

Abstract
Objective
Alterations in sphingolipid and ceramide metabolism have been associated with various diseases, including nonalcoholic fatty liver disease (NAFLD). Acid sphingomyelinase (ASM) converts the membrane lipid sphingomyelin to ceramide, thereby affecting membrane composition and domain formation. We investigated the ways in which the Asm knockout (Smpd1−/−) genotype affects diet-induced NAFLD.
Methods
Smpd1−/− mice and wild type controls were fed either a standard or Western diet (WD) for 6 weeks. Liver and adipose tissue morphology and mRNA expression were assessed. Quantitative proteome analysis of liver tissue was performed. Expression of selected genes was quantified in adipose and liver tissue of obese NAFLD patients.
Results
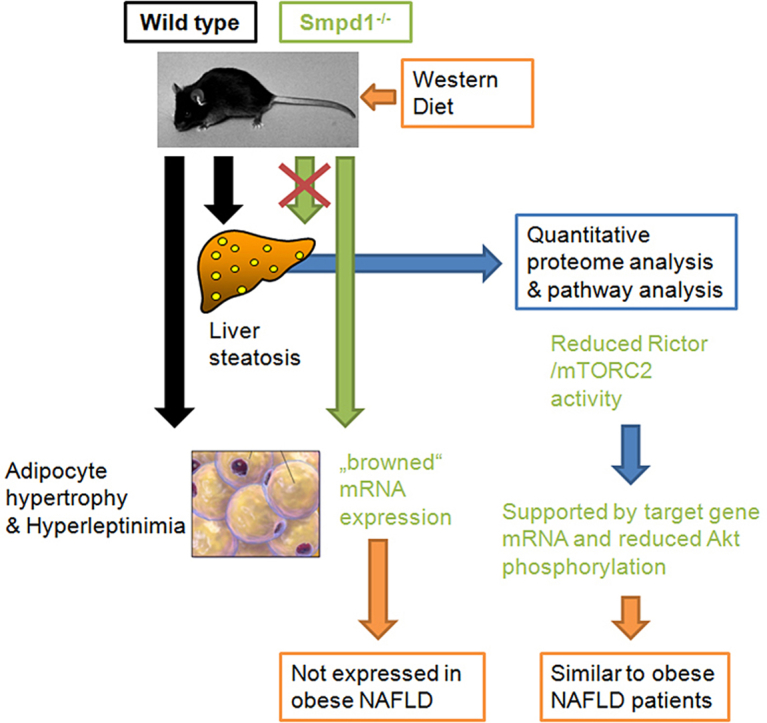
Although Smpd1−/− mice exhibited basal steatosis with normal chow, no aggravation of NAFLD-type injury was observed with a Western diet. This protective effect was associated with the absence of adipocyte hypertrophy and the increased expression of genes associated with brown adipocyte differentiation. In white adipose tissue from obese patients with NAFLD, no expression of these genes was detectable. To further elucidate which pathways in liver tissue may be affected by Smpd1−/−, we performed an unbiased proteome analysis. Protein expression in WD-fed Smpd1−/− mice indicated a reduction in Rictor (mTORC2) activity; this reduction was confirmed by diminished Akt phosphorylation and altered mRNA expression of Rictor target genes.
Conclusion
These findings indicate that the protective effect of Asm deficiency on diet-induced steatosis is conferred by alterations in adipocyte morphology and lipid metabolism and by reductions in Rictor activation.
Keywords: Ceramide, NAFLD, Rictor, Western diet
Graphical abstract
Highlights
-
•
Smpd1 knockout protects from dietary induced liver steaosis and abrogates hypertrophy of visceral adipocytes in mice.
-
•
Expression of genes related to browning is absent in human adipose tissue from obese individuals.
-
•
Protection from dietary-induced liver steatosis is associated with reduced Rictor/mTORC2 activity in Smpd1 knockout mice.
1. Introduction
Western societies are experiencing an increase in the prevalence of obesity, which leads to a parallel increase in the prevalence of related diseases, such as the metabolic syndrome, type II diabetes, and nonalcoholic fatty liver disease (NAFLD) [1]. NAFLD is primarily associated with excessive accumulation of fat in the liver and ranges from simple steatosis to nonalcoholic steatohepatitis (NASH), which is accompanied by cell death, tissue inflammation, and a high risk of the development of fibrosis, with further progression to cirrhosis or hepatocellular carcinoma [2]. Obesity is also associated with the development of systemic inflammation and insulin resistance as part of the metabolic syndrome, which promotes the progression of NAFLD. In particular, cytokines derived from adipose tissue, also known as adipokines, play an important role in this systemic disease [3].
Hypertrophy of adipose tissue alters adipokine secretion and enhances the release of free fatty acids (FAAs) into the circulation [4], [5]. This effect seems to be caused specifically by visceral adipocyte hypertrophy, which is associated with serum inflammation markers and altered adipokine secretion, indicating an interaction between adipose tissue and liver tissue in the metabolic syndrome [6], [7], [8]. Thus, these changes in the concentrations of circulating lipid components and in (hepato)cellular lipid metabolism seem to play an important role in the formation of NAFLD.
Ceramides are sphingolipids that are important components of cell membranes. The ceramide content of a membrane affects fluidity and the signal transduction pathways regulating cell differentiation or apoptosis. The effects of ceramide are mediated by the formation of ceramide-rich microdomains that facilitate the trapping and clustering of receptors and, thereby, the induction of apoptosis [9], [10]. Altered ceramide levels in cell membranes have been associated with many human diseases, such as neurodegenerative and skin disorders, pulmonary and cardiovascular diseases, and hormonal disorders and liver diseases [10], [11], [12], [13], [14].
Ceramide can be derived from membrane-bound sphingomyelin that is cleaved by an enzyme called acid sphingomyelinase (ASM), coded by the SMPD1 gene. The conversion of sphingomyelin to ceramide within cell membranes is essential for various signaling pathways [15], [16]. ASM deficiency has also been discussed as a possible mechanism in the development of obesity, the metabolic syndrome, diabetes, and various liver diseases, such as steatosis or fibrosis [14], [17], [18], [19]. It has also been reported that sphingolipids, especially ceramide, play a pivotal role in obesity and the metabolic syndrome [20], [21]. Boini and colleagues found that excessive accumulation of sphingolipids, ceramide, and the metabolites of ceramide contribute to the development of obesity and associated kidney damage in mice fed a high-fat diet (HFD). Treatment with amitriptyline, a functional ASM antagonist, diminishes both the steatosis associated with HFD and the accumulation of fat in this murine model.
A protective effect against diet-induced liver steatosis has also been observed in Asm knockout (Smpd1−/−) mice and in Asm and low-density lipoprotein receptor (Ldlr) double knockout mice [22], [23]. Although the results of these studies indicate that ASM deficiency may protect from diet-induced liver steatosis by reducing autophagy and endoplasmic reticulum stress, many other processes may also be involved. Therefore, we aimed for a broader view of processes potentially affected by ASM knockout, processes that may even protect from steatosis. In addition, we specifically investigated the contribution of adipose tissue to the development of NAFLD. We found that reductions in the activation of rapamycin-insensitive companion of mTOR (Rictor, or mTORC2) in the liver and alterations in adipocyte physiology may contribute to the protective effect of Smpd1−/− against diet-induced steatosis.
