010.407. [DOI] [PubMed] [Google Scholar]
- 140.Fernandez de Mattos S., Villalonga P., Clardy J., Lam E.W. FOXO3a mediates the cytotoxic effects of cisplatin in colon cancer cells. Mol. Cancer Ther. 2008;7:3237–3246. doi: 10.1158/1535-7163.MCT-08-0398. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 141.Xia W., Bacus S., Hegde P., et al. A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc. Natl. Acad. Sci. USA. 2006;103:7795–7800. doi: 10.1073/pnas.0602468103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Krol J., Francis R.E., Albergaria A., et al. The transcription factor FOXO3a is a crucial cellular target of gefitinib (Iressa) in breast cancer cells. Mol. Cancer Ther. 2007;6:3169–3179. doi: 10.1158/1535-7163.MCT-07-0507. [DOI] [PubMed] [Google Scholar]
- 143.McGovern U.B., Francis R.E., Peck B., et al. Gefitinib (Iressa) represses FOXM1 expression via FOXO3a in breast cancer. Mol. Cancer Ther. 2009;8:582–591. doi: 10.1158/1535-7163.MCT-08-0805. [DOI] [PubMed] [Google Scholar]
- 144.Pellicano F., Scott M.T., Helgason G.V., Hopcroft L.E., Allan E.K., Aspinall-O'Dea M., et al. The Antiproliferative Activity of Kinase Inhibitors in Chronic Myeloid Leukemia Cells Is Mediated by FOXO Transcription Factors. Stem Cells. 2014;32:2324–2337. doi: 10.1002/stem.1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Weng S-C., Kashida Y., Kulp S.K., et al. Sensitizing estrogen receptor–negative breast cancer cells to tamoxifen with OSU-03012, a novel celecoxib-derived phosphoinositide-dependent protein kinase-1/Akt signaling inhibitor. Mol. Cancer Ther. 2008;7:800–808. doi: 10.1158/1535-7163.MCT-07-0434. [DOI] [PubMed] [Google Scholar]
- 146.Kim A., Lee J.E., Lee S.S., et al. Coexistent mutations of KRAS and PIK3CA affect the efficacy of NVP-BEZ235, a dual PI3K/MTOR inhibitor, in regulating the PI3K/MTOR pathway in colorectal cancer. Int. J. Cancer. 2013;133:984–996. doi: 10.1002/ijc.28073. [DOI] [PubMed] [Google Scholar]
- 147.Brunet A., Sweeney L.B., Sturgill J.F., et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 148.Khongkow M., Olmos Y., Gong C., et al. SIRT6 modulates paclitaxel and epirubicin resistance and survival in breast cancer. Carcinogenesis. 2013;34:1476–1486. doi: 10.1093/carcin/bgt098. [DOI] [PubMed] [Google Scholar]
- 149.Thakur B.K., Lippka Y., Dittrich T., Chandra P., Skokowa J., Welte K. NAMPT pathway is involved in the FOXO3a-mediated regulation of GADD45A expression. Biochem. Biophys. Res. Commun. 2012;420:714–720. doi: 10.1016/j.bbrc.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 150.Wang F., Chan C.H., Chen K., Guan X., Lin H.K., Tong Q. Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to Skp2-mediated FOXO3 ubiquitination and degradation. Oncogene. 2012;31:1546–1557. doi: 10.1038/onc.2011.347. [DOI] [PubMed] [Google Scholar]
- 151.Hoffmann G., Breitenbücher F., Schuler M., Ehrenhofer-Murray A.E. A Novel Sirtuin 2 (SIRT2) Inhibitor with p53-dependent Pro-apoptotic Activity in Non-small Cell Lung Cancer. J. Biol. Chem. 2014;289:5208–5216. doi: 10.1074/jbc.M113.487736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Peck B., Chen C.Y., Ho K.K., et al. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol. Cancer Ther. 2010;9:844–855. doi: 10.1158/1535-7163.MCT-09-0971. [DOI] [PubMed] [Google Scholar]
- 153.Bhat U.G., Halasi M., Gartel A.L. FoxM1 is a general target for proteasome inhibitors. PLoS One. 2009;4:e6593. doi: 10.1371/journal.pone.0006593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kwok J.M., Myatt S.S., Marson C.M., Coombes R.C., Constantinidou D., Lam E.W. Thiostrepton selectively targets breast cancer cells through inhibition of forkhead box M1 expression. Mol. Cancer Ther. 2008;7:2022–2032. doi: 10.1158/1535-7163.MCT-08-0188. [DOI] [PubMed] [Google Scholar]
- 155.Hegde N.S., Sanders D.A., Rodriguez R., Balasubramanian S. The transcription factor FOXM1 is a cellular target of the natural product thiostrepton. Nat. Chem. 2011;3:725–731. doi: 10.1038/nchem.1114. [DOI] [PubMed] [Google Scholar]
- 156.Kainz K.P., Krenn L., Erdem Z., et al. 2-deprenyl-rheediaxanthone B isolated from Metaxya rostrata induces active cell death in colorectal tumor cells. PLoS One. 2013;8:e65745. doi: 10.1371/journal.pone.0065745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kalinichenko V.V., Gusarova G.A., Kim I.M., et al. Foxf1 haploinsufficiency reduces Notch-2 signaling during mouse lung development. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004;286:L521–L530. doi: 10.1152/ajplung.00212.2003. [DOI] [PubMed] [Google Scholar]