

Abstract
The success of Mycobacterium tuberculosis (M. tuberculosis) as a pathogen is largely contributes to its ability to manipulate the host immune responses. The genome of M. tuberculosis encodes multiple immune-modulatory proteins, including several members of the multi-genic PE_PPE family. Despite of intense research, the roles of PE_PGRS proteins in mycobacterial pathogenesis remain elusive. The function of M. tuberculosis PE_PGRS41, characterized by an extended and unique C-terminal domain, was studied. Expression of PE_PGRS41 in Mycobacterium smegmatis, a non-pathogenic species intrinsically deficient of PE_PGRS, severely impaired the resistance of the recombinant to multiple stresses via altering the cell wall integrity. Macrophages infected by M. smegmatis harboring PE_PGRS41 decreased the production of TNF-α, IL-1β and IL-6. In addition, PE_PGRS41 boosted the survival of M. smegmatis within macrophage accompanied with enhanced cytotoxic cell death through inhibiting the cell apoptosis and autophagy. Taken together, these results implicate that PE_PGRS41 is a virulence factor of M. tuberculosis and sufficient to confer pathogenic properties to M. smegmatis.
Tuberculosis (TB), caused by Mycobacterium tuberculosis, remains a global burden of morbidity and mortality. There were 10.4 million new cases and 1.4 million deaths reported in 2015 by the World Health Organization (www.whoint/tb/publications/global_report/). M. tuberculosis survives and persists in the host by modulating host immune responses. Upon infection, M. tuberculosis cell envelope-associated factors interact with the innate immune cells1, to tip the balance between the host and pathogen2. The deciphering of the M. tuberculosis genome uncovers a unique multigenic PE_PGRS proteins with a highly conserved PE domain and a variable PGRS domain3. PE is named after N-terminal Pro(P)-Glu(E) sequence, and PGRS means polymorphic GC-rich repetitive sequence. PE_PGRS proteins restrict to virulent mycobacteria4, such as M. tuberculosis, Mycobacterium bovis, Mycobacterium marinum, Mycobacterium ulcerans and Mycobacterium kansasii. Mycobacterial PE_PGRS are cell surface proteins implicated in virulence5,6 and pathogen-host interaction5,7.
To further elucidate the role of PE_PGRS family proteins in molecular pathogenesis of tuberculosis, we generated and characterized a prototypical member of M. tuberculosis PE_PGRS family protein PE_PGRS41, also known as acid and phagosome regulated protein C (aprC). PE_PGRS41 modulates pH-driven adaptation to the macrophage phagosome8. We investigated whether M. tuberculosis PE_PGRS41 could modulate signaling pathways of the host innate immune system and sought to identify crucial host target molecules involved in such interaction. M. smegmatis expressing PE_PGRS41 affected macrophage activation, and significantly decreased the expression and production of TNF-α, IL-1β and IL-6 by comparison to control strains. We found that LPS priming of macrophages immediately prior to infection with PE_PGRS41 expressing M. smegmatis, enhances TNF-α, IL-1β and IL-6 mRNA levels and secretion. Furthermore, M. tuberculosis PE_PGRS41 blocked innate defense response against Mycobacteria by decreasing cell apoptosis and autophagy and increasing cell death of macrophage, resulted in increased intracellular survival of recombinant M. smegmantis expressing PE_PGRS41 within macrophage. Thus, our findings revealed M. tuberculosis PE_PGRS41 alteres the immune environment of the host cells, which might be involved in the pathogenesis of mycobacterial disease and hence influenced host cell responses to M. tuberculosis infection.