2. Material and methods
2.1. Animals and sample collection
Four- to six-week-old C57Bl/6 and Smpd1−/− mice [11] were fed a standard diet (SD; n = 6 animals per group) or a Western diet rich in carbohydrates and fat (WD; TD.88137, details are given in Supplementary Table 1; ssniff Spezialdiäten, Soest, Germany) ad libitum for six weeks (n = 6 animals per group). Food intake was not measured. After six weeks, mice were sacrificed, blood was drawn from the vena cava and centrifuged, and serum was stored at −80 °C. Total protein, albumin, total bilirubin, aspartate aminotransferase (AST), alanine aminotransferase (ALT), and lactate dehydrogenase (LDH) levels were measured with a Spotchem II system (Akray, Kyoto, Japan). Liver tissue and white adipose tissue were collected for isolation of RNA and protein and for histopathological processing. All mice were bred and housed in the Central Animal Facility (ZTL) of the University Hospital Essen, University of Duisburg-Essen (Germany), according to the recommendations of the Federation of European Laboratory Animal Science Associations (FELASA). All procedures were approved by the State Agency for the Protection of Nature, the Environment, and Consumers, North Rhine-Westphalia (Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen; LANUV NRW).
2.2. Histopathology and sample handling
Liver and adipose tissues were stored in a 4.5% formalin solution, embedded in paraffin, and sectioned. Staining was performed as previously described [24]. The size of adipocytes was determined in paraffin-embedded sections stained with hematoxylin and eosin (H&E), as described previously [6].
Liver and adipose tissues for the isolation of RNA and protein were immediately frozen in liquid nitrogen. Total RNA was isolated by TRIzol extraction (Invitrogen, Darmstadt, Germany) and purified with the RNeasy Mini Kit (Qiagen, Hilden, Germany). Reverse transcription was performed with the QuantiTect RT kit (Qiagen) with 1 μg of total RNA.
2.3. Patients
The study protocol conformed to the revised Declaration of Helsinki (Edinburgh, 2000) and was approved by the local Institutional Review Board (Ethik-Kommission am Universitätsklinikum Essen; file number 09-4252). Before enrollment, all patients provided written informed consent for participation in the study.
Data from liver and matched adipose tissue samples were collected from morbidly obese patients with biopsy-proven NAFLD who were undergoing bariatric surgery. The samples were analyzed and compared with four non-steatotic liver samples. All enrolled patients underwent physical and ultrasound examinations, a complete set of laboratory studies, and liver biopsy. Supplementary Tables 2 and 3 present detailed demographic and clinical information.
Subjects reporting excessive alcohol consumption (>20 g/day for men or >10 g/day for women) and those with other known causes of secondary fatty liver disease (e.g., viral hepatitis, metabolic liver disease, toxic liver disease) were excluded from the study. An experienced pathologist (HAB) used the NAFLD Activity Score (NAS) to determine the degree of NAFLD in wedge liver biopsy samples obtained during a surgical procedure and stained with H&E [25]. The mean adipocyte diameter in representative slides was calculated from multiple (>50) individual measurements of adipocyte diameter (ImageJ, National Institutes of Health, Bethesda, MD, USA), as previously described [6].
2.4. Quantitative real-time polymerase chain reaction
Gene expression levels were measured by quantitative real-time polymerase chain reaction (qRT-PCR) with succinate dehydrogenase complex subunit A (Sdha) as a reference gene; oligonucleotide sequences are presented in Supplementary Table 4. Relative gene expression was calculated from the threshold cycles in relation to the reference gene and to untreated controls. Reactions were performed on a CFX96 Touch qPCR System (Biorad Laboratories, Munich, Germany), as previously described [26].
2.5. Quantitative proteome analysis
Mass spectrometry–based proteome analysis was performed according to a previously described protocol [27], [28]. Briefly, liver tissue samples were lysed in 30 mM Tris HCl, 2 M thiourea, 7 M urea, and 4% CHAPS detergent (pH 8.5) and subjected to in-gel digestion with trypsin. Trypsinized peptides were then analyzed online with an Ultimate 3000 RSLCnano system coupled to an Orbitrap Elite mass spectrometer (both from Thermo Fisher Scientific, Bremen, Germany). Liquid chromatography–tandem mass spectrometry (LC-MS/MS) data were analyzed with Proteome Discoverer software (version 1.4; Thermo Fisher Scientific) for protein identification and Progenesis LC-MS (version 4.1; Nonlinear Dynamics Ltd., Newcastle upon Tyne, UK) for protein quantification based on two unique peptides per protein. Statistical significance was determined with ANOVA and Tukey's honestly significant difference test for post hoc analysis with RStudio software (version 0.99; RStudio Inc., Boston, MA, USA). Proteins with fold changes higher than 1.5 or lower than −1.5 and with p values lower than 0.05 were considered to be differentially expressed between two experimental groups and were used for further analysis. In total, six biological replicates were analyzed per experimental group.
2.6. Pathway analysis
Data from quantitative proteomics experiments were analyzed with Ingenuity® Pathway Analysis (IPA; QIAGEN, Redwood City, CA, USA; www.qiagen.com/ingenuity). The proteins differentially expressed between the experimental groups were analyzed for enriched canonical pathways and associated upstream regulators. Upstream regulators and canonical pathways with z-scores higher than 2 were considered to be activated, and those with z-scores lower than −2 were considered to be inhibited.
2.7. Western blotting
Protein lysates generated during proteomics experiments were used for Western blotting. Protein concentrations were assessed with Pierce BCA kit (Thermo Fisher, Braunschweig, Germany), and 30 μg of total protein was separated with sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Immunoblotting was performed according to standard procedures with the following primary antibodies: Nrf2/Nef2l2 (Cell Signaling Technology, Danvers, MA, USA); pERK (Cell Signaling Technology); or ERK (Santa Cruz Biotechnology, La Jolla, CA, USA). For a loading control, we used β-Actin (13E5) (Cell Signaling Technology).
2.8. Biochemical assays and enzyme-linked immunosorbent assays
Biochemical assays for FFAs in serum were performed with the Fatty Acid Quantification Kit (Biovision/BioCat GmbH, Heidelberg, Germany). Serum leptin and adiponectin levels were determined with Quantikine mouse enzyme-linked immunosorbent assay (ELISA) kits for leptin and adiponectin (R&D Systems Inc., Minneapolis, MN, USA).
2.9. Ceramide content in liver tissue
Liver samples were subjected to cryoconservation in liquid nitrogen and were lysed in TN3 buffer. The samples were extracted in CHCl3:CH3OH:1N HCl (100:100:1; v/v/v), and the organic phase was collected and dried. Diacylglycerol (DAG) was degraded in 0.1 N methanolic potassium hydroxide (KOH) at 37 °C for 60 min; the samples were re-extracted, and the organic phase was dried and resuspended in 20 μL of a detergent solution containing 7.5% (w/v) n-octyl glucopyranoside and 5 mM cardiolipin in 1 mM diethylenetriaminepentaacetic acid (DETAPAC). The samples were sonicated for 10 min in a bath sonicator to facilitate micelle formation. Phosphorylation of ceramide was initiated by the addition of 70 μL of a buffer consisting of 0.1 M imidazole/HCl (pH 6.6), 0.1 M NaCl, 25 mM MgCl2, 2 mM ethylene glycol tetraacetic acid (EGTA), 2.8 mM dithiothreitol (DTT), 1 mM adenosine triphosphate (ATP), 10 μCi [32P]γATP, and DAG kinase, according to the vendor's instructions (GE Healthcare Europe, Freiburg, Germany). The samples were incubated for 60 min at room temperature; the incubation was stopped by extraction in 1 mL CHCl3:CH3OH:1N HCl (100:100:1; v/v/v), 170 μL buffered saline solution (135 mM NaCl, 1.5 mM CaCl2, 0.5 mM MgCl2, 5.6 mM glucose, and 10 mM HEPES [pH 7.2]), and 30 μL of a 100-mM ethylenediaminetetraacetic acid (EDTA) solution. The organic phase was collected, dried, and dissolved in 20 μL CHCl3:CH3OH (1:1; v/v). Lipids were separated on Silica G60 thin-layer chromatography (TLC) plates with CHCl3:CH3OH:CH3COOH (65:15:5; v/v/v); each plate was dried and analyzed by autoradiography. Ceramide spots were identified by co-migration with a C16-ceramide standard, scraped from the plate, and quantified by liquid scintillation counting. Ceramide amounts were determined from a standard curve using C16-ceramide.
2.10. Statistical analysis
Statistical significance was determined by two-way ANOVA with the Bonferroni correction for multiple comparisons with Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA). For patient data, unpaired two-tailed t-tests were performed. Statistical significance set at the level of p ≤ 0.05. If not otherwise stated, all data are presented as means ± SEM. Correlations between parametric data were measured with the Pearson product-moment correlation coefficient; correlations between nonparametric data were measured with Spearman's rank correlation coefficient.
3. Results
3.1. A Western diet does not aggravate steatosis in Smpd1−/− mice
Consuming excess calories in the form of fat and carbohydrates leads to higher serum levels of FFA because the storage capacity of adipocytes is exceeded [7], resulting in fat accumulation in liver tissue and, subsequently, NAFLD. To test the role of Asm in diet-induced steatosis of liver tissue, we fed WT mice and Smpd1−/− mice either a SD or WD ad libitum for as long as six weeks so that we could focus on the early stages of NAFLD. Histologic assessment of liver tissue sections confirmed robust steatosis in WD-fed WT mice (Figure 1). SD-fed Smpd1−/− mice exhibited a mild steatotic phenotype (Figure 1B), probably because of the accumulation of sphingomyelin. However, this phenotype was not aggravated by WD feeding (Figure 1). Steatosis was not as severe in Smpd1−/− mice as in WD-fed WT mice. In addition, the liver-to-body weight ratio (LBWR) was significantly higher in Smpd1−/− mice than in corresponding WT mice (p = 0.0003; Figure 1E). In particular, the LBWR was higher in WD-fed Smpd1−/− mice than in WD-fed WT mice (p < 0.001).
Figure 1.

Acid sphingomyelinase deficiency ameliorates diet-induced hepatocellular steatosis. (A) After 6 weeks of eating either a standard diet (SD) or a Western diet (WD), liver tissue from acid sphingomyelinase deficient (Smpd1−/−) mice and C57BL/6 mice (wild-type, WT) was sectioned and stained with hematoxylin and eosin (H&E). SD-fed Smpd1−/− mice exhibited slight steatosis, which seems to be mostly based on formation of foam cells (macrophages with lipid accumulation). Although WD-fed WT animals exhibited robust steatosis of hepatocytes, WD did not aggravate hepatic steatosis in Smpd1−/− mice. Foam cell formation seemed to be slightly increased. (B) Smpd−/− animals exhibited a slightly higher liver-to-body weight ratio, which increased with WD consumption. This effect did not occur in WT animals.
3.2. Serum indicators of liver damage are unaltered by short-term steatosis
Liver damage was determined by liver serum indicators. No significant differences in routine liver damage variables were found between the groups (Suppl. Fig. 1). Albumin production was significantly lower in Smpd1−/− mice than in WT mice (Suppl. Fig. 1E; p < 0.04), and WD feeding significantly reduced serum albumin levels independent of genotype (p < 0.02). Short-term WD feeding did not affect serum markers of liver damage in WT or Smpd1−/− mice.
3.3. Western diet feeding increases profibrotic gene expression but not fibrosis in liver tissue of Smpd1−/− mice
Steatosis may progress to steatohepatitis and finally to fibrosis and cirrhosis, changes that are indicated by the expression of typical markers such as transforming growth factor beta 1 (Tgfβ1), collagen type I-alpha 1 (Col1a1), and α-smooth muscle actin (Acta2). Our results show that mRNA expression of both the profibrotic growth factor Tgfβ1 (Suppl. Fig. 2A) and Col1a1 (Suppl. Fig. 2B) was significantly higher in liver tissue from WD-fed Smpd1−/− than in WT mice. No alterations were observed in the expression of Acta2, a marker of hepatic stellate cell activation, or in collagen deposition, as assessed by Sirius Red staining (Suppl. Fig. 2C, D).
3.4. Altered adipocyte physiology may contribute to diminished steatosis in Smpd1−/− mice fed a Western diet
To identify the molecular mechanisms that protect Smpd1−/− mice from steatosis, we investigated visceral white adipose tissue. Adipose tissues store energy in the form of fat in adipocytes. In addition, white adipose tissue has been shown to be an important endocrine organ, producing and secreting specific adipokines that influence liver inflammation and the progression of NAFLD [6], [29]. Thus, we investigated the adipose tissue and factors released from adipocytes in the present model. Smpd1−/− animals either lacked or had a reduced amount of adipose tissue [30]. All of the data presented below on the expression of mRNA in adipocytes and adipose tissue refer to WT animals and those Smpd1−/− animals that had at least a minimal amount of adipose tissue (SD, n = 4; WD, n = 5).
Although WD consumption led to a significant increase in the size of adipocytes in WT mice, it did not lead to adipocyte hypertrophy in Smpd1−/− mice (Figure 2A,B). The levels of FFAs in sera from WD-fed WT mice were higher than those in SD-fed WT mice (Figure 2C), although this effect was not statistically significant. Smpd1−/− mice exhibited higher serum levels of FFAs than did WT mice (p < 0.05), but WD feeding did not further increase serum FFA levels in Smpd1−/− mice. Serum adiponectin concentrations were similar in SD-fed WT and Smpd1−/− mice (Figure 2D). WD feeding led to a slight, statistically significant reduction irrespective of genotype (p = 0.008). Levels of leptin, an adipokine that signals satiety, were increased in WD-fed WT mice, but this increase was not observed in WD-fed Smpd1−/− mice (p < 0.01; Figure 2E).
Figure 2.

Acid sphingomyelinase deficiency protects from diet-induced adipocyte hypertrophy. (A) Adipose tissue from acid sphingomyelinase–deficient (Smpd−/−) mice and C57BL/6 mice (wild-type, WT) fed either a standard diet (SD) or a Western diet (WD) for 6 weeks was sectioned and stained with hematoxylin and eosin (H&E). WD consumption led to hypertrophy of adipocytes in WT mice but not in Smpd−/− mice. (B) Mean adipocyte diameter was significantly larger in WD-fed WT mice than in WD-fed Smpd−/− mice. (C) Although SD-fed Smpd−/− mice exhibited higher serum concentrations of free fatty acids (FFAs), no statistically significant differences were observed between the WD-fed groups because of higher FFA concentrations in WT mice. (D) Serum adiponectin concentrations were lower in WD mice than in SD mice, irrespective of genotype. (E) WD-fed WT mice exhibited substantial hyperleptinemia; this condition did not occur in Smpd−/− mice.
These findings show that WD-fed Smpd1−/− mice do not exhibit adipocyte hypertrophy or hyperleptinemia.
