

Abstract
Background: In the past decade, the identification and characterization of antiviral genes with the ability to interfere with virus replication has established cell-intrinsic innate immunity as a third line of antiviral defense in addition to adaptive and classical innate immunity. Understanding how cellular factors have evolved to inhibit HIV-1 reveals particularly vulnerable points of the viral replication cycle. Many, but not all, antiviral proteins share type I interferon-upregulated expression and sensitivity to viral counteraction or evasion measures. Whereas well-established restriction factors interfere with early post-entry steps and release of HIV-1, recent research has revealed a diverse set of proteins that reduce the infectious quality of released particles using individual, to date poorly understood modes of action. These include induction of paucity of mature glycoproteins in nascent virions or self-incorporation into the virus particle, resulting in poor infectiousness of the virion and impaired spread of the infection.
Conclusion: A better understanding of these newly discovered antiviral factors may open new avenues towards the design of drugs that repress the spread of viruses whose genomes have already integrated.
Keywords: HIV-1 Nef, IFITM, interferon, interferon-stimulated genes, particle infectivity, SERINC, 90K
Assembly and maturation of HIV-1 particles
Like all intracellularly replicating pathogens, HIV-1 undergoes extensive interplay with cellular machineries and benefits from interactions with essential cofactors which enable and support HIV-1 replication. Well-known examples include the HIV-1 receptor CD4, chemokine coreceptors CCR5 and CXCR4, the Tat-interacting CyclinT1 that enhances processivity of the RNA polymerase II during LTR-mediated viral transcription and the nuclear mRNA export factor CRM1. On the contrary, eukaryotic cells dispose of a set of antiviral genes, whose expression is often induced or enhanced by type I interferons (IFNs), and whose products can effectively interfere with distinct steps of the HIV-1 replication cycle. The impacts of anti-HIV-1 factors acting early post-entry and during virus release include SAMHD1, APOBEC3G, Mx2 and tetherin, respectively, and are discussed extensively elsewhere.
Following integration of the proviral DNA into the host cell´s chromosome and LTR-driven transcription, newly synthesized viral proteins and the viral genomic RNA are transported to the plasma membrane in a coordinated fashion, followed by particle assembly and egress. The composition of these particles is tightly controlled and requires the proper function of viral structural, enzymatic, and accessory proteins, but also cellular factors. A budded virion is confined by a plasma membrane-derived lipid bilayer containing approximately 7-14 trimers of Envelope glycoproteins [1, 2]. Gag, besides constituting the most abundant protein within virions, is essential in recruiting the viral genomic RNAs and other essential proteins, including protease, integrase and reverse transcriptase. Of note, a ribosomal frameshifting mechanism ensures incorporation of these enzymatic proteins which are expressed from a Gag-Pol polyprotein at a low frequency of 5%. Incorporated accessory proteins include Vpr, Vif and Nef. A final, protease-mediated series of Gag and Gag-Pol cleavage and rearrangement events renders immature virions fully infectious, resulting in the virons´ interior typical morphological transition from an electron-dense ring lining the surrounding membrane to a cone-shaped appearance in electron microscopy. These rearrangements furthermore trigger the clustering of the sparse Env trimers on the virus surface, an event that might contribute to virus maturation and infectiousness [3]. Importantly, the right configuration of particles is crucial for assuring a high infectious quality, which is reflected by their ability to efficiently infect new target cells. Prevention of APOBEC3G incorporation by HIV-1 Vif provides a compelling example for the fact that modulation of the (cellular or viral) protein composition of nascent virions can have dramatic (positive or negative) effects on the infectiousness of HIV-1 particles. Alternatively, alteration of the virus membrane fluidity, restriction of the accessibility of the HIV-1 Env glycoprotein to the HIV-1 receptor/coreceptor complex by steric hindrance through an incorporated protein or inhibition of the maturation-induced clustering of HIV-1 Env trimers could represent mechanisms of cellular defense that target particle infectivity. Despite these theoretical possibilities, until recently no antiviral protein was known that directly reduces particle infectivity.