Results
PE_PGRS41 affects the growth of M. smegmatis upon exposure to hygromycin
To investigate the effect of PE_PGRS41 on the macrophage response to bacterial infection, we generated recombinant M. smegmatis strains. The PE_PGRS41 gene (1086 bp) was amplified from M. tuberculosis H37Rv genome using specifical primers (Table 1) and used to construct recombinant Ms_PE_PGRS41 (Fig. 1A). The Ms_PE_PGRS41 strain was engineered to express a His-tagged PE_PGRS41 protein from a recombinant PALACE vector, while the Ms_Vec strain harbored the vector alone. Both Ms_Vec and Ms_PE_PGRS41, which were grown in Middlebrook 7H9 medium in the presence of hygromycin (Hyg). PE_PGRS41 successfully expressed in M. smegmatis (Fig. 1B) and localized to cell wall component of M. smegmatis (Fig. 1C). To test the effect of PE_PGRS41 on recombinant strains, the growth rates of Ms_Vec and Ms_PE_PGRS41 were detected. No growth difference was detected in Ms_Vec and Ms_PE_PGRS41 strain with/without acetamide (Ace) or Hyg (Fig. 1S), demonstrating that the expression of PE_PGRS41 does not affect the growth of M. smegmatis. Notably, although both Ms_Vec and Ms_PE_PGRS41 habored the PALACE vector with anti-Hyg resistance gene, the cell size of recombinant Ms_PE_PGRS41 became smaller than Ms_Vec when exposure to Hyg after induction by Ace (Fig. 1D). Moreover, Hyg exposure delayed the growth of Ms_PE_PGRS41 strains while with negligible effect upon Ms_Vec after Ace induction (Fig. 1E). These data suggest that PE_PGRS41 might affect the growth of M. smegmatis upon exposure to antibiotic.
Table 1. Primers used in this study.
| Primer | Sequence (5′–3′) |
|---|---|
| IL-1β (F) | CTGAAGGCCCGAATGCACCAG |
| IL-1β (R) | GCAAAGGTGGTGTCAGTATC |
| TNF-α (F) | GTAATGCTCCTCCCTACTTC |
| TNF-α (R) | GCAAAGGTGGTGTCAGTATC |
| IL-6 (F) | TCTCTGGTGACATGAAGAAGCT |
| IL-6 (R) | GCAAAGGTGGTGTCAGTATC |
| IL-10 (F) | AGACTCTGCTTCCTGATTGG |
| IL-10 (R) | GTGAGTGTCCCTGCTGGTC |
| ATG-5 (F) | AAGCAACTCTGGATGGGATTG |
| ATG-5 (R) | CAGGATCAATAGCAGAAGGAC |
| ATG-10 (F) | CCCAGCAGGAACATCCAATA |
| ATG-10 (R) | AGGCTCAGCCATGATGTGAT |
| ATG-12 (F) | AGTAGAGCGAACACGAACCATCC |
| ATG-12 (R) | AAGGAGCAAAGGACTGATTCACATAA |
| ATG-4c (F) | CAGCTGTGGTTGCTCACATTT |
| ATG-4c (R) | CTAAGTAGTCGGTGTTGGTTC |
| ATG-8 (F) | TCGGAGAAGACCTTCAAG |
| ATG-8 (R) | CATGTTGACATGGTCAGG |
| DRAM2H (F) | AAGCAAGTTCATGCTCTGATC |
| DRAM2H (R) | CCAGATAACCAACAACAGTCTG |
| β-actin (F) | TTCCTTCCTGGGCATGGAGTCC |
| β-actin (R) | TGGCGTACAGGTCTTTGCGG |
Figure 1. The effect of PE_PGRS41 on the growth of M. smegmatis.

(A) PCR amplification of PE_PGRS41 encoding sequence from M. tuberculosis genome approximately 1086 bp. (B) M. tuberculosis PE_PGRS41 was expressed in M. smegmatis and detected using Western blotting. Cell lysates of Ms_Vec and Ms_PE_PGRS41 were subjected to Western blot to determine the expression of His-tagged PE_PGRS41 protein in M. smegmatis by anti-His antibody. (C) Cell fractionation experiments were performed to determine the sub-cellular localization of PE_PGRS41, GroEL2 protein serves as a cytoplasm marker of M. smegmatis, CW represents cell wall; CP represents cytoplasm. (D) The morphology of Ms_Vec and Ms_PE_PGRS41 were detected after induction by acetamide in the presence of hygromycin. (E) Ms_Vec and Ms_PE_PGRS41 were grown in Middlebrook 7H9 medium supplemented with 0.05% Tween 80, 1% acetamide and 0.2% glycerinum, with hygromycin (100 μg/ml). The OD600 was determined at an interval of 4 h.