3.5. Levels of mRNA expression of genes associated with browning of adipocytes and cell cycle control are elevated in Smpd1−/− mice
On the basis of the observation of altered adipocyte physiology and adipokine release, we hypothesized that the expression of genes associated with lipid metabolism is altered in adipose tissue of Smpd1−/− mice. Thus, we investigated the expression of metabolism-associated genes and fatty acid transporters. In adipose tissue from Smpd1−/− mice, the mRNA expression of caveolin 1 (Cav1), a central fatty acid transporter of adipocytes, was lower (n.s.) than in WT animals, irrespective of diet (Figure 3A). WD consumption increased the expression of solute carrier family 27 member 1 (Slc27a1) mRNA (p = 0.004; Figure 3B), independent of genotype.
Figure 3.

Acid sphingomyelinase deficiency affects adipocyte expression of genes related to beta-oxidation and cell differentiation in adipose tissue. RNA was isolated from adipose tissue from acid sphingomyelinase–deficient mice (Smpd−/−) mice and C57BL/6 (wild-type, WT) mice fed either a standard diet (SD) or a Western diet (WD) for 6 weeks. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed to assess mRNA expression. (A) Caveolin1 (Cav1) expression did not differ between the groups. (B) WD consumption increased the expression of the fatty acid transporter solute carrier family 27 member 1 (Slc27a1, fatty acid transport protein 1), irrespective of genotype. The expression of genes associated with β-oxidation (C, carnitine palmitoyltransferase 1a, Cpt1a; D, peroxisome proliferation-activated receptor, gamma, coactivator 1 alpha, Ppargc1a) and adipocyte differentiation (E, cyclin-dependent kinase inhibitor 1A, Cdkn1a) was significantly higher in Smpd−/− mice than in WT mice, independent of dietary group (p = 0.03; p < 0.04). (F) WD consumption enhanced apolipoprotein E (apoE) expression in adipose tissue from WT and Smpd−/− mice.
The expression of genes catalyzing molecules within β-oxidation, e.g., carnitine palmitoyltransferase 1a (Cpt1a; Figure 3C; p = 0.03) and peroxisome proliferator–activated receptor gamma coactivator 1 alpha (Ppargc1a; Figure 3D; p < 0.04), was significantly higher in Smpd1−/− mice than in WT mice, independent of dietary group. Of note, elevations in the expression of both genes have been associated with differentiation of brown adipocytes [31], [32], [33], [34]. The mRNA expression of cyclin-dependent kinase inhibitor 1A (Cdkn1a), which is involved in cell cycle arrest, senescence, and adipogenesis [34], [35], was significantly higher in Smpd1−/− animals (p < 0.04; Figure 3E). WD consumption enhanced the expression of apolipoprotein E (apoE), a molecule important for the catabolism of triglycerides and lipoproteins [36], in adipose tissue from WT and Smpd1−/− mice (p < 0.05, Figure 3F). Although the effect was larger in adipose tissue from Smpd1−/− mice than in adipose tissue from WT mice, this difference was not statistically significant.
These findings show that the mRNA expression profile of adipose tissue from Smpd1−/− mice differs significantly from that of adipose tissue from WT mice, in particular regarding genes of adipocyte differentiation.
3.6. Expression of genes that may protect from NAFLD is lacking in human adipose tissue
To relate the described findings in Smpd1−/− mice to humans, we analyzed the ceramide content of liver tissue from obese subjects and control subjects (Supplementary Table 2). Additionally, we analyzed gene expression in adipose and liver tissues from obese patients (Supplementary Table 3). The ceramide content in liver tissue was significantly lower in obese subjects than in healthy control subjects (Suppl. Fig. 3A). Obese subjects were grouped by maximum diameter of adipocytes (<125 μm or ≥125 μm). As we have previously shown, the NAS was significantly lower in patients with adipocyte diameters smaller than 125 μm compared to patients with adipocyte diameters of 125 μm or larger (Suppl. Fig. 3B). The mRNA expression of PPARGC1A, CDKN1A, and APOE, the expression of which has been found to be increased in adipose tissues of Smpd−/− mice, was not detectable in human adipose tissue, although these genes were expressed in liver tissue (not shown). CPT1A mRNA expression in adipose tissue did not differ by size of adipocytes (Suppl. Fig. 3C). Because we found no changes in the expression of genes of adipocyte differentiation in human adipose tissue, we assessed the expression of abhydrolase domain containing 5 (ABHD5 or CGI-58), a lipid droplet–associated protein that may be involved in long-chain fatty acid oxidation. Expression of ABHD5 was significantly higher in the patients with larger adipocytes (≥125 μm; Suppl. Fig. 3D). However, this difference was not found in liver tissue (not shown). This finding suggests that human adipocyte biology in obesity does not resemble the situation in Smpd1−/− mice. However, more severe liver injuries are observed in patients with larger adipocytes.
3.7. Quantitative proteome analysis of liver tissue identifies Rictor as a possible downstream mediator of the antisteatotic effects of the Smpd−/− genotype
To explore the differences between the genotypes in the effects of SD or WD on a wide unbiased range of regulatory processes, we determined protein regulation by label-free quantitative proteome analysis with mass spectrometry. Relevant alterations in canonical pathways between WD-fed WT and WD-fed Smpd1−/− mice are shown in Figure 4A. We performed IPA to identify upstream regulators in sets of altered proteins. The strongest indications of affected regulators (Figure 4B, Supplementary Table 5) were detected for Rictor (down-regulated in WD-fed Smpd1−/− mice; Figure 4C), nuclear factor, erythroid derived 2, like 2 (Nfe2l2; up-regulated in Smpd1−/− mice), leptin (up-regulated in Smpd1−/− mice), peroxisome proliferator–activated receptor alpha and gamma (Ppara and Pparg; both down-regulated in Smpd1−/− mice). To confirm these findings, we assessed the protein expression of Nfe2l2, which is involved in the activation of genes regulating the inflammatory response, in liver tissue with Western blot analyses (Figure 4D). In liver tissue from WD-fed Smpd1−/− mice, Nfe2l2 expression was lower than in liver tissue from WD-fed WT mice. Rictor regulates the activation of Akt by phosphorylating Akt at Serin473. Western blot analysis for phosphorylation of Akt-Ser473 showed that Akt phosphorylation was lower in WD-fed Smpd1−/− mice than in WD-fed WT mice (Figure 4E). Both quantitative proteome analysis and Akt phosphorylation suggest lower Rictor activity in WD-fed Smpd1−/− mice.
Figure 4.

Reduced Rictor activity in liver tissue is associated with protection from diet-induced liver steatosis by acid sphingomyelinase knockout. Quantitative proteome analysis was performed to identify alterations between acid sphingomyelinase–deficient mice (Smpd−/−) and C57BL/6 (wild-type, WT) mice consuming a standard diet (SD) or a Western diet (WD). (A) Among canonical pathways whose activation was stronger in WD-fed Smpd−/− mice than in WD-fed WT mice, Rho signaling pathways were prominent. (B) Ingenuity® Pathway Analysis, performed to detect upstream regulators from sets of altered proteins, found that the activity of rapamycin-insensitive companion of mTOR (Rictor) was lower and that of nuclear factor, erythroid derived 2, like 2 (Nfe2l2/Nrf2) was higher in WD-fed Smpd−/− mice than in WD-fed WT mice (see also Supplementary Table 5). (C) Combined Log2 ratio of genes found altered in quantitative proteome analysis and identified as downstream targets of Rictor related to SD-fed WT mice. A significant upregulation of Rictor target genes was observed for WD-fed Smpd−/− mice, indicating reduced Rictor activity (targets are negatively regulated by Rictor). (D) Protein expression of Nfe2l2/NRF2 in liver tissue, as determined by Western blot, was lower in WD-fed Smpd−/− mice than in WD-fed WT animals. (E) Because Rictor regulates the activation of Akt by phosphorylation at Serin473, we performed Western blot analysis for Akt-Ser473 phosphorylation. We found that Akt phosphorylation was lower in WD-fed Smpd−/− mice than in WD-fed WT mice, a finding confirming the findings of quantitative proteome analysis.
3.8. Confirmation of altered Rictor signaling by qRT-PCR assessment of relevant target genes
To further support the findings of quantitative proteome analysis, we analyzed the mRNA expression levels of various target genes for altered regulatory pathways or factors. Selected target genes for Rictor were proteasome subunit alpha type 1 (Psma1), ATPase H+ transporting V0 subunit D1 (Atp6v0d1), nicotinamide adenine dinucleotide (NADH) dehydrogenase 1 alpha subcomplex 6 (Ndufa6), and fatty acid binding protein 5 (Fabp5). Because Rictor inhibits the expression of these targets, we expected to find increased expression of these genes with reduced Rictor activation in Smpd1−/− mice, as observed for quantitative proteome analysis (Figure 4C). The aldo-keto reductase family 1, member A1 (Akr1a1) and glucan (1,4-alpha-), branching enzyme 1 (Gbe1) genes served as exemplary target genes for Nfe2l2 with regard to mRNA expression. For Ppara, we chose the targets acyl-CoenzymeA oxidase 1, palmitoyl (Acox1), fatty acid synthase (Fasn), Fabp4, and Fabp5. Acox1, Fasn, and Fabp4 served as targets for Pparg. We observed no statistically significant alterations in the mRNA expression of Pparg, Acox1, Fasn, Psma1, Akr1a1, or Gbe1 (Suppl. Fig. 4). In contrast, the expression of Fabp4 and Fabp5 was significantly higher in the liver of WD-fed Smpd1−/− mice than in the liver of WD-fed WT mice (Figure 5C,D; p < 0.05 for Fabp4; p < 0.01 for Fabp5). The expression of Ndufa6 did not differ significantly between Smpd1−/− mice and WT mice irrespective of diet (Figure 5A). Atp6v0d1 expression was slightly lower in SD-fed Smpd1−/− mice than in SD-fed WT mice (Figure 5B). WD consumption further reduced Atp6v0d1 expression in Smpd1−/− mice but increased Atp6v0d1 expression in WT animals. Genes associated with fatty acid metabolism or transport were differentially expressed in WD-fed WT mice and WD-fed Smpd1−/− mice and may be associated with the protective effect of diet-induced steatosis. Furthermore, the regulation of downstream targets and effectors seems to be consistent with diminished Rictor activation in WD-fed Smpd1−/− mice.
Figure 5.

Alterations in the expression of genes related to fatty acid transport and oxidation in liver tissue are associated with protection against diet-induced liver steatosis by acid sphingomyelinase knockout. We used quantitative real-time polymerase chain reaction (qRT-PCR) to quantify RNA in liver tissue from acid sphingomyelinase–deficient (Smpd−/−) mice and C57BL/6 (wild-type, WT) mice fed a standard diet (SD) or a Western diet (WD). (A) Ndufa6 mRNA expression was not significantly different between Smpd−/− mice and WT mice irrespective of diet. (B) Expression of ATPase H+ transporting V0 subunit D1 (Atp6v0d1), a target of Rictor, whose activation was found to be altered, according to the results of quantitative proteome analysis, was slightly lower in SD-fed Smpd−/− mice than in SD-fed WT mice. WD consumption diminished Atp6v0d1 expression in Smpd−/− mice but increased its expression in WT mice; this difference was statistically significant. (C, D) The expression of Fabp4 (target gene of Ppara and Pparg) and Fabp5 (target gene of Rictor, Ppara, and Pparg) mRNA target genes was significantly higher in the liver of WD-fed Smpd−/− mice than in the liver of WD-fed WT mice. (E, F) In liver tissue samples from obese subjects (see Suppl. Table 3), mRNA expression of FABP4 and FABP5 was significantly higher in patients with maximal adipocyte size (≥125 μm).
Our observations of gene expression were tested in part in human NAFLD (in liver tissue of obese subjects; Suppl. Table 3). The mRNA expression of FABP4 and FABP5 was significantly higher in patients with adipocytes larger than 125 μm in diameter (Figure 5E,F). Moreover, the expression of FABP4 and FABP5 exhibited statistically significant positive correlations with NAS as a measure of liver injury (FABP4, Spearman r = 0.4146, p = 0.003; FABP5, Spearman r = 0.4976, p = 0.0004).
4. Discussion
ASM deficiency results in the lysosomal storage of sphingolipids, which disrupts a variety of signal transduction pathways [37] and causes various cardiovascular, neurological, infectious, metabolic, and hepatic diseases [10], [23]. ASM inhibition has been described as a potential therapeutic agent that may reduce the progression of liver diseases [23] and may reduce liver toxicity due to defective transduction of death receptor and apoptosis signals [9], [22], [38]. Although some effects of ASM knockout have been demonstrated in the context of hepatic steatosis, fibrosis, diabetes, and fat disorder [20], [22], [23], [39], the interaction of the liver with adipose tissue and its derived cytokines, also known as adipokines, has not been described. With this study, we aimed to determine whether the interaction between liver tissue and adipose tissue in Asm-deficient mice protects them from diet-induced steatosis apart from other mechanisms, such as endoplasmic reticulum (ER) stress and autophagy. We found that adipocyte hypertrophy, which is associated with reduced leptin release and altered adipocyte expression profiles, does not occur in WD-fed Smpd1−/− mice. In addition, we found that Rictor may be a regulator within the liver and may confer protection from diet-induced liver steatosis in a murine model.
As has been previously shown [30], we found that Smpd1−/− mice are resistant to WD-induced steatosis after a feeding duration of 6 weeks and that, instead of accumulating lipids by hepatocytes (steatosis), Asm-deficient mice accumulate lipids within foam cells in various tissues. We also observed elevated mRNA expression of early pro-fibrogenic genes, although we found no signs of actual collagen deposition or stellate cell activation. These seemingly contradictory results may indicate an early response to dietary stress that may not lead to established fibrosis. These observations are in line with previous studies demonstrating that the Smpd1−/− genotype exerts a protective effect against fibrosis and HSC activation with longer durations of HFD consumption [23], [39], [40]. Because the genes activated early in fibrosis were clearly up regulated, the feeding period we chose may have been too short to allow the formation of fibrosis. Additional studies are necessary for addressing our contradictory findings related to fibrogenesis in Smpd1−/− mice. Taken together, however, our findings show that Smpd1−/− mice are protected from diet-induced liver steatosis.
Current lines of research have brought attention to the interaction between adipose tissue and the liver as a driving force for NAFLD and the progression to NASH. The current study showed that WD-fed Smpd1−/− mice do not exhibit adipocyte hypertrophy in visceral adipose tissue. This observation is supported by the finding of reductions in body weight among patients with Niemann-Pick disease, a condition that results from deficient ASM activity. A study using an Asm−/−/ldrl−/− double knockout mouse model found that these animals exhibit lower body weight and do not accumulate fat in white adipose tissue after eating a WD for ten weeks [22]. Because ldlr−/− animals exhibit hypertrophy in visceral adipocytes when eating a HFD, our findings indicate that their resistance to adipocyte hypertrophy is probably due to Asm deficiency alone. In addition, WD-induced increases in the release of leptin were found in WT animals but not in Smpd1−/− mice. This finding was associated with an mRNA expression profile that suggested altered adipocyte differentiation or proliferation, hinting at a “browning” of adipocytes. Indeed, increased energy expenditure or fatty acid oxidation could explain the resistance of these mice to both adipocyte hypertrophy and liver steatosis. In contrast, the mRNA expression of fatty acid transporters and inflammatory genes in adipose tissue did not differ significantly between WD-fed Smpd1−/− mice and WD-fed WT mice. These findings show that one mechanism that may protect Smpd1−/− mice from liver steatosis is an altered visceral adipocyte profile leading to diminished leptin release and changes in fatty acid metabolism.
To translate these findings from Smpd1−/− mice to humans, we analyzed ceramide content in the liver and mRNA expression in adipose tissue from obese subjects. Liver ceramide content was lower in obese subjects than in control subjects of normal weight, a finding that corresponds to diminished ASM activity. Adipose tissue expression of PPARGC1A, CDKN1A, and APOE mRNA was not detectable, irrespective of the extent of adipocyte hypertrophy. When patients were grouped by adipocyte size, we observed no difference in the mRNA expression of CPT1. The lack of expression of PPARGC1A and APOE indicates diminished breakdown or mitochondrial oxidation of lipids. Unaltered CPT1 expression hints at unchanged β-oxidation in adipocytes despite increased availability of long-chain fatty acids. Unfortunately, it was not possible to obtain exact measures of adipose tissue metabolic rates or fatty acid composition. CGI-58 is associated with lipid droplets and activates lipolysis by adipocyte triglyceride lipase (ATGL) in adipocytes. Because mRNA expression of CGI-58 was significantly higher in adipose tissue from patients with larger adipocytes, lipolysis may be affected not by adipocyte size but rather by oxidation of fatty acids. This finding merits deeper analysis of human adipocyte biology in various states of nutrient (over-)supply.
Because it is still not clear what causes the protection from WD-induced steatosis in Smpd1−/− mice, we performed quantitative proteome analysis for unbiased identification of alterations in proteins and pathways. IPA showed that Rictor, among other regulators, was downregulated because of many significantly regulated target proteins. Reductions in Akt phosphorylation at Serin473 indirectly confirmed this assumption. The mRNA expression of target genes was also partly elevated in Smpd1−/− mice, supporting the finding of lower Rictor activation in WD-fed Smpd1−/− mice than in WD-fed WT mice. Quantitative proteome analysis showed that many proteins associated with fatty acid transport, fatty acid oxidation, and glucose metabolism were affected, converging on the few known pathways in IPA.
These findings show that the reductions in the activity of Rictor may contribute to the protective effect of the Smpd1−/− genotype against steatosis in WD-fed mice. Currently, the only known connection between ceramide metabolism and mTORC2 has been demonstrated in yeast [41]. This finding suggests that Rictor and mammalian target of rapamycin complex 2 (mTORC2) are promising targets in ceramide- or ASM-associated diseases.
Rictor is an integral part of mTORC2. Both mTORC1 and mTORC2 are central regulators of multiple cellular functions. In particular, mTORC2 has been associated with metabolic functions such as autophagy, glycolysis, and lipogenesis, but also with actin cytoskeleton formation and insulin/Akt signaling. A liver-specific knockout of Rictor induces hepatic insulin resistance and leads to diminished phosphorylation of Akt and Pkc and, subsequently, to reductions in glucose flux and maturation of Srebp-1c [42]. A diminished association of Rictor with mTORC and diminished mTORC2 activity has also been linked to hepatic glucose intolerance [43]. Similar findings have been observed with adipose tissue–specific Rictor knockout, which leads to impaired tolerance of glucose and insulin, hyperinsulinemia, and increased body size [44], [45]. A more recent study on adipose tissue–specific RICTOR knockout mice demonstrated that hepatic glucose and lipid metabolism were altered [46]. Moreover, HFD feeding in adipose tissue–specific RICTOR knockout mice did not lead to increased body weight or aggravate insulin resistance. It was concluded that adipose tissue ablation of RICTOR mimics the effects of HFD and that RICTOR dependent de novo lipogenesis in adipose tissue could be an early target during development of insulin resistance. This is in line with our finding that the effects of WD on adipose tissue were reduced in an Smpd1−/− model, leading to reduced activity of Rictor in the liver. Another possible explanation may be that reduction of RICTOR in the lineage of brown adipocytes protects against WD-induced obesity and seems to increase energy expenditure at thermoneutrality [47], although Rictor knockout also inhibited the differentiation of brown adipocytes [47]. Thyroid hormone signaling also seems to co-activate FOXO1 target genes via deacetylation of RICTOR [48]. Diminished RICTOR activity leads to reduced AKT phosphorylation, FOXO1 phosphorylation, and subsequently increased nuclear localization and DNA binding of FOXO1. These recent findings suggest mTORC2/RICTOR as an important regulating element in different metabolic signaling pathways. RICTOR might also be a promising target to prevent tumor formation in metabolic conditions [49]. Knockout of FASN in a murine model of hepatocarcinogenesis and in human cell lines abolished AKT-dependent carcinogenesis. This was associated with diminished mTORC2 activity and Rictor knockout could reproduce the effect of FASN knockout. Taken together current findings of other groups and our own data suggest RICTOR as a possibly valuable target to counter metabolic alterations in adipose tissue and liver.
In summary, we have shown that the protective effect of ASM knockout could be associated with altered adipocyte morphology and metabolism. Although it is known that the effects of RICTOR knockout in specific tissues may lead to insulin resistance and lipolysis, reductions in RICTOR activation in adipose tissue could be beneficial with regard to energy expenditure and the reduction of adipocyte hypertrophy. Unfortunately, it was not within the scope of the presented work to assess RICTOR activity in murine or human adipose tissue. It remains to be determined whether diminished but not completely abrogated hepatic mTORC2 activity could protect against WD-induced steatosis.
Conflict of interest
All authors declare that they have no conflict of interest.
Authors' contributions
Study concept and design: SS, JPS, EG, AC; Acquisition of data: SS, JPS, DAM, MS, SJ, LW, ACar, LPB; Analysis and interpretation of data: SS, JPS, DAM; Drafting of the manuscript: SS; Critical revision of the manuscript for important intellectual content: JPS, EG, AC; Statistical analysis: JPS, DAM; obtained funding: GG, AC; Technical or material support: HAB, BS, EG; Study supervision: AC.
Funding
This work was supported by Deutsche Forschungsgemeinschaft (DFG) grant 267/8-1 to A.C.
Acknowledgments
We thank Martin Pronadl and Rudolf Ott from the Clinic of Surgery II at Alfried Krupp Hospital Essen, Germany, for collecting tissue and serum samples during bariatric surgical procedures and clinical follow-up of the enrolled patients. We are also grateful to Mrs. Dorothe Möllmann of the Institute of Pathology, University Hospital, University Duisburg-Essen, for preparing tissue sections and performing H&E staining. Furthermore, we gratefully acknowledge Kristin Rosowski and Stephanie Tautges for their excellent technical assistance during proteomics experiments. We also would like to thank Dr. Flo Witte for her expert writing assistance.
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molmet.2017.03.002.
Contributor Information
Svenja Sydor, Email: Svenja.sydor@uni-due.de.
Jan-Peter Sowa, Email: Jan-peter.sowa@uk-essen.de.
Dominik A. Megger, Email: dominik.megger@rub.de.
Martin Schlattjan, Email: martin.schlattjan@uk-essen.de.
Sami Jafoui, Email: Sami.Jafoui@uk-essen.de.
Lena Wingerter, Email: Lena.wingerter@hotmail.de.
Alexander Carpinteiro, Email: Alexander.carpinteiro@uk-essen.de.
Hideo A. Baba, Email: hideo.baba@uk-essen.de.
Lars P. Bechmann, Email: lars.bechmann@uk-essen.de.
Barbara Sitek, Email: barbara.sitek@rub.de.
Guido Gerken, Email: Guido.gerken@uk-essen.de.
Erich Gulbins, Email: Erich.gulbins@uk-essen.de.
Ali Canbay, Email: ali.canbay@med.ovgu.de.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
References
- 1.[cited 2017 Feb 24]. WHO | Obesity and overweight [Internet]. WHO. Available from: http://www.who.int/mediacentre/factsheets/fs311/en/.
- 2.Kim C.H., Younossi Z.M. Nonalcoholic fatty liver disease: a manifestation of the metabolic syndrome. Cleveland Clinic Journal of Medicine. 2008;75:721–728. doi: 10.3949/ccjm.75.10.721. [DOI] [PubMed] [Google Scholar]
- 3.Gregor M.F., Hotamisligil G.S. Inflammatory mechanisms in obesity. Annual Review of Immunology. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 4.Unger R.H. Lipid overload and overflow: metabolic trauma and the metabolic syndrome. Trends in Endocrinology and Metabolism: TEM. 2003;14:398–403. doi: 10.1016/j.tem.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 5.Jernås M., Palming J., Sjöholm K., Jennische E., Svensson P.-A., Gabrielsson B.G. Separation of human adipocytes by size: hypertrophic fat cells display distinct gene expression. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2006;20:1540–1542. doi: 10.1096/fj.05-5678fje. [DOI] [PubMed] [Google Scholar]
- 6.Wree A., Schlattjan M., Bechmann L.P., Claudel T., Sowa J.-P., Stojakovic T. Adipocyte cell size, free fatty acids and apolipoproteins are associated with non-alcoholic liver injury progression in severely obese patients. Metabolism. 2014;63:1542–1552. doi: 10.1016/j.metabol.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 7.Garaulet M., Hernandez-Morante J.J., Lujan J., Tebar F.J., Zamora S. Relationship between fat cell size and number and fatty acid composition in adipose tissue from different fat depots in overweight/obese humans. International Journal of Obesity (2005) 2006;30:899–905. doi: 10.1038/sj.ijo.0803219. [DOI] [PubMed] [Google Scholar]
- 8.Wajchenberg B.L., Giannella-Neto D., da Silva M.E., Santos R.F. Depot-specific hormonal characteristics of subcutaneous and visceral adipose tissue and their relation to the metabolic syndrome. Hormone and Metabolic Research. 2002;34:616–621. doi: 10.1055/s-2002-38256. [DOI] [PubMed] [Google Scholar]
- 9.Gulbins E., Grassmé H. Ceramide and cell death receptor clustering. Biochimica et Biophysica Acta. 2002;1585:139–145. doi: 10.1016/s1388-1981(02)00334-7. [DOI] [PubMed] [Google Scholar]
- 10.Smith E.L., Schuchman E.H. The unexpected role of acid sphingomyelinase in cell death and the pathophysiology of common diseases. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2008;22:3419–3431. doi: 10.1096/fj.08-108043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fanzo J.C., Lynch M.P., Phee H., Hyer M., Cremesti A., Grassme H. CD95 rapidly clusters in cells of diverse origins. Cancer Biology & Theraphy. 2003;2:392–395. doi: 10.4161/cbt.2.4.442. [DOI] [PubMed] [Google Scholar]
- 12.Jernigan P.L., Hoehn R.S., Grassmé H., Edwards M.J., Müller C.P., Kornhuber J. Sphingolipids in Major Depression. Neurosignals. 2015;23:49–58. doi: 10.1159/000442603. [DOI] [PubMed] [Google Scholar]
- 13.Carpinteiro A., Becker K.A., Japtok L., Hessler G., Keitsch S., Požgajovà M. Regulation of hematogenous tumor metastasis by acid sphingomyelinase. EMBO Molecular Medicine. 2015;7:714–734. doi: 10.15252/emmm.201404571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nojima H., Freeman C.M., Gulbins E., Lentsch A.B. Sphingolipids in liver injury, repair and regeneration. Biological Chemistry. 2015;396:633–643. doi: 10.1515/hsz-2014-0296. [DOI] [PubMed] [Google Scholar]
- 15.Holland W.L., Brozinick J.T., Wang L.-P., Hawkins E.D., Sargent K.M., Liu Y. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metabolism. 2007;5:167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Summers S.A. Ceramides in insulin resistance and lipotoxicity. Progress in Lipid Research. 2006;45:42–72. doi: 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Samad F., Badeanlou L., Shah C., Yang G. Adipose tissue and ceramide biosynthesis in the pathogenesis of obesity. Advance in Experimental Medicine and Biology. 2011;721:67–86. doi: 10.1007/978-1-4614-0650-1_5. [DOI] [PubMed] [Google Scholar]
- 18.Larsen P.J., Tennagels N. On ceramides, other sphingolipids and impaired glucose homeostasis. Molecular Metabolism. 2014;3:252–260. doi: 10.1016/j.molmet.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorden D.L., Myers D.S., Ivanova P.T., Fahy E., Maurya M.R., Gupta S. Biomarkers of NAFLD progression: a lipidomics approach to an epidemic. Journal of Lipid Research. 2015;56:722–736. doi: 10.1194/jlr.P056002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Błachnio-Zabielska A.U., Pułka M., Baranowski M., Nikołajuk A., Zabielski P., Górska M. Ceramide metabolism is affected by obesity and diabetes in human adipose tissue. Journal of Cellular Physiology. 2012;227:550–557. doi: 10.1002/jcp.22745. [DOI] [PubMed] [Google Scholar]
- 21.Boini K.M., Zhang C., Xia M., Poklis J.L., Li P.-L. Role of sphingolipid mediator ceramide in obesity and renal injury in mice fed a high-fat diet. The Journal of Pharmacology and Experimental Therapeutics. 2010;334:839–846. doi: 10.1124/jpet.110.168815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deevska G.M., Rozenova K.A., Giltiay N.V., Chambers M.A., White J., Boyanovsky B.B. Acid sphingomyelinase deficiency prevents diet-induced hepatic triacylglycerol accumulation and hyperglycemia in mice. The Journal of Biological Chemistry. 2009;284:8359–8368. doi: 10.1074/jbc.M807800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia-Ruiz C., Mato J.M., Vance D., Kaplowitz N., Fernández-Checa J.C. Acid sphingomyelinase-ceramide system in steatohepatitis: a novel target regulating multiple pathways. Journal of Hepatology. 2015;62:219–233. doi: 10.1016/j.jhep.2014.09.023. [DOI] [PubMed] [Google Scholar]
- 24.Beilfuss A., Sowa J.-P., Sydor S., Beste M., Bechmann L.P., Schlattjan M. Vitamin D counteracts fibrogenic TGF-β signalling in human hepatic stellate cells both receptor-dependently and independently. Gut. 2015;64:791–799. doi: 10.1136/gutjnl-2014-307024. [DOI] [PubMed] [Google Scholar]
- 25.Kleiner D.E., Brunt E.M., Van Natta M., Behling C., Contos M.J., Cummings O.W. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 26.Sydor S., Gu Y., Schlattjan M., Bechmann L.P., Rauen U., Best J. Steatosis does not impair liver regeneration after partial hepatectomy. Laboratory Investigations: a Journal of Technical Methods and Pathology. 2013;93:20–30. doi: 10.1038/labinvest.2012.142. [DOI] [PubMed] [Google Scholar]
- 27.Megger D.A., Bracht T., Kohl M., Ahrens M., Naboulsi W., Weber F. Proteomic differences between hepatocellular carcinoma and nontumorous liver tissue investigated by a combined gel-based and label-free quantitative proteomics study. Molecular & Cellular Proteomics: MCP. 2013;12:2006–2020. doi: 10.1074/mcp.M113.028027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Padden J., Megger D.A., Bracht T., Reis H., Ahrens M., Kohl M. Identification of novel biomarker candidates for the immunohistochemical diagnosis of cholangiocellular carcinoma. Molecular & Cellular Proteomics: MCP. 2014;13:2661–2672. doi: 10.1074/mcp.M113.034942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Balistreri C.R., Caruso C., Candore G. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediators of Inflammation. 2010;2010:802078. doi: 10.1155/2010/802078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horinouchi K., Erlich S., Perl D.P., Ferlinz K., Bisgaier C.L., Sandhoff K. Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nature Genetics. 1995;10:288–293. doi: 10.1038/ng0795-288. [DOI] [PubMed] [Google Scholar]
- 31.Brown N.F., Hill J.K., Esser V., Kirkland J.L., Corkey B.E., Foster D.W. Mouse white adipocytes and 3T3-L1 cells display an anomalous pattern of carnitine palmitoyltransferase (CPT) I isoform expression during differentiation. Inter-tissue and inter-species expression of CPT I and CPT II enzymes. The Biochemical Journal. 1997;327(Pt 1):225–231. doi: 10.1042/bj3270225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ström K., Hansson O., Lucas S., Nevsten P., Fernandez C., Klint C. Attainment of brown adipocyte features in white adipocytes of hormone-sensitive lipase null mice. PloS One. 2008;3:e1793. doi: 10.1371/journal.pone.0001793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Townsend K.L., An D., Lynes M.D., Huang T.L., Zhang H., Goodyear L.J. Increased mitochondrial activity in BMP7-treated brown adipocytes, due to increased CPT1- and CD36-mediated fatty acid uptake. Antioxidants & Redox Signaling. 2013;19:243–257. doi: 10.1089/ars.2012.4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hallenborg P., Petersen R.K., Feddersen S., Sundekilde U., Hansen J.B., Blagoev B. PPARγ ligand production is tightly linked to clonal expansion during initiation of adipocyte differentiation. Journal of Lipid Research. 2014;55:2491–2500. doi: 10.1194/jlr.M050658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu X., Malki A., Cao Y., Li Y., Qian Y., Wang X. Glucose- and triglyceride-lowering dietary penta-O-galloyl-α-d-glucose reduces expression of PPARγ and C/EBPα, induces p21-mediated G1 phase cell cycle arrest, and inhibits adipogenesis in 3t3-L1 preadipocytes. Experimental and Clinical Endocrinology & Diabetes: Official Journal, German Society of Endocrinology [and] German Diabetes Association. 2015;123:308–316. doi: 10.1055/s-0035-1548789. [DOI] [PubMed] [Google Scholar]
- 36.Lasrich D., Bartelt A., Grewal T., Heeren J. Apolipoprotein E promotes lipid accumulation and differentiation in human adipocytes. Experimental Cell Research. 2015;337:94–102. doi: 10.1016/j.yexcr.2015.07.015. [DOI] [PubMed] [Google Scholar]
- 37.Morales A., Lee H., Goñi F.M., Kolesnick R., Fernandez-Checa J.C. Sphingolipids and cell death. Apoptosis: an International Journal on Programmed Cell Death. 2007;12:923–939. doi: 10.1007/s10495-007-0721-0. [DOI] [PubMed] [Google Scholar]
- 38.Lin T., Genestier L., Pinkoski M.J., Castro A., Nicholas S., Mogil R. Role of acidic sphingomyelinase in Fas/CD95-mediated cell death. The Journal of Biological Chemistry. 2000;275:8657–8663. doi: 10.1074/jbc.275.12.8657. [DOI] [PubMed] [Google Scholar]
- 39.Fucho R., Martínez L., Baulies A., Torres S., Tarrats N., Fernandez A. ASMase regulates autophagy and lysosomal membrane permeabilization and its inhibition prevents early stage non-alcoholic steatohepatitis. Journal of Hepatology. 2014;61:1126–1134. doi: 10.1016/j.jhep.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moles A., Tarrats N., Morales A., Domínguez M., Bataller R., Caballería J. Acidic sphingomyelinase controls hepatic stellate cell activation and in vivo liver fibrogenesis. The American Journal of Pathology. 2010;177:1214–1224. doi: 10.2353/ajpath.2010.091257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aronova S., Wedaman K., Aronov P.A., Fontes K., Ramos K., Hammock B.D. Regulation of ceramide biosynthesis by TOR complex 2. Cell Metabolism. 2008;7:148–158. doi: 10.1016/j.cmet.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hagiwara A., Cornu M., Cybulski N., Polak P., Betz C., Trapani F. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metabolism. 2012;15:725–738. doi: 10.1016/j.cmet.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 43.Zhang C., Cooper D.E., Grevengoed T.J., Li L.O., Klett E.L., Eaton J.M. Glycerol-3-phosphate acyltransferase-4-deficient mice are protected from diet-induced insulin resistance by the enhanced association of mTOR and rictor. American Journal of Physiology, Endocrinology and Metabolism. 2014;307:E305–E315. doi: 10.1152/ajpendo.00034.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cybulski N., Polak P., Auwerx J., Rüegg M.A., Hall M.N. mTOR complex 2 in adipose tissue negatively controls whole-body growth. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:9902–9907. doi: 10.1073/pnas.0811321106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar A., Lawrence J.C., Jung D.Y., Ko H.J., Keller S.R., Kim J.K. Fat cell-specific ablation of rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes. 2010;59:1397–1406. doi: 10.2337/db09-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang Y., Wallace M., Sanchez-Gurmaches J., Hsiao W.-Y., Li H., Lee P.L. Adipose tissue mTORC2 regulates ChREBP-driven de novo lipogenesis and hepatic glucose metabolism. Nature Communications. 2016 Apr 21;7:11365. doi: 10.1038/ncomms11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hung C.-M., Calejman C.M., Sanchez-Gurmaches J., Li H., Clish C.B., Hettmer S. Rictor/mTORC2 loss in the Myf5 lineage reprograms brown fat metabolism and protects mice against obesity and metabolic disease. Cell Reports. 2014;8:256–271. doi: 10.1016/j.celrep.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh B.K., Sinha R.A., Zhou J., Tripathi M., Ohba K., Wang M.-E. Hepatic FOXO1 target genes are Co-regulated by thyroid hormone via RICTOR protein deacetylation and MTORC2-AKT protein inhibition. The Journal of Biological Chemistry. 2016 Jan 1;291:198–214. doi: 10.1074/jbc.M115.668673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li L., Pilo G.M., Li X., Cigliano A., Latte G., Che L. Inactivation of fatty acid synthase impairs hepatocarcinogenesis driven by AKT in mice and humans. Journal of Hepatology. 2016 Feb;64:333–341. doi: 10.1016/j.jhep.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.