

Abstract
Cellular function phenotype is regulated by various microRNAs (miRs), including miR-135a. However, how miR-135a is involved in the calcification in senescent vascular smooth muscle cells (VSMCs) is not clear yet. In the present study, we first identified the significantly altered miRNAs in VSMCs, then performed consecutive passage culture of VSMCs and analyzed the expression of miR-135a and calcification genes in the senescent phase. Next, the effects of the miR-135a inhibition on calcification and calcification genes were analyzed. The luciferase assay was used to validate the target protein of miR-135a. The western blotting was used to determine the effects of miR-135a on Krüppel-like factor 4 (KLF4) and signal transducer and activator of transcription 3 protein (STAT3) expression, as well as the relationship between KLF4 and STAT3. Finally, the quantified cellular calcification was measured to examine the involvement of miR-135a, KLF4 and STAT3 in VSMCs calcification. Our results showed that miR-135a was significantly altered in VSMCs. Cell calcification and calcification genes were greatly altered by miR-135a inhibition. KLF4 was validated as the target RNA of miR-135a. Expression of KLF4 and STAT3 were both significantly decreased by over expressed miR-135a, while the inhibition of miR-135a and KLF4 siRNA both decreased the STAT3 protein levels. Moreover, the inhibition of miR-135a dramatically increased the calcium concentration, but co-treatment with KLF4 or STAT3 siRNA both decreased the calcium concentration. The present study identified miR-135a as a potential osteogenic differentiation suppressor in senescent VSMCs and revealed that KLF4/STAT3 pathway, at least partially, was involved in the mechanism.
Keywords: Vascular smooth muscle cell, miR-135a, calcification, KLF4, STAT3
1. INTRODUCTION
Atherosclerosis is one of the most common diseases of the aging population. Vascular calcification is a common feature of atherosclerotic lesions and contributes to cardiovascular complications due to the loss of aortic resilience and function, which is a leading cause of morbidity and mortality especially in senior patients [1]. Vascular calcification appears to be initiated by the release of vesicular structures from Vascular Smooth Muscle Cells (VSMCs) that contain hydroxyapatite [2]. The transition of VSMCs towards an osteoblast-like phenotype promotes the release of the vesicular structures. The mineralization within these structures can be promoted by expression of osteoblastic proteins. Components of the osteoblastic transition of VSMCs include osteoblastic morphogens, the Bone Morphogenetic Proteins (BMP)-2 and BMP-4, transcription factors and bone proteins such as alkaline phosphatase (ALP) and osteocalcin (OC) [1, 3].
MicroRNAs (miRs) are small non-encoding RNAs serving as important post-transcriptional gene regulators. The key function of miRs is to control cell proliferation and differentiation of various cell types. A growing number of studies have demonstrated that the pathogenic change in tissues, such as cardiac hypertrophy, heart failure, cardiac fibrosis, and vascular atherosclerosis, was associated with variations in the expression of miRs [4, 5]. Circulating levels of some miRs were down-regulated in patients with coronary artery disease [6], and the patients with type 2 diabetes had less circulating endothelial miR-126 [7]. In vitro experiments showed that miR-125b [1, 8], miR-145 [9] and miR-146a [10] played a regulative role in the aberrant VSMC proliferation, which is a key pathological process of proliferative vascular diseases, such as atherosclerosis or vascular calcification.
MicroRNA-135a (miR-135a) is a highly conserved micro RNA, which has been proven to contribute to colorectal pathogenesis through its regulation of adenomatous polyposis coli gene [11]. It could promote the growth and invasion of colorectal cancer cell via Metastatic Suppressor 1 (MTSS1) [12], induce breast cancer cell migration and invasion by targeting homeobox A10 (HOXA10) and contribute to the development of portal vein tumor thrombus [13, 14]. Moreover, ubiquitous loss of miR-135a expression is critical for the over expression of HOXA10 in epithelial ovarian cancer cells, which is implicated in epithelial ovarian carcinogenesis [13]. However, the role of miR-135a in senescent VSMC calcification and vascular disease has not been explored until recently.
Krüppel-like factor 4 (KLF4) is a zinc-finger transcription factor that plays a key role in cellular differentiation and proliferation [15]. Recent studies have revealed that KLF4 is expressed in several vascular cell types [16, 17]. miR-146a targeted the KLF4 3'-untranslated region and promoted VSMC proliferation and vascular neointimal hyperplasia, as revealed in both in vitro and in vivo studies [10]. However, the role of miR-135a and its relation with KLF4 in the calcification of senescent VSMCs has not been explored yet. Signal transducer and activator of transcription 3 protein (STAT3) is a member of the STAT protein family. STAT3 can be activated in response to many cytokines and growth factors and mediate the expression of various genes in response to cell stimuli, and thus plays a key role in cellular processes such as calcification [18]. The analysis of senescent VSMCs can be an effective approach to understanding the behavior of KLF4 in vascular calcification. Therefore, in this study, we aimed to determine how endogenous miR-135a is involved in VSMCs function phenotype in osteogenic differentiation and the underlying mechanism regarding KLF4 and STAT3 pathway.