PE_PGRS41 sensitizes M. smegmatis response to multiple antibiotics
M. tuberculosis cell wall serves as an effective permeable barrier of antibiotics9. PE_PGRS41 was a cell wall associated protein of mycobacteria (Fig. 1C). To study whether PE_PGRS41 functions in cell wall permeability, recombinant Ms_PE_PGRS41 and Ms_Vec were treated with ten anti-tuberculosis drugs as described in the method and the MIC of each antibiotic was detected (Table 2). Both Ms_PE_PGRS41 and Ms_Vec displayed comparable susceptibility to ofloxacin (Ofl), tetracycline (Tet), streptomycin (Str), isoniazid (INH) and chloramphenicol (Chl). The MIC values of Ofl, Tet, Str, INH and Chl for Ms_Vec and Ms_PE_PGRS41 were 2 μg/ml, 1 μg/ml, 0.125 μg/ml, 4 μg/ml and 32 μg/ml, respectively. However, Ms_PE_PGRS41 was susceptible to ciprofloxacin (Cip), rifampicin (Rif), especially vancomycin (Van) and norfloxacin (Nor). The MIC values of Cip and Rif for Ms_Vec were 1 μg/ml and 16 μg/ml, while 0.5 μg/ml and 8 μg/ml for Ms_PE_PGRS41, respectively. The MIC values of Ms_PE_PGRS41 for Van and Nor were 4 and 8 fold lower than Ms_Vec, respectively (Table 2 and Fig. 2). These results suggest a novel function of PE family protein PE_PGRS41 in the susceptibility to antibiotics.
Table 2. The MIC of recombinant Ms_Vec and Ms_PE_PGRS41 to different antibiotics.
| Antibiotics (ug/ml) | Ms_Vec | Ms_PE_PGRS41 |
|---|---|---|
| Isoniazid (INH) | 4 | 4 |
| Norfloxacin (NOR) | 16 | 2 |
| Ciprofloxacin (Cip) | 1 | 0.5 |
| Rifampicin (Rif) | 16 | 8 |
| Gentamicin (Gen) | 1 | 0.5 |
| Tetracycline (Tet) | 1 | 1 |
| Ofloxacin (Ofl) | 2 | 2 |
| Vancomycin (Van) | 5 | 1.25 |
| Streptomycin (Str) | 0.125 | 0.125 |
| Chloramphenicol (Chl) | 32 | 32 |
Figure 2.

M. tuberculosis PE_PGRS41 expression in M. smegmatis decreased bacterial survival following exposure to various antibiotics, including vancomycin (A), norfloxacin (B), isoniazid (C), and ciprofloxacin (D). Ms_Vec and Ms_PE_PGRS41 were exposed to different concentration of vancomycin, norfloxacin, isoniazid, and ciprofloxacin for 5 h, then ten-fold dilution spotted the bacteria on MB 7H9 supplemented with 1% agar, the bacterial number of Ms_Vec and Ms_PE_PGRS41 were counted after 3 days cultivation.