Within the cytokine family of interferons, type I and type III IFNs exert antiviral properties, whereas type II IFNs have been considered more immunomodulatory. Whereas type III IFNs signal via a specific type III IFN receptor, which is probably not expressed at high levels on primary HIV-1 target cells in vivo [4-6], type I IFNs robustly induce upregulated expression of antiviral IFN-stimulated genes (ISGs) in cultured primary HIV-1 target cells, including CD4+ T-cells and macrophages. ISGs constitute a large group of several hundred genes, from which only a few of them have been analyzed in-depth for potential antiviral activity. This review highlights recent research that identifies processes safeguarding HIV-1 ´s particle infectivity being highly vulnerable. Particle infectivity is negatively modulated by a diverse set of cellular proteins, comprising ISGs and non-ISGs, each of them with an individual mode of action and different sensitivity to viral counteracting strategies.
90K/LGALS3BP
90K (according to its appearance as a 90 kDa species in SDS-PAGE)/lectin galectin soluble 3 binding protein (LGALS3BP) is a secretory glycoprotein [7]. It was initially identified as a tumor antigen which is upregulated in the context of breast [8] and lung [9] carcinoma. Physiological functions of extracellular 90K comprise modulation of cell migration and adhesion by interaction with multiple cell surface receptors, including galectin-3, and modulation of the activity of immune cells [10]. In healthy individuals, 90K protein reaches concentrations up to the µg/ml range in diverse body fluids. Since it shares mRNA upregulation in CD4+ T-cells of HIV-1 infected individuals [11] and type I IFN-induced expression [10, 12, 13] with established restriction factors of HIV-1, it was included in a functional screen of candidate ISGs for antiviral activity (Goffinet, unpublished data). 90K acts antiviral against HIV-1 by reducing the particle infectivity of newly produced HIV-1 virions [14]. This defect is accompanied by impaired incorporation of HIV-1 Env glycoproteins into virus particles, probably explaining the reduced infectious quality of de novo-synthesized virions, and defective processing of the HIV-1 Env gp160 precursor into the mature proteins gp120 and gp41. Addition of exogenous, soluble 90K to the producer cell culture fails to inhibit particle infectivity, suggesting that 90K exerts its antiviral effect intracellularly and that outside-in signaling of extracellular 90K is dispensable for its antiviral function. Analysis of a set of modified variants of 90K revealed that the two central protein-binding domains BTB-POZ and IVR are required and sufficient for 90K´s antiviral activity, since deletion of the N-terminal scavenger receptor cysteine rich (SRCR)-like domain and the C-terminal domain did not abolish antiviral activity.
The exact mode of action of 90K remains elusive to date. Experimental data argues against a virion incorporation of 90K. Given that 90K and HIV-1 Env share subcellular localization and posttranslational maturation in the secretory pathway, and that 90K interferes with HIV-1 Env incorporation, a model in which 90K targets HIV-1 Env, either via direct or indirect interaction, has been favored. However, Co-IP experiments failed so far to provide evidence for a detectable interaction of 90K with HIV-1 Env. Thus, an indirect scenario, in which 90K inhibits the function or expression of a cellular or a viral factor that is required for efficient HIV-1 Env incorporation cannot be excluded. Although 90K expression induces a processing defect of the HIV-1 Env gp160 precursor, resulting in high cell-associated levels of uncleaved polyprotein and only a small fraction of mature gp120 and gp41, it remains unclear whether the processing defect is the direct cause for poor incorporation of mature glycoproteins or whether these two inhibitory actions can be genetically and/or functionally uncoupled. Future work is needed to clarify the exact mode of action of 90K.
In human target cells of HIV-1, 90K protein expression is highly cell-type specific. It is readily detectable in primary monocyte-derived macrophages [14]. Importantly, siRNA-mediated knockdown of 90K facilitates the spread of HIV-1 in cultures of primary macrophages and increases the particle infectivity of newly produced virions, indicating that endogenous expression levels of 90K contribute to the control of HIV-1 spread in this important HIV-1 target cell type. In striking contrast, 90K protein is undetectable in primary CD4+ T-cells and in all analyzed immortalized T-cell lines, even using very sensitive ELISA assays and despite type I IFN-mediated upregulation of 90K mRNA, suggesting that 90K expression is tightly repressed at the post-transcriptional level in T-cells. Given the specific expression of 90K in macrophages within those HIV-1 target cell types that fully support virus replication, prediction of the in vivo importance of 90K is difficult. Expression levels of 90K protein in the serum correlate with viral loads, and high 90K expression levels at the viral set-point are predictive of a negative outcome of the infection [15-17]. Although this seems to contradict a protective role of 90K for the host during HIV-1 infection, a positive correlation of ISG expression and viral loads has been long-appreciated [11, 18, 19]. This observation raises the possibility that sustained upregulation of type I IFNs during the chronic phase of HIV-1 infection indirectly promotes virus-beneficial effects in addition to the direct antiviral effects of type I IFN-induced ISGs which may be protective during acute HIV-1 infection. In addition, it is important to take into consideration that levels of soluble 90K may not necessarily reflect the level of the cell-associated, antiviral protein fraction. Along this line, potential HIV-1 evasion mechanisms still need to be defined.
Keeping the density of glycoprotein molecules on the virus surface low is a property of some viruses that is particularly pronounced for HIV-1, and is thought to provide a selective advantage during immune evasion [20]. It is unclear whether the drawback of reduction of virion infectivity, induced by 90K-mediated paucity of Env embedded in particles, is outweighed by improved immune escape. More work is warranted to fully understand the impact of 90K on HIV-1 infection.
Interferon induced transmembrane (IFITM) proteins
IFITM proteins are small two-pass transmembrane proteins which are located predominantly at the plasma membranes (IFITM1) or at endosomal and lysosomal membranes (IFITM2 and 3). They belong to the CD225/dispanin protein superfamily, which exists throughout the kingdoms, including bacteria, invertebrates and primates [21]. Additional family members, including IFITM5, whose expression is restricted to osteoblasts [22], and 10 additional, recently discovered human ifitm genes [21] probably do not contribute to inhibition of HIV-1 infection or haven´t yet been tested for antiviral activity, respectively. IFITM1, 2 and 3 are almost ubiquitously expressed and their expression is further upregulated by type I IFNs [22, 23]. IFITM proteins contribute to regulation of early development, cell adhesion, and control of cell growth [24]. Virologists´ interest in IFITM proteins steeply raised upon the first description of their antiviral activity against Influenza A virus and flaviviruses, including West Nile virus and Dengue virus [22], indicating a broad antiviral activity of these small factors. Further targets of IFITM proteins are coronaviruses, Hepatitis C virus, Ebola virus and Vesicular Stomatitis virus (VSV) [23, 25, 26], as well as bunyaviruses [27] and reoviruses [28]. It appears that expression of IFITM proteins in target cells is sufficient for inhibition of these viruses. Specifically, IFITM1, 2 and 3 interfere with glycoprotein-dependent fusion of the viral and endosomal membrane, preventing the delivery of viral genomes to the cytoplasm and resulting in trapping of virions in endosomes and lysosomal degradation [22]. Strikingly, a minor SNP in human ifitm3, rs12252-C, which leads to expression of an N-terminally truncated IFITM3 protein, strongly associates with a poor prognosis during pandemic H1N1/09 Influenza A infection [29]. These findings highlight the important contribution of IFITM3 in defending humans against viral pathogenic infections and establish IFITM3 as a bona-fide antiviral factor.
The impact of IFITM proteins on single-round HIV-1 infection has been discussed controversially. Using RNAi, an antiviral activity of IFITM3 was found to be unlikely to exist since no inhibition of cell-free HIV-1 was observed in HeLa CD4 cells which endogenously express IFITM3 [22]. Although IFITM proteins scored positive in a large-scale screen for antiviral activity against HIV-1, the detected antiviral effect must probably be attributed to the VSV-G-mediated entry that was employed by the VSV-G/HIV-1 pseudotypes that lacked authentic HIV-1 Env [30]. Modest consequences of IFITM2 and IFITM3 overexpression and absent antiviral activity of IFITM1 have been reported using a BlaM-Vpr-based HIV-1 fusion assay [31, 32], suggesting that fusion of cell-free HIV-1 is only weakly affected. In contrast to the moderate effect of IFITM proteins in single-round HIV-1 infection, stable expression of individual IFITM proteins in a T-cell line effectively impedes viral spread in cell culture using a replication-competent HIV-1 [31-35], implying more potent effector functions of IFITM proteins in a system in which cell-to-cell transmission predominates. Indeed, findings from two independent groups recently shed some light into a potential mechanism [35, 36]. Whereas IFITM2 or 3 expressed on membranes of target cells fail to exert antiviral activity in the context of cell-to-cell transmission, the antiviral capacity is more obvious when IFITM3 is expressed in the infected donor cells that transmit the virus to IFITM-negative target cells. The antiviral effect potentiates when IFITM is expressed on both donor and target cells. Specifically, IFITM proteins are incorporated into membranes of nascent virions and thereby decrease their particle infectivity by impairing fusion [35, 36], apparently not at the expense of Env incorporation and also not in an Env-dependent manner [35, 36]. The exact mode of action of virus-associated IFITMs, whether virion incorporation of IFITM proteins is governed by specific determinants or occurs randomly, and whether IFITM proteins are incorporated in virus particles of other families, remain yet to be investigated. In the context of other viruses, multiple, partially contradicting models have been debated. It has been suggested that IFITM proteins inhibit the fusogenicity of membranes in which they locate. Specifically, IFITM3 may restrict influenza A virus entry by restricting viral membrane hemifusion [37] or by blocking the formation of fusion pores at a post-hemifusion stage [38]. An alternative hypothesis says that IFITM3 disrupts cholesterol homeostasis, leading to accumulation of cholesterol in intracellular compartments [39]. Alternatively, viral incorporation of IFITM proteins may cause exclusion and inclusion of yet to be defined specific cellular factors with antiviral activity as a consequence. Although evidence for direct viral antagonists of IFITM proteins is lacking, HIV-1 may be able to overcome IFITM-imposed antiviral activity, since evasion mutations in Vpu and Env were reported after passage of HIV-1 in an IFITM1-expressing T-cell line [33].
Nef-mediated Antagonism of Serine Inco-rporator (SERINC) Proteins
Among the accessory genes of primate lentiviruses, Nef displays probably the highest degree of multifunctionality. As evidenced by infections with inactivated nef mutations or deletions in rhesus macaques and humans, an intact nef gene is required in vivo for sustained viremia and development of immunodeficiency [40-42]. Although Nef does not exert any enzymatic activity, it fulfills a multitude of functions that culminate in alteration of cell signaling and activation by interfering with the cellular vesicular trafficking machinery. Association of Nef with membranes is governed by its myristoylation; it is also incorporated in virus particles. Downregulation of several cell surface receptors, including CD4 and coreceptors, as well as MHC class I and II receptors and modulation of transcriptional activity of infected T-cells are established functions of HIV-1 Nef. Another long-appreciated activity of Nef is the enhancement of HIV-1 particle infectivity. Production of HIV-1 devoid of an intact nef gene results in impaired particle infectivity, ranging from 3 to 40-fold reduced titers, depending on the producer cell type, target cell type and viral isolate [43, 44]. The most pronounced requirement for Nef is seen in lymphoid cells. Nef-mediated enhancement of particle infectivity requires its expression in the producer cell, is highly conserved within primate lentiviruses [45] and is maintained during disease progression [46]. The fact that Nef is incorporated into virions and cleaved by HIV-1 protease during maturation of particles may support a specific function of virus-incorporated Nef that manifests itself early during the next round of infection. Its membrane association may be sufficient to result in virion packaging, since Nef is also found in orthologous retroviruses when co-expressed in the producer cell. Recently, the mechanism of Nef-mediated infectivity enhancement has been elucidated [47, 48]. Using differential mRNA sequencing in different producer cell lines that differ in their requirement for Nef for optimal HIV-1 infectivity [47], and mass spectrometry-based approaches [48], respectively, members of the serine incorporator (SERINC) family were identified as potent cellular inhibitors of retrovirion particle infectivity in the absence of Nef. SERINC proteins are highly conserved, multi-pass transmembrane proteins expressed predominantly at the plasma membrane with poorly elucidated cellular functions besides that they mediate serine incorporation into phosphatidylserine and sphingolipids [49]. SERINC5, which is abundantly expressed in PBMCs, and SERINC3 were reported to inhibit HIV-1 in the absence of Nef in a dose-dependent and synergistic fashion that appears to require SERINC incorporation in the virion. SERINC5 inhibition acts specifically against Env-containing particles, since VSV-G pseudotypes were largely insensitive to SERINC5 [47, 48]. Nef was shown to counteract SERINC activity by downregulating it from the cell surface via clathrin-dependent endocytosis and sequestering it in endosomes, thus precluding their incorporation into virion particles. Interestingly, the prototypic retrovirus MLV has evolved its own antagonist (Glyco-gag) to counteract SERINC5 [47, 48], underlining the importance of this antiviral factor. Interestingly, expression of SERINC3 and SERINC5 are not induced or stimulated by type I interferons, in contrast to the targets of most other viral antagonists [47]. The antiviral activity of SERINC5 appears to be caused predominantly by post-entry defects, since the detectable slight inhibition of virus-cell fusion does probably not fully account for the far more pronounced infectivity decrease imposed by physiological levels of SERINC5 expression [47, 48]. Future work is needed to fully understand the antiviral mode of action of SERINC proteins, and to decipher whether other enveloped viruses are prone to SERINC-mediated inhibition, and how exactly Nef counteracts SERINC proteins. Notably, a better understanding of the SERINC-imposed antiviral block to infection and the potential Nef-SERINC interface might be translated into the design of a small antiviral molecule that interrupts Nef-SERINC interaction and allows full-blown SERINC-mediated HIV-1 inhibition.
CONCLUSION
In the past decade, discovery and characterization of antiviral restriction factors has catapulted our understanding on cell-intrinsic immunity. Whereas many antiviral factors were found to target early post-entry steps of the HIV-1 replication cycle, recent findings have uncovered the potential of cellular proteins 90K and members of the IFITM and SERINC protein family to reduce the particle infectivity of HIV-1. They act by modifying the essential particle composition or by incorporating into nascent virions, respectively. Their existence may, at least partially, contribute to the inherently high frequency of uninfectious HIV-1 particles [50-52]. Extensive investigation is required to fully understand their mode of action and respective virally-encoded antagonistic or evasion strategies, and opens up an exciting new area of research, with potential translation into the design of new antiviral treatment strategies.
Fig. (1).
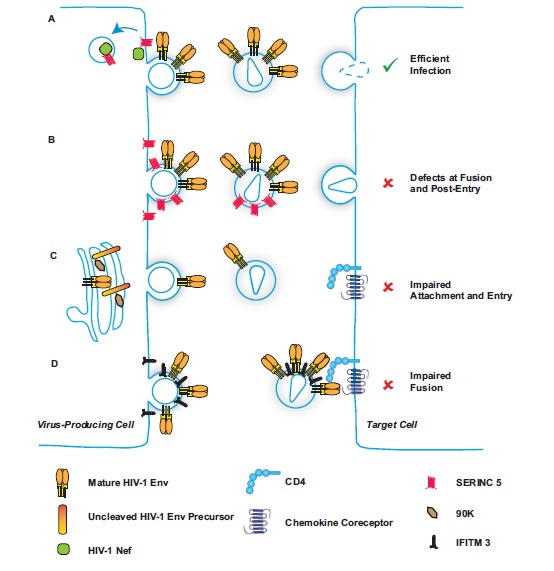
Particle infectivity is modulated by viral and cellular factors. (A) In the course of wild-type HIV-1 infection, HIV-1 Nef downregulates antiviral SERINC5 from the cell surface in a clathrin-dependent manner, thus preventing incorporation of this small transmembrane protein into nascent virions. (B) In the absence of HIV-1 Nef expression, SERINC5 incorporates into budding particles, inducing defects at fusion and post-entry following infection of the target cell. (C) Cellular 90K expression in the virus-producing cell induces a defect in posttranslational maturation of HIV-1 Env and prevents efficient viral incorporation of cleaved HIV-1 Env, resulting in poor ability of particles to attach and enter new target cells. (D) IFITM3 incorporates into HIV-1 virions and prevents fusion with the target cells during cell-to-cell transmission.
ACKNOWLEDGEMENTS
This work was supported by grants from the DFG (Collaborative Research Center 900: Microbial Persistence and its Control), from the Helmholtz Center of Infection Research, and from the Margarete von Wrangell Habilitation Program to CG. I thank Martin Das for graphical assistance.
ABBREVIATIONS
- HIV
Human immunodeficiency virus
- IFITM
Interferon-induced transmembrane
- IFN
Interferon
- ISG
Interferon-stimulated gene
- LGALS3BP
Lectin galectin soluble 3 binding protein
- LTR
Long terminal repeats
- SDS-PAGE
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SERINC
Serine incorporator
- TCR
T-cell receptor
CONFLICT OF INTEREST
The author confirms that this article content has no conflict of interest.
REFERENCES
- 1.Chertova E., Bess J.W., Jr, Crise B.J., et al. Envelope glycoprotein incorporation, not shedding of surface envelope glycoprotein (gp120/SU), Is the primary determinant of SU content of purified human immunodeficiency virus type 1 and simian immunodeficiency virus. J. Virol. 2002;76(11):5315–5325. doi: 10.1128/JVI.76.11.5315-5325.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu P., Liu J., Bess J., Jr, et al. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature. 2006;441(7095):847–852. doi: 10.1038/nature04817. [DOI] [PubMed] [Google Scholar]
- 3.Chojnacki J., Staudt T., Glass B., et al. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science. 2012;338(6106):524–528. doi: 10.1126/science.1226359. [DOI] [PubMed] [Google Scholar]
- 4.Lasfar A., Lewis-Antes A., Smirnov S.V., et al. Characterization of the mouse IFN-lambda ligand-receptor system: IFN-lambdas exhibit antitumor activity against B16 melanoma. Cancer Res. 2006;66(8):4468–4477. doi: 10.1158/0008-5472.CAN-05-3653. [DOI] [PubMed] [Google Scholar]
- 5.Sommereyns C., Paul S., Staeheli P., Michiels T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008;4(3):e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Witte K., Gruetz G., Volk H.D., et al. Despite IFN-lambda receptor expression, blood immune cells, but not keratinocytes or melanocytes, have an impaired response to type III interferons: implications for therapeutic applications of these cytokines. Genes Immun. 2009;10(8):702–714. doi: 10.1038/gene.2009.72. [DOI] [PubMed] [Google Scholar]
- 7.Koths K., Taylor E., Halenbeck R., Casipit C., Wang A. Cloning and characterization of a human Mac-2-binding protein, a new member of the superfamily defined by the macrophage scavenger receptor cysteine-rich domain. J. Biol. Chem. 1993;268(19):14245–14249. [PubMed] [Google Scholar]
- 8.Iacobelli S., Arno E., D'Orazio A., Coletti G. Detection of antigens recognized by a novel monoclonal antibody in tissue and serum from patients with breast cancer. Cancer Res. 1986;46(6):3005–3010. [PubMed] [Google Scholar]
- 9.Linsley P.S., Horn D., Marquardt H., et al. Identification of a novel serum protein secreted by lung carcinoma cells. Biochem. 1986;25(10):2978–2986. doi: 10.1021/bi00358a037. [DOI] [PubMed] [Google Scholar]
- 10.Ullrich A., Sures I., D'Egidio M., et al. The secreted tumor-associated antigen 90K is a potent immune stimulator. J. Biol. Chem. 1994;269(28):18401–18407. [PubMed] [Google Scholar]
- 11.Rotger M., Dang K.K., Fellay J., et al. Genome-wide mRNA expression correlates of viral control in CD4+ T-cells from HIV-1-infected individuals. PLoS Pathog. 2010;6(2):e1000781. doi: 10.1371/journal.ppat.1000781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brakebusch C., Sures I., Jallal B., et al. Isolation and functional characterization of the human 90K promoter. Genomics. 1999;57(2):268–278. doi: 10.1006/geno.1999.5760. [DOI] [PubMed] [Google Scholar]
- 13.Waddell S.J., Popper S.J., Rubins K.H., et al. Dissecting interferon-induced transcriptional programs in human peripheral blood cells. PLoS One. 2010;5(3):e9753. doi: 10.1371/journal.pone.0009753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lodermeyer V., Suhr K., Schrott N., et al. 90K, an interferon-stimulated gene product, reduces the infectivity of HIV-1. Retrovirology. 2013;10:111. doi: 10.1186/1742-4690-10-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Natoli C., Dianzani F., Mazzotta F., et al. 90K protein: a new predictor marker of disease progression in human immunodeficiency virus infection. J Acq Immun Def Synd. 1993;6(4):370–375. [PubMed] [Google Scholar]
- 16.Briggs N.C., Natoli C., Tinari N., D'Egidio M., Goedert J.J., Iacobelli S.A. 90-kDa protein serum marker for the prediction of progression to AIDS in a cohort of HIV-1+ homosexual men. AIDS Res. Hum. Retroviruses. 1993;9(9):811–816. doi: 10.1089/aid.1993.9.811. [DOI] [PubMed] [Google Scholar]
- 17.Longo G., Natoli C., Rafanelli D., et al. Prognostic value of a novel circulating serum 90K antigen in HIV-infected haemophilia patients. Br. J. Haematol. 1993;85(1):207–209. doi: 10.1111/j.1365-2141.1993.tb08674.x. [DOI] [PubMed] [Google Scholar]
- 18.Hyrcza M.D., Kovacs C., Loutfy M., et al. Distinct transcriptional profiles in ex vivo CD4+ and CD8+ T cells are established early in human immunodeficiency virus type 1 infection and are characterized by a chronic interferon response as well as extensive transcriptional changes in CD8+ T cells. J. Virol. 2007;81(7):3477–3486. doi: 10.1128/JVI.01552-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sedaghat A.R., German J., Teslovich T.M., et al. Chronic CD4+ T-cell activation and depletion in human immunodeficiency virus type 1 infection: type I interferon-mediated disruption of T-cell dynamics. J. Virol. 2008;82(4):1870–1883. doi: 10.1128/JVI.02228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klein J.S., Bjorkman P.J. Few and far between: how HIV may be evading antibody avidity. PLoS Pathog. 2010;6(5):e1000908. doi: 10.1371/journal.ppat.1000908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sallman A.M., Bringeland N., Fredriksson R., Schioth H.B. The dispanins: a novel gene family of ancient origin that contains 14 human members. PLoS One. 2012;7(2):e31961. doi: 10.1371/journal.pone.0031961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brass A.L., Huang I.C., Benita Y., et al. The IFITM Proteins Mediate Cellular Resistance to Influenza A H1N1 Virus, West Nile Virus, and Dengue Virus. Cell. 2009;139(7):1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wrensch F., Karsten C.B., Gnirss K., et al. Interferon-Induced Transmembrane Protein-Mediated Inhibition of Host Cell Entry of Ebolaviruses. J. Infect. Dis. 2015;212:S210–S218. doi: 10.1093/infdis/jiv255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siegrist F, Ebeling M, Certa U. The small interferon-induced transmembrane genes and proteins. 2011. [DOI] [PubMed]
- 25.Huang I.C., Bailey C.C., Weyer J.L., et al. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 2011;7(1):e1001258. doi: 10.1371/journal.ppat.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weidner J.M., Jiang D., Pan X.B., Chang J., Block T.M., Guo J.T. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J. Virol. 2010;84(24):12646–12657. doi: 10.1128/JVI.01328-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mudhasani R., Tran J.P., Retterer C., et al. IFITM-2 and IFITM-3 but not IFITM-1 restrict Rift Valley fever virus. J. Virol. 2013;87(15):8451–8464. doi: 10.1128/JVI.03382-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anafu A.A., Bowen C.H., Chin C.R., Brass A.L., Holm G.H. Interferon-inducible transmembrane protein 3 (IFITM3) restricts reovirus cell entry. J. Biol. Chem. 2013;288(24):17261–17271. doi: 10.1074/jbc.M112.438515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Everitt A.R., Clare S., Pertel T., et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature. 2012;484(7395):519–U146. doi: 10.1038/nature10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schoggins J.W., Wilson S.J., Panis M., et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472(7344):481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu J., Pan Q.H., Rong L.W., Liu S.L., Liang C. The IFITM Proteins Inhibit HIV-1 Infection. J. Virol. 2011;85(5):2126–2137. doi: 10.1128/JVI.01531-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jia R., Pan Q., Ding S., et al. The N-terminal region of IFITM3 modulates its antiviral activity by regulating IFITM3 cellular localization. J. Virol. 2012;86(24):13697–13707. doi: 10.1128/JVI.01828-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding S., Pan Q., Liu S.L., Liang C. HIV-1 mutates to evade IFITM1 restriction. Virology. 2014;454-455:11–24. doi: 10.1016/j.virol.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jia R., Ding S., Pan Q., Liu S.L., Qiao W., Liang C. The C-terminal sequence of IFITM1 regulates its anti-HIV-1 activity. PLoS One. 2015;10(3):e0118794. doi: 10.1371/journal.pone.0118794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Compton A.A., Bruel T., Porrot F., et al. IFITM proteins incorporated into HIV-1 virions impair viral fusion and spread. Cell Host Microbe. 2014;16(6):736–747. doi: 10.1016/j.chom.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tartour K., Appourchaux R., Gaillard J., et al. IFITM proteins are incorporated onto HIV-1 virion particles and negatively imprint their infectivity. Retrovirology. 2014;11:103. doi: 10.1186/s12977-014-0103-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li K., Markosyan R.M., Zheng Y.M., et al. IFITM proteins restrict viral membrane hemifusion. PLoS Pathog. 2013;9(1):e1003124. doi: 10.1371/journal.ppat.1003124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Desai T.M., Marin M., Chin C.R., Savidis G., Brass A.L., Melikyan G.B. IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog. 2014;10(4):e1004048. doi: 10.1371/journal.ppat.1004048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amini-Bavil-Olyaee S., Choi Y.J., Lee J.H., et al. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe. 2013;13(4):452–464. doi: 10.1016/j.chom.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deacon N.J., Tsykin A., Solomon A., et al. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science. 1995;270(5238):988–991. doi: 10.1126/science.270.5238.988. [DOI] [PubMed] [Google Scholar]
- 41.Kestler H.W., III, Ringler D.J., Mori K., et al. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell. 1991;65(4):651–662. doi: 10.1016/0092-8674(91)90097-i. [DOI] [PubMed] [Google Scholar]
- 42.Kirchhoff F., Greenough T.C., Brettler D.B., Sullivan J.L., Desrosiers R.C. Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N. Engl. J. Med. 1995;332(4):228–232. doi: 10.1056/NEJM199501263320405. [DOI] [PubMed] [Google Scholar]
- 43.Chowers M.Y., Spina C.A., Kwoh T.J., Fitch N.J., Richman D.D., Guatelli J.C. Optimal infectivity in vitro of human immunodeficiency virus type 1 requires an intact nef gene. J. Virol. 1994;68(5):2906–2914. doi: 10.1128/jvi.68.5.2906-2914.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller M.D., Warmerdam M.T., Gaston I., Greene W.C., Feinberg M.B. The human immunodeficiency virus-1 nef gene product: a positive factor for viral infection and replication in primary lymphocytes and macrophages. J Experiment Med. 1994;179(1):101–113. doi: 10.1084/jem.179.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Munch J., Rajan D., Schindler M., et al. Nef-mediated enhancement of virion infectivity and stimulation of viral replication are fundamental properties of primate lentiviruses. J. Virol. 2007;81(24):13852–13864. doi: 10.1128/JVI.00904-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carl S., Greenough T.C., Krumbiegel M., et al. Modulation of different human immunodeficiency virus type 1 Nef functions during progression to AIDS. J. Virol. 2001;75(8):3657–3665. doi: 10.1128/JVI.75.8.3657-3665.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosa A., Chande A., Ziglio S., et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature. 2015;526(7572):212–217. doi: 10.1038/nature15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Usami Y., Wu Y., Gottlinger H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature. 2015;526(7572):218–223. doi: 10.1038/nature15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Inuzuka M., Hayakawa M., Ingi T. Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J. Biol. Chem. 2005;280(42):35776–35783. doi: 10.1074/jbc.M505712200. [DOI] [PubMed] [Google Scholar]
- 50.Dimitrov D.S., Willey R.L., Sato H., Chang L.J., Blumenthal R., Martin M.A. Quantitation of human immunodeficiency virus type 1 infection kinetics. J. Virol. 1993;67(4):2182–2190. doi: 10.1128/jvi.67.4.2182-2190.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Munch J., Rucker E., Standker L., et al. Semen-derived amyloid fibrils drastically enhance HIV infection. Cell. 2007;131(6):1059–1071. doi: 10.1016/j.cell.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 52.Rusert P., Fischer M., Joos B., et al. Quantification of infectious HIV-1 plasma viral load using a boosted in vitro infection protocol. Virology. 2004;326(1):113–129. doi: 10.1016/j.virol.2004.05.022. [DOI] [PubMed] [Google Scholar]