2. MATERIALS AND METHODS
2.1. Isolation of VSMCs
Rat VSMCs were prepared from abdominal aorta of eight-week-old male Sprague-Dawley rats (180-200g, obtained from Shanghai Animal Center). First, rats were anesthetized by intraperitoneal injection of sodium pentobarbital (obtained from Shanghai Animal Center, 25 mg/kg), then sacrificed by quick abdominal aorta exsanguination. The thoracic aortas were then isolated and placed in Krebs-Henseleit solution (containing NaCl 118.0 mM, KCl 4.7 mM, CaCl2 2.5 mM, MgSO4 1.2 mM, NaHCO3 25.0 mM, glucose 11.0 mM, obtained from Shanghai Animal Center) at 37°C. After that, the smooth muscle layer was chopped into small fragments in 0.5 mL fetal bovine serum (FBS) (HyClone, Logan, UT, USA), then transferred to a flask and maintained at 37°C for 4 h. The flask was turned over gently and incubated for 5 days with DMEM (Dulbecco′s modified Eagle′s medium, Invitrogen, USA). VSMCs migrated out of the explants 5 days later and passage was performed 10 days after isolation.
2.2. Cultures of VSMCs
Cells were maintained in DMEM, supplemented with 10% FBS, 50 μg/ml penicillin and 50 μg/ml streptomycin (Invitrogen, USA) at 37°C in a humidified atmosphere containing 5% CO2. The medium was changed every 2 days. After 6-9 days, when the cells had formed a confluent monolayer, they were made quiescent by incubating in serum-free medium 24 h prior to experimental treatments. The cells were harvested by the addition of 0.25% trypsin-1 mM ethylenediaminetetraacetic acid (EDTA) (Invitrogen, USA). The cell culture was continued up to 16 passages.
2.3. Count of VSMCs
The total cell number and the population doubling time (PDT) of each passage were measured. VSMCs were plated at a density of 2×103 cells/cm2 in 6-well plates in growth medium. The cells were allowed to attach to the bottom of the wells (1-2 h). The medium was then changed to serum-free and Ca2+-free DMEM containing 50 μg/ml penicillin, 50 μg/ml streptomycin, 2 mM L-glutamine, 0.2% BSA and 0.5 mM CaCl2. After trypsinization, the cells were counted manually in a counting chamber. The following formula was used to calculate PDT: PDT = (T2-T1)*log (2)/Log (P2/P1) (Td = doubling time, P1 = the number of cell at T1, P2 = the number of cell at T2).
2.4. miRNA Preparation and Real-Time Polymerase Chain Reaction (PCR)
Total RNA from cells was extracted by using RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocols. miR-135a was reverse transcribed using a specific stem-loop primer (Applied Biosystems, Carlsbad, CA, USA) and quantified by real-time PCR with the TaqMan MicroRNA assay kit (Applied Biosystems). U6 was used for the normalization of miR-135a expression.
2.5. Calcification Assays
Similarly to Villa-Bellosta and Sorribas [19], VSMCs were incubated in MEM with 0.2% FCS, 2.5 mM Pi from a stock of KH2PO4 and K2HPO4 (Sigma, St. Louis, USA) to induce the calcification. Mineralized matrix formation was qualitatively visualized by Alizarin red staining [20]. VSMCs were fixed in 4% paraformaldehyde (Sigma, St. Louis, USA) and stained with 40 mmol/L Alizarin red dye (Sigma-Aldrich, Munich, Germany) for 30 min at room temperature. Excess dye was removed by washing the plates with distilled water. For the quantitative determination of calcium content, VSMC cultures were incubated overnight in 0.6 mol/L HCl, and the dissolved calcium was quantified using a colorimetric Calcium Assay kit (Abnova, Taipei, Taiwan), as previously described [21].
2.6. Alkaline Phosphatase (ALP) Activity Assay
Cellular ALP activity was measured by the use of a colorimetric assay based on the utilization of p-nitrophenyl phosphatase hydrolysed by ALP into a yellow colored product. Cells were lysed using 1% Triton X-100-containing Tris-HCl buffer (pH 7.0), then centrifuged for 5 min at 12000×g to remove cellular debris. Aliquots of each sample were incubated with ALP substrate buffer (100 mmol/L diethanolamine, 150 mmol/L NaCl, 2 mmol/L MgCl2, and 2.5 μg/mL p-nitrophenyl phosphate, Sigma, St. Louis, USA) for 10 min at 37°C, measured at 405 nm according to the manufacturer's instructions on a plate reader, and normalized to the total protein content determined by the Bicinchoninic Acid (BCA) method from the same protein extracts.
2.7. Microarray Data Analysis and Luciferase Activity Assay
Raw data was extracted using the Agilent Feature Extraction software (V9.3.5) and was normalized using quantile normalization in the R language (version 2.14.0, http://www.r-project.org/). Values were then log2 transformed and median centered across miRNAs and samples. The luciferase assay was used to validate KLF4 as a target of miR-135a. Approximately 75,000 VSMCs were seeded in 12-well plates, then transfected with the wild type (psiCHECK2-KLF4-UTR), mutant (psiCHECK2-KLF4-UTR-mut) KLF4 3′UTR or the vector alone (co-transfected using pGL3-control) according to the manufacturer's instructions. After 48 h, cells were lysed, then the firefly and Renilla luciferase activities were measured using the dual luciferase reporter assay kit (Promega, MA, USA) on a luminometer (Orion II, Luminometer, Germany). To check the specificity of the miR-135a effect, VSMCs were co-transfected with wild type psiCHECK2-KLF4-3′UTR or over expressed miR-135a before the luciferase activities were measured. Control cells were transfected with the scramble sequence. Renilla reporter luciferase activity was normalized to the firefly luciferase activity.
2.8. Transfection of miRNA
Cells were seeded in 6-well plates and transfected at 70-80% confluence with a miR-135a inhibitor or the negative control using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The inhibitor and corresponding negative control were all products from Genepharma (Shanghai, China).
2.9. Lentiviral Expression Vector of Anti-miR-135a and Transfection
5×105 cells were seeded per well in 6-well plates one day before transfection. The next day, Lenti-anti-miR135a (Shanghai Genepharma, China) was added into VSMCs with 1 ml fresh DMEM/F12 containing 10% FBS and 5 μg/ml Polybrene (Sigma, USA). 12 h later, the medium was replaced with fresh culture medium. Three days later, the cells were collected for subsequent culture. The experiment was repeated six times.
2.10. Luciferase Reporter Assay for KLF4-3′-UTR Targeting
Luciferase vectors including the 3’-UTR of KLF4 containing the KLF4-miR-135a response elements were purchased from Genepharma (Shanghai, China). For the luciferase reporter experiment, VSMCs were seeded in 12-well plates and cotransfected with the luciferase reporter vectors and the miR-135a mimic or negative control. Luciferase activity was measured using the Dual-Light Chemiluminescent Reporter Gene Assay System (Applied Biosystems).
2.11. Statistical Analysis
The data were reported as mean ± standard error of mean (SEM). All statistical analyses were performed using SPSS 17.0 by one-way ANOVA followed by Tukey’s multiple comparison. P <0.05 (two-sided) was considered significant.
3. RESULTS
3.1. Consecutive Passage Culture of VSMCs and the Expression of miR-135a and Calcified Genes in Different Passage
The consecutive passage culture was used to determine the proliferation ability and the passage of senescence phase of VSMCs. Compared with the typical “peak and valley” morphological appearance under microscope in logarithmic phase, VSMCs had an enlarged, flattened and stellar shape in senescence phase. As shown in Fig. (1A), cells of passages 5 to 10 were in the proliferation phase, while the senescence phase occurred in passages 14 to 16. The population doubling time (PDT) of VSMCs in proliferation phase was about 2 days. The cell growth slowed down and the cell number decreased in senescence phase of passage 14 to 16. Fig. (1B) shows that the expression of miR-135a was gradually lowered as senility occurred (&p<0.05 compared to passage 10). Fig. (1C and 1D) showed that the mRNA expression of ALP and OC were significantly up-regulated at senescence phase (passages 13-15) compared with the proliferation phase (&p<0.05 compared to passage 10).
Fig. (1).
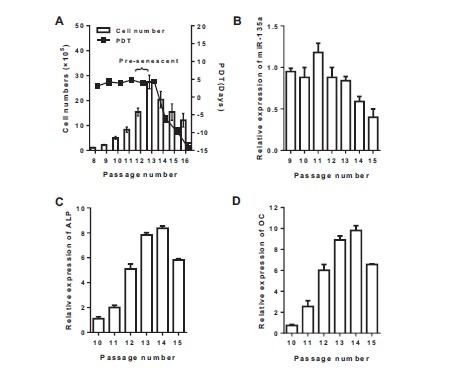
Cell number, the Population Doubling Time (PDT) and mRNA expression in VSMCs. A: The consecutive passage culture was used to determine the total cell number and the Population Doubling Time (PDT) of VSMCs. B, C and D: mRNA expression of miR-135a, Alkaline Phosphatase (ALP) and Osteocalcin (OC) in VSMCs of 9 to 15 passage was measured by real-time PCR. Values are expressed as mean ± SEM, n = 6 in each group.
3.2. Effects of the Inhibition of miR-135a Expression on Cellular Calcification, ALP Activity and Calcification Genes
To analyze the role of miR-135a on cellular calcification, we inhibited miR-135a expression in VSMCs. As shown in Fig. (2A), inhibition of endogenous miR-135a in VSMCs promoted cellular calcification as demonstrated by increased matrix mineralization. ALP activity, mRNA expression of ALP and OC were also significantly increased by miR-135a inhibition (Fig. 2B, 2C and 2D, &p<0.05 compared with control).
Fig. (2).
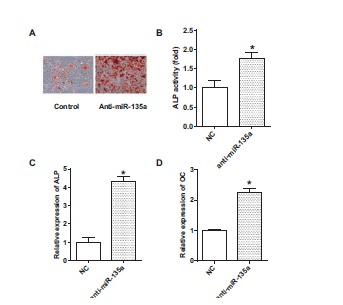
Inhibition of miR-135a on calcification, ALP activity and mRNA expression of Alkaline Phosphatase (ALP) and Osteocalcin (OC). A: Mineralized matrix formation was assessed by AlizarinRed S staining; B: Alkaline Phosphatase (ALP) activity was measured in cell cultures; C and D: The mRNA expression of Alkaline Phosphatase (ALP) or Osteocalcin (OC) was measured by real-time PCR. Cells were in passage 14. Values are expressed as mean ± SEM, n = 6 in each group.*P< 0.05, versus control group.
3.3. The Target of miR-135a and the Protein Expression of KLF4 and STAT3
To determine whether miR-135a regulated KLF4 through the predicted binding sites in its 3′-UTR (Fig. 3A), we designed two luciferase constructs by incorporating wild-type or mutant 3′-UTR of KLF4 individually, which constitutively expressed luciferase unless repressed by the incorporated 3′-UTR. As shown in Fig. (3B), co-transfection of VSMCs with the pMIR-REPORT construct containing mutant KLF4 3′-UTR and PLemiR-135a did not show much difference compared with control (P >0.05), but co-transfection with luciferase construct containing wild-type KLF4 3′-UTR and PLemiR-135a resulted in a nearly 80% decline in the luciferase activity (P< 0.05 compared with control). Western blotting (Fig. 3C) showed that the expression of KLF4 and STAT3 were both significantly decreased while co-transfected with the luciferase construct containing over expressed miR-135a compared with control group (p< 0.05). Fig. (3D) shows that the inhibition of miR-135a and KLF4 siRNA interference both decreased the STAT3 protein levels (p< 0.05 compared with control).
Fig. (3).
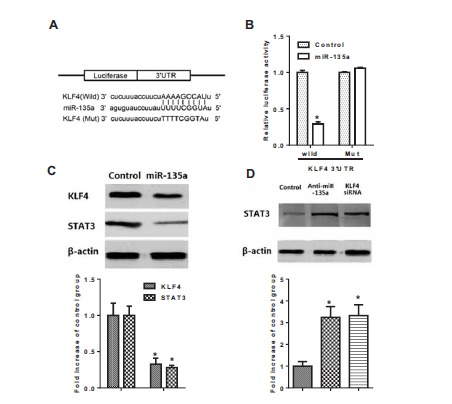
Luciferase activity assay and Western blotting in VSMCs. A: Schematic representation of the putative miR-135a binding site in the KLF4 3′UTR in Target scan. B: Luciferase activity assay of KLF4; C: Protein expression of KLF4 and STAT3 measured by Western Blotting. D: Protein expression of STAT3 measured by Western Blotting. Cells were in passage 14. Values are expressed as mean ± SEM, n = 6 in each group. *P< 0.05, versus control group.
3.4. The Quantified Cellular Calcification Under Different Treatments
As shown in Fig. (4), the normal calcium concentration in VSMCs was about 200 μg Ca2+/mg protein. With the inhibition of miR-135a, the calcium concentration was dramatically increased to about 1000 μg Ca2+/mg protein (p<0.05 compared with control). KLF4 or STAT3 siRNA interference both decreased the calcium concentration to nearly 500 μg Ca2+/mg protein, which were significantly different from the anti-miR-135a group (p< 0.05). The treatment of KLF4 or STAT3 siRNA interference alone did not dramatically alter the calcium concentration compared with control (p> 0.05).
Fig. (4).
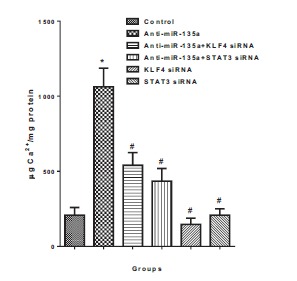
Calcification of VSMCs under different treatment. Cells were in passage 14. Values are expressed as mean ± SEM, n = 6 in each group. *P< 0.05, versus control group. #P< 0.05, versus Anti-miR-135a group.
4. DISCUSSION
The present study showed that miR-135a was significantly altered in senescent VSMCs. Cell calcification and calcification genes, including ALP and OC, were greatly altered by miR-135a inhibition. The luciferase assay validated KLF4 as the target protein of miR-135a, and Western blotting revealed that the expression of KLF4 and STAT3 were both significantly decreased by over expressed miR-135a, while the inhibition of miR-135a and KLF4
siRNA both decreased the STAT3 protein levels. Moreover, the inhibition of miR-135a dramatically increased the calcium concentration, but co-treatment with KLF4 or STAT3 siRNA both decreased the calcium concentration. The present study identified miR-135a as a potential osteogenic differentiation suppressor in senescent VSMCs and revealed that KLF4/STAT3 pathway, at least partially, was involved in the mechanism.
Calcifying aortic valve disease (CAVD) or arteriosclerosis due to calcifications is a major health problem of up to 50% of people over age 75 years. In addition, cardiovascular calcification acts as an independent risk factor for cardiovascular morbidity and mortality [1, 22]. Although aging was popularly recognized as a potent cardiovascular risk factor, the mechanisms underlying the association between aging and calcification remain unknown. miRs were reported to be involved in cardiovascular calcification [8-10]. Many studies have explored the role of miRs in vascular smooth muscle cell calcification, not just miR-135a. Gui et al. suggested that increased expression of several miRs, including miR-135a, miR-762, miR-714, and miR-712, might be involved in VSMC calcification by disrupting Ca(2+) efflux proteins [23]. In another study, inorganic phosphate (Pi) treatment increased calcification and migration of HVSMCs and reduced cell proliferation and the amount of the actin cytoskeleton. Significant downregulation of miRNA-143 (miR-143) and miR-145 and concomitant upregulation of KLF-4 and -5 and versican was observed in those cells [24]. In contrast, miR-221 and miR-222 mimics induced significant changes in ectonucleotide phosphodiesterase 1 and Pit-1 expression, suggesting that these miRNAs may promote VSMC calcification through cellular inorganic phosphate and pyrophosphate levels [25]. Liao et al. proved that overexpression of miR-133a inhibited VSMC transdifferentiation into osteoblast-like cells as evidenced by a decrease in ALP activity, osteocalcin secretion, mineralized nodule formation and Runx2 expression, which was identified as a direct target of miR-133a [26]. Wen et al. suggested that overexpression of miR-125b could inhibit β-glycerophosphoric acid-induced osteogenic markers expression and calcification of VSMCs [27], demonstrating the important role of miR-125b in the regulation of transdifferentiation and calcification of VSMCs. They found out that miR-125b targeted Ets1 and regulated its protein expression in VSMCs, while the downregulation of Ets1 expression inhibited β-glycerophosphoric acid-induced VSMCs phenotypic transition and calcification. Providing our result that miR-135a mediates VSMCs calcification through KLF4/STAT3 pathway, this study suggested that unlike miR-135a, miR-125b could regulate VSMCs calcification through a different pathway. Different microRNAs have different targets while regulating VSMC calcification. The interaction between these pathways involved in this process is worthy of investigation. Taken together, these results indicate that the roles of different miRs in the osteogenic differentiation of VSMCs are controversial. The mechanism of the suppression or acceleration of VSMC calcification by different microRNAs deserves intense exploration.
In this study, we defined a novel miR-135a-dependent mechanism during the progression of vascular calcification. Altered expression of miR-135a has been observed in several cancers, which indicated that increased expression of miR-135a could promote cell apoptosis and inhibit cell proliferation [28, 29]. miR-135a was also reported to play a critical role in maintaining stem cell pluripotency via the regulation of sirtuin1 (SIRT1) in mouse embryonic stem cells [30]. Moreover, up-regulated miR-135a promoted portal vein tumor thrombosis by repressing MTSS1 expression in vitro and in vivo and hepatocellular carcinoma metastasis [14]. In addition, miR-135a may mediate the regulation of vascular calcification by other substances, such as statins, a cornerstone for treating atherosclerotic cardiovascular disease [31]. Several studies have reported the impact of statins on vascular calcification. Kizu et al. reported that statins inhibited in vitro calcification of human VSMCs induced by inflammatory mediators and depressed expression of ALP in HVSMCs [32]. Later, Son et al. suggested that the Gas6/Axl-PI3K/Akt pathway might play a central role in the effect of statins on calcification of VSMCs [33]. The relationship between miRNAs and the effect of statins has been documented. For example, Li et al. suggested that miRNAs mediated the protective effect of statins in unstable angina patients [34], while others reported the participation of regulatory miRs 221/222 on nitric oxide (NO) release induction mediated by statins [35]. The potential role of miR-135a in mediating the effects of statins on vascular calcification deserves further investigation.
Our results revealed that as senility occurred, the expression of miR-135a gradually lowered, but the mRNA expression of ALP and OC were significantly up-regulated. Moreover, the inhibition of endogenous miR-135a in VSMCs promoted cellular calcification as demonstrated by increased matrix mineralization. The ALP activity, mRNA expression of ALP and OC were also significantly increased by the inhibition of miR-135a, indicating the relationship between miR-135a and ALP and OC. Taken together, these findings indicated that the decrease of miR-135a in the senescent phase of VSMCs caused cell osteogenic differentiation, increased ALP activity and regulated the mRNA expression of ALP and OC.
Many studies have shown the regulation of KLF4 on osteogenic differentiation. Yoshida et al. showed that Klf4 regulated the transcription of osteogenic genes in SMCs, while siRNA-mediated knockdown of Klf4 attenuated high phosphate-induced increases in expression of osteopontin and ALP in cultured SMCs [36]. The mechanism might involve decreased expression of SMC differentiation marker genes such as SM α-actin, SM-myosin heavy chain, and SM22α. Chen et al. reported that expression of KLF4 is closely correlated to the growth-arrest and the first step of odontoblast and ameloblast differentiation [37]. While recent insights indicated that KLF4 was indispensable for vascular homeostasis, it plays an exacted role in proliferation and angiogenesis [38, 39]. Mounting evidence indicates that KLF4 was highly expressed in VSMCs and acts as critical regulator of vascular homeostasis [40-42]. Luciferase results in the present study showed that co-transfection of VSMCs with the pMIR-REPORT construct containing mutant KLF4 3′-UTR and PLemiR-135a did not show much difference, but co-transfection with this luciferase construct containing wild-type KLF4 3′-UTR and PLemiR-135a resulted in a nearly 80% decline in the luciferase activity, indicating that the direct target of miR-135a is KLF4. In addition, Western blotting demonstrated that protein KLF4 were significantly decreased while VSMCs were co-transfected with the luciferase construct containing over expressed miR-135a. Taken together, these results showed that miR-135a could directly down-regulated the KLF4 expression.
However, how KLF4 regulates cell calcification needs further investigation. Here we focused on STAT3, a key mediator of the expression of a variety of genes in response to cell stimuli. Previous study has found out that IL-6/sIL-6R stimulation accelerated the ROR2/WNT5A pathway in cells in a STAT3-dependent manner, resulting in augmented calcification [18]. STAT3 is involved in IL-6-mediated ROR2 mRNA expression and is essential for IL-6/sIL-6R-induced osteoblast-like differentiation and mineralization [18]. A previous study has reported the crosstalk between KLF4 and activated STAT3 in the regulation of axon regeneration [43]. We found that expression of STAT3 was significantly decreased while co-transfected with the luciferase construct containing over expressed miR-135a, and the inhibition of miR-135a and KLF4 siRNA both decreased the STAT3 protein levels. These findings revealed that miR-135a could down-regulate the STAT3 expression through KLF4. Finally, to confirm the role of miR-135a, KLF4 and STAT3 on VSMC calcification, we performed a cellular calcification quantification. With the inhibition of miR-135a, the calcium concentration was dramatically increased, but the co-treatment with KLF4 or STAT3 siRNA both decreased the calcium concentration, which was significantly different from the Anti-miR-135a group.
The prevalence of vascular calcification increases as glomerular filtration rate (GFR) declines and calcification occurs years earlier in patients with chronic kidney disease and is associated with significant morbidity and mortality. [44]. Although no study has yet measured miR-135a expression in this population, some studies have revealed the involvement of miRs in vascular damage in experimental chronic kidney disease [45], such as miR-126, -143 and -223. As miR-135a plays an important role in vascular calcification, the relationship between it and the chronic kidney disease requires further study. It would be interesting to explore the role of miR-135a and identify it as a possible target to prevent vascular calcification of patients with chronic kidney disease.
5. CONCLUSION
Our results identified miR-135a as a potential osteogenic differentiation suppressor. In senescent VSMCs, miR-135a was decreased, inducing an up-regulation of calcification genes. KLF4 and STAT3 were both regulating targets of miR-135a. The present study identified miR-135a as a potential osteogenic differentiation suppressor in senescent VSMCs and revealed that the KLF4/STAT3 pathway, at least partially, was involved in the mechanism.
ACKNOWLEDGEMENTS
Lin Lin, Yue He and Bei-Li Xi designed the project and wrote the paper. Yi Qu conducted the experiments. Hong-Chao Zheng, Qian Chen, Jun Li, Ying Hu, Ming-Hao Ye and Ping Chen provided technical assistance and materials.
CONFLICT OF INTEREST
This project was supported by the Young Scholars Fund of 2014 Shanghai Xuhui District Medical Science and Technology Foundation (Grant No. SHXH201423).
REFERENCES
- 1.Goettsch C., Rauner M., Pacyna N., Hempel U., Bornstein S.R., Hofbauer L.C. miR-125b regulates calcification of vascular smooth muscle cells. Am. J. Pathol. 2011;179(4):1594–1600. doi: 10.1016/j.ajpath.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mathew S., Tustison K.S., Sugatani T., Chaudhary L.R., Rifas L., Hruska K.A. The mechanism of phosphorus as a cardiovascular risk factor in CKD. J. Am. Soc. Nephrol. 2008;19(6):1092–1105. doi: 10.1681/ASN.2007070760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reynolds J.L., Joannides A.J., Skepper J.N., et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J. Am. Soc. Nephrol. 2004;15(11):2857–2867. doi: 10.1097/01.ASN.0000141960.01035.28. [DOI] [PubMed] [Google Scholar]
- 4.Pan Z.W., Lu Y.J., Yang B.F. MicroRNAs: a novel class of potential therapeutic targets for cardiovascular diseases. Acta Pharmacol. Sin. 2010;31(1):1–9. doi: 10.1038/aps.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Urbich C., Kuehbacher A., Dimmeler S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc. Res. 2008;79(4):581–588. doi: 10.1093/cvr/cvn156. [DOI] [PubMed] [Google Scholar]
- 6.Fichtlscherer S., De Rosa S., Fox H., et al. Circulating microRNAs in patients with coronary artery disease. Circ. Res. 2010;107(5):677–684. doi: 10.1161/CIRCRESAHA.109.215566. [DOI] [PubMed] [Google Scholar]
- 7.Zampetaki A., Kiechl S., Drozdov I., et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ. Res. 2010;107(6):810–817. doi: 10.1161/CIRCRESAHA.110.226357. [DOI] [PubMed] [Google Scholar]
- 8.Wen P., Cao H., Fang L., et al. miR-125b/Ets1 axis regulates transdifferentiation and calcification of vascular smooth muscle cells in a high-phosphate environment. Exp. Cell Res. 2014;322(2):302–312. doi: 10.1016/j.yexcr.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 9.Ohnaka M., Marui A., Yamahara K., et al. Effect of microRNA-145 to prevent vein graft disease in rabbits by regulation of smooth muscle cell phenotype. J. Thorac. Cardiovasc. Surg. 2014;148(2):676–682. doi: 10.1016/j.jtcvs.2013.11.054. [DOI] [PubMed] [Google Scholar]
- 10.Sun S.G., Zheng B., Han M., et al. miR-146a and Kruppel-like factor 4 form a feedback loop to participate in vascular smooth muscle cell proliferation. EMBO Rep. 2011;12(1):56–62. doi: 10.1038/embor.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagel R., le Sage C., Diosdado B., et al. Regulation of the adenomatous polyposis coli gene by the miR-135 family in colorectal cancer. Cancer Res. 2008;68(14):5795–5802. doi: 10.1158/0008-5472.CAN-08-0951. [DOI] [PubMed] [Google Scholar]
- 12.Zhou W., Li X., Liu F., et al. MiR-135a promotes growth and invasion of colorectal cancer via metastasis suppressor 1 in vitro. Acta Biochim. Biophys. Sin. (Shanghai) 2012;44(10):838–846. doi: 10.1093/abbs/gms071. [DOI] [PubMed] [Google Scholar]
- 13.Tang W., Jiang Y., Mu X., Xu L., Cheng W., Wang X. MiR-135a functions as a tumor suppressor in epithelial ovarian cancer and regulates HOXA10 expression. Cell. Signal. 2014;26(7):1420–1426. doi: 10.1016/j.cellsig.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 14.Liu S., Guo W., Shi J., et al. MicroRNA-135a contributes to the development of portal vein tumor thrombus by promoting metastasis in hepatocellular carcinoma. J. Hepatol. 2012;56(2):389–396. doi: 10.1016/j.jhep.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 15.Lee H.Y., Ahn J.B., Rha S.Y., et al. High KLF4 level in normal tissue predicts poor survival in colorectal cancer patients. World J. Surg. Oncol. 2014;12:232. doi: 10.1186/1477-7819-12-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshida T., Hayashi M. Role of Kruppel-like factor 4 and its binding proteins in vascular disease. J. Atheroscler. Thromb. 2014;21(5):402–413. doi: 10.5551/jat.23044. [DOI] [PubMed] [Google Scholar]
- 17.Turner E.C., Huang C.L., Govindarajan K., Caplice N.M. Identification of a Klf4-dependent upstream repressor region mediating transcriptional regulation of the myocardin gene in human smooth muscle cells. Biochim. Biophys. Acta. 2013;1829(11):1191–1201. doi: 10.1016/j.bbagrm.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 18.Fukuyo S., Yamaoka K., Sonomoto K., et al. IL-6-accelerated calcification by induction of ROR2 in human adipose tissue-derived mesenchymal stem cells is STAT3 dependent. Rheumatology (Oxford) 2014;53(7):1282–1290. doi: 10.1093/rheumatology/ket496. [DOI] [PubMed] [Google Scholar]
- 19.Villa-Bellosta R., Sorribas V. Phosphonoformic acid prevents vascular smooth muscle cell calcification by inhibiting calcium-phosphate deposition. Arterioscler. Thromb. Vasc. Biol. 2009;29(5):761–766. doi: 10.1161/ATVBAHA.108.183384. [DOI] [PubMed] [Google Scholar]
- 20.Verberckmoes S.C., Persy V., Behets G.J., et al. Uremia-related vascular calcification: More than apatite deposition. Kidney Int. 2007;71(4):298–303. doi: 10.1038/sj.ki.5002028. [DOI] [PubMed] [Google Scholar]
- 21.Martín-Pardillos A., Sosa C., Sorribas V. Arsenic increases Pi-mediated vascular calcification and induces premature senescence in vascular smooth muscle cells. Toxicol. Sci. 2013;131(2):641–653. doi: 10.1093/toxsci/kfs313. [DOI] [PubMed] [Google Scholar]
- 22.Roosens B., Bala G., Droogmans S., et al. Occurrence of cardiovascular calcifications in normal, aging rats. Exp. Gerontol. 2012;47(8):614–619. doi: 10.1016/j.exger.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 23.Gui T., Zhou G., Sun Y., et al. MicroRNAs that target Ca(2+) transporters are involved in vascular smooth muscle cell calcification. Lab. Invest. 2012;92(9):1250–1259. doi: 10.1038/labinvest.2012.85. [DOI] [PubMed] [Google Scholar]
- 24.Rangrez A.Y., M'Baya-Moutoula E., Metzinger-Le Meuth V., et al. Inorganic phosphate accelerates the migration of vascular smooth muscle cells: evidence for the involvement of miR-223. PLoS One. 2012;7(10):e47807. doi: 10.1371/journal.pone.0047807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mackenzie N.C., Staines K.A., Zhu D., Genever P., Macrae V.E. miRNA-221 and miRNA-222 synergistically function to promote vascular calcification. Cell Biochem. Funct. 2014;32(2):209–216. doi: 10.1002/cbf.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao X.B., Zhang Z.Y., Yuan K., et al. MiR-133a modulates osteogenic differentiation of vascular smooth muscle cells. Endocrinology. 2013;154(9):3344–3352. doi: 10.1210/en.2012-2236. [DOI] [PubMed] [Google Scholar]
- 27.Wen P., Cao H., Fang L., et al. miR-125b/Ets1 axis regulates transdifferentiation and calcification of vascular smooth muscle cells in a high-phosphate environment. Exp. Cell Res. 2014;322(2):302–312. doi: 10.1016/j.yexcr.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 28.Earle J.S., Luthra R., Romans A., et al. Association of microRNA expression with microsatellite instability status in colorectal adenocarcinoma. J. Mol. Diagn. 2010;12(4):433–440. doi: 10.2353/jmoldx.2010.090154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greco S., De Simone M., Colussi C., et al. Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J. 2009;23(10):3335–3346. doi: 10.1096/fj.08-128579. [DOI] [PubMed] [Google Scholar]
- 30.Saunders L.R., Sharma A.D., Tawney J., et al. miRNAs regulate SIRT1 expression during mouse embryonic stem cell differentiation and in adult mouse tissues. Aging (Albany, N.Y.) 2010;2(7):415–431. doi: 10.18632/aging.100176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nissen S.E., Nicholls S.J., Sipahi I., et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006;295(13):1556–1565. doi: 10.1001/jama.295.13.jpc60002. [DOI] [PubMed] [Google Scholar]
- 32.Kizu A., Shioi A., Jono S., Koyama H., Okuno Y., Nishizawa Y. Statins inhibit in vitro calcification of human vascular smooth muscle cells induced by inflammatory mediators. J. Cell. Biochem. 2004;93(5):1011–1019. doi: 10.1002/jcb.20207. [DOI] [PubMed] [Google Scholar]
- 33.Son B.K., Kozaki K., Iijima K., et al. Gas6/Axl-PI3K/Akt pathway plays a central role in the effect of statins on inorganic phosphate-induced calcification of vascular smooth muscle cells. Eur. J. Pharmacol. 2007;556(1-3):1–8. doi: 10.1016/j.ejphar.2006.09.070. [DOI] [PubMed] [Google Scholar]
- 34.Li J., Chen H., Ren J., et al. Effects of statin on circulating microRNAome and predicted function regulatory network in patients with unstable angina. BMC Med. Genomics. 2015;8:12. doi: 10.1186/s12920-015-0082-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cerda A., Fajardo C.M., Basso R.G., Hirata M.H., Hirata R.D. Role of microRNAs 221/222 on statin induced nitric oxide release in human endothelial cells. Arq. Bras. Cardiol. 2015;104(3):195–201. doi: 10.5935/abc.20140192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshida T., Yamashita M., Hayashi M. Kruppel-like factor 4 contributes to high phosphate-induced phenotypic switching of vascular smooth muscle cells into osteogenic cells. J. Biol. Chem. 2012;287(31):25706–25714. doi: 10.1074/jbc.M112.361360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Z., Couble M.L., Mouterfi N., Magloire H., Chen Z., Bleicher F. Spatial and temporal expression of KLF4 and KLF5 during murine tooth development. Arch. Oral Biol. 2009;54(5):403–411. doi: 10.1016/j.archoralbio.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 38.Zheng X., Li A., Zhao L., et al. Key role of microRNA-15a in the KLF4 suppressions of proliferation and angiogenesis in endothelial and vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2013;437(4):625–631. doi: 10.1016/j.bbrc.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 39.Liu Y., Zheng B., Zhang X.H., Nie C.J., Li Y.H., Wen J.K. Localization and function of KLF4 in cytoplasm of vascular smooth muscle cell. Biochem. Biophys. Res. Commun. 2013;436(2):162–168. doi: 10.1016/j.bbrc.2013.05.067. [DOI] [PubMed] [Google Scholar]
- 40.Atkins G.B., Jain M.K. Role of Kruppel-like transcription factors in endothelial biology. Circ. Res. 2007;100(12):1686–1695. doi: 10.1161/01.RES.0000267856.00713.0a. [DOI] [PubMed] [Google Scholar]
- 41.Mun G.I., Boo Y.C. A regulatory role of Kruppel-like factor 4 in endothelial argininosuccinate synthetase 1 expression in response to laminar shear stress. Biochem. Biophys. Res. Commun. 2012;420(2):450–455. doi: 10.1016/j.bbrc.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 42.Zhou G., Hamik A., Nayak L., et al. Endothelial Kruppel-like factor 4 protects against atherothrombosis in mice. J. Clin. Invest. 2012;122(12):4727–4731. doi: 10.1172/JCI66056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qin S., Zou Y., Zhang C.L. Cross-talk between KLF4 and STAT3 regulates axon regeneration. Nat. Commun. 2013;4:2633. doi: 10.1038/ncomms3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palit S., Kendrick J. Vascular calcification in chronic kidney disease: role of disordered mineral metabolism. Curr. Pharm. Des. 2014;20(37):5829–5833. doi: 10.2174/1381612820666140212194926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taïbi F., Metzinger-Le Meuth V., M'Baya-Moutoula E., et al. Possible involvement of microRNAs in vascular damage in experimental chronic kidney disease. Biochim. Biophys. Acta. 2014;1842(1):88–98. doi: 10.1016/j.bbadis.2013.10.005. [DOI] [PubMed] [Google Scholar]