PE_PGRS41 alters the cell wall permeability of M. smegmatis
Recombinant Ms_PE_PGRS41 was more sensitive to multiple antibiotics, including norfloxacin, rifampicin, vancomycin and Ciprofloxacin. To examine whether cell permeability alteration plays a role in the increased sensitivity of PE_PGRS41 recombinant, we used fluorescence spectroscopy to measure the whole-well accumulations of Nile Red and ethidium bromide (EtBr), representatives of the hydrophobic and hydrophilic compounds, respectively10,11. The results showed that EtBr accumulated more rapidly and to higher levels in Ms_PE_PGRS41 than the Ms_Vec, indicating an increase in cell wall permeability (Fig. 3A), while no difference was detected in Nile red accumulation between above mentioned two recombinant strains (Fig. 3B). Consistent with a defective cell wall, Ms_PE_PGRS41 was more sensitive to different acid stress and the detergent sodium dodecyl sulphate (SDS) and acid stress. The survival ability of Ms_PE_PGRS41 was significant reduced by comparison to control strains Ms_Vec after 6 and 9 h treatment with acid stress (Fig. 3C). The survival of Ms_Vec was 10- to 100- fold increased in comparison to that of Ms_PE_PGRS41 when exposed to SDS for indicated time, suggesting defective cell wall integrality in Ms_PE_PGRS41 (Fig. 3D). These data suggest the PE_PGRS41 can alter the cell wall integrity and permeability of M. smegmatis.
Figure 3. PE_PGRS41 expression in M. smegmatis results in increased cell wall permeability.

(A) Mid-log-phase cultures of Ms_Vec and Ms_PE_PGRS41 were incubated in 7H9 containing 2 μg/ml ethidium bromide for indicated time. (B) Mid-log-phase cultures of Ms_Vec and Ms_PE_PGRS41 were incubated in 7H9 with 2 μM Nile red stain for indicated time. (C) The growth of Ms_Vec and Ms_PE_PGRS41 after treatment with different pH gradient for indicated time. The Ms_Vec and Ms_PE_PGRS41 strains were centrifuged, re-suspended to 5 ml MB 7H9 at an OD600 of 0.5, 10-fold serial dilutions of Ms_Vec and Ms_PE_PGRS41 were spotted on MB 7H10. (D) Mid-log-phase cultures of Ms_Vec and Ms_PE_PGRS41 were incubated in MB 7H9 supplemented with 0.05% SDS for indicated time. And then the recombinant strains were plated onto 7H10 plates by serially ten-fold dilution, the bacterial numbers were counted after 3–4 days of cultivation.
PE_PGRS41 enhances intracellular survival of M. smegmatis within macrophages
Several PE_PGRS proteins are responsible for the virulence M. tuberculosis and contribute to its ability to grow in a macrophage12,13,14. As the first step to determine the role of PE_PGRS41 in mycobacterial virulence, we performed macrophage infection experiments and compared the intracellular survival of the Ms_Vec and Ms_PE_PGRS41. PMA-differentiated THP-1 macrophages and murine RAW264.7 cells were infected with recombinant Ms_Vec and Ms_PE_PGRS41 strains at an MOI of 10 (10 bacterium for 1 macrophage cells) at 37 °C for 4 h, followed by washing and gentamicin treatment to remove extracellular bacteria. The intracellular growth of bacteria was assayed by enumerating the CFU at different time points post infection. Inside macrophages, Ms_PE_PGRS41 survived better than Ms_Vec during the course of infection in both RAW264.7 cells (Fig. 4A) and THP-1 macrophages (Fig. 4B). To confirm this phenotype is not because of difference in entry of macrophages, the entry of macrophages was measured at an MOI of 25 to allow a more accurate estimate of entry. Immediately following the infection (4 h at 37 °C), macrophage cells were washed three times to remove extracellular bacteria, and the CFU recovered from macrophages was enumerated. The result showed that the Ms_Vec appears to enter macrophages at a rate equivalent to the Ms_PE_PGRS41 (Fig. 2S). The data suggest that recombinant M. smegmatis strains expressing PE_PGRS41 enhances survival in both murine and human macrophage cell lines, indicating a potential role for PE_PGRS41 in bacterial persistence.
Figure 4. PE_PGRS41 increased the intracellular survival of M. smegmatisin macrophage after infection.

(A) Mouse RAW264.7 cells were infected with Ms_Vec and Ms_PE_PGRS41 at an MOI of 10. (B) PMA-differentiated human THP-1 macrophages were infected with Ms_Vec or Ms_PE_PGRS41 at an MOI of 10. After 6, 24, 48, and 72 h infection, the macrophages were washed and lysed using 0.01% SDS. Lysates were plated on MB 7H10 medium to detect the bacterial number.
PE_PGRS41 promotes the death of macrophage
Infection of macrophages with M. tuberculosis can induce necrosis, defined by cell lysis. One of the consequences of infection by M. tuberculosis is the induction of macrophage cell death either by apoptosis or by necrosis15, with different impact on the immunopathology of TB. Necrosis is believed to benefit the M. tuberculosis survival16. To determine whether necrosis of macrophage plays a role in PE_PGRS41 promoted intracellular survival, macrophages were infected with recombinant strains Ms_Vec or Ms_PE_PGRS41, LDH release in the culture supernatants was determined. LDH release from infected macrophage increased following 6 h infection with both Ms_PE_PGRS41 and the control strain Ms_Vec, Macrophages infected with Ms_PE_PGRS41 released more LDH compared to Ms_Vec after 24 h infection and later time points (Fig. 5), suggesting Ms_PE_PGRS41can precipitate the macrophage cell death.
Figure 5. PE_PGRS41 promotes cell death of macrophage.
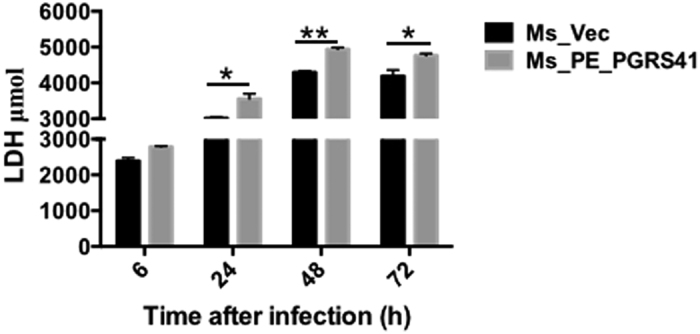
Macrophages were infected with Ms_Vec or Ms_PE_PGRS41 at an MOI of 10. After 6, 24, 48, and 72 h infection, culture supernatants were collected and the release of LDH was measured. Data are shown as means ± SEM of triplicate wells.
PE_PGRS41 inhibits macrophage pro-inflammatory cytokines production
Pro-inflammatory cytokines such as TNF-α, IL-6 and IL-1β are essential for the recruitment and activation of cells of the immune system in response to bacterial infection. To assess the immuno-modulatory potential of PE_PGRS41, we estimated the levels of several cytokines known to regulate the intracellular fate of M. tuberculosis17. We next investigated whether PE_PGRS41 modulates the expression of inflammatory cytokines in macrophages infected with Ms_Vec and Ms_PE_PGRS41. Expression of M. tuberculosis PE_PGRS41 in M. smegmatis inhibited the expression and production of pro-inflammatory cytokines TNF-α, IL-6 and IL-1β (Fig. 6), while the expression and secretion of anti-inflammatory cytokine IL-10 was increased in Ms_PE_PGRS41 infected macrophage in comparition to Ms_Vec infected macrophage (Fig. 3S). IL-10 inhibits the production of host-protective pro-inflammatory cytokines18 and is an inhibitor of early mycobacterial clearance19. We consistently observed a decrease in levels of TNF-α, IL-6 and IL-1β along with increased IL-10 transcript levels in infected macrophages. These data suggests that PE_PGRS41 plays a critical role in regulating cytokines expression.
Figure 6. Overexpression of M. tuberculosis PE_PGRS41 in M. smegmatis diminishes the abundance of pro-informatory cytokines.

PMA-differentiated THP-1 cells (2 × 106/well/2 ml) were infected with recombinant Ms_Vec and Ms_PE_PGRS41 strains. After 6, 24, and 48 h infection, the infected cells and supernatant were collected. RT-PCR was used for analyze TNF-α mRNA (A), IL-6 mRNA (B), IL-1β mRNA (C). ELISA was used for detect the production of TNF-α (D), IL-6 (E), IL-1β (F).
PE_PGRS41 suppresses the activation of macrophage
PE_PGRS41 of M. tuberculosis was found to inhibit the transcription of pro-inflammatory cytokines such as TNF-α, IL-6 and IL-1β. Lipopolysaccharide (LPS), a structural component of the outer membrane of Gram-negative bacteria, is one of the most effective stimulators of the immune system20. To determine whether this phenotype is due to inhibited activation of macrophage, we treated THP-1 cells with LPS (100 ng/mL) and then determined whether PE_PGRS41 suppresses cytokines will be released from LPS-activated macrophage. We found LPS priming can recover the expression of the repressed the cytokines (TNF-α, IL-6 and IL-1β) after Ms_PE_PGRS41 infection (Fig. 6). When THP-1 cells were infected with Ms_PE_PGRS41 combined with a stimulatory dose of LPS, TNF-α (Fig. 7A), IL-6 (Fig. 7B) and IL-1β (Fig. 7C) expression were significantly increased compared to the response to LPS or Ms_PE_PGRS41 infection alone. This suggests that PE_PGRS41 can repress the activation of macrophage, resulting in decreased production of inflammatory cytokines in macrophages. Nitric oxide is potent antimicrobial agent acquired by innate cells and NO production is a critical defense mechanism in determining the outcome of TB infection, since it reduces the survival of M. tuberculosis21. To identify factors contributing to the increased intra-macrophage survival of the recombinant M. smegmatis strains, we measured the levels of inducible nitric oxide synthase 2 (iNOS), which is a key determinant of intracellular bacillary burden in host cells22,23. The expression level of iNOS in Ms_PE_PGRS41 infected THP-1 cells was detected by Western blotting, and the level of NO was measured by Griess reagent method. We observed comparable iNOS expression in Ms_Vec and Ms_PE_PGRS41 infected THP-1 macrophages after 6, 24, 48 and 72 h infection (Fig. 4S). There was no significant difference for the NO production between the PE_PGRS41 overexpression M. smegmatis and control (Fig. 4S).
Figure 7. LPS priming recovers the cytokines production in Ms_PE_PGRS41 infected macrophages.

LPS was added into culture 2 h prior to infection. The expression of TNF-α mRNA (A), IL-6 mRNA (B), and IL-1β mRNA (C) in THP-1 cells infected for 6, 24 and 48 h with M. smegmatis harbored PALACE (Ms_Vec) or PE_PGRS41-overexpressing M. smegmatis (Ms_PE_PGRS41).
PE_PGRS41 represses cell apoptosis of macrophage
TNF-α and IL-1β provides protection against M. tuberculosis infection perhaps through the induction of host cell apoptosis24,25,26,27. Because we observed decreased transcript levels and production of TNF-α and IL-1β in Ms_PE_PGRS41 infected macrophages, we examined the hallmarks of apoptosis using fluorescence microscopy and Flow cytometry. The infected THP-1 cells stained with the Annexin V and PI that detects phosphatidylserine exposure on the outer leaflet of the plasma membrane and late apoptotic cells. Results showed that PE_PGRS41 overexpression markedly decreases the M. smegmatis infection-induced apoptosis of macrophage THP-1 cells when compared with Ms_Vec infected THP-1 cells (Fig. 8A). Flow cytometry validated this result (Fig. 8B). Several PE proteins-induced apoptosis has been shown to be dependent on activation of the caspase cascade with cleavage of caspases 328. Moreover, the cleavage caspase-3 and caspase-9 were decreased in MS_PE_PGRS41 infected macrophage in comparion to Ms_Vec infected macrophage (Fig. 8C). These results may provides us that PE_PGRS41 inhibits cell apoptosis depends on caspase cascade.
Figure 8. PE_PGRS41 depresses cell apoptosis of macrophage.

Macrophages were infected with Ms_Vec and Ms_PE_PGRS41 for indicated time points, cells were collected and the level of apoptosis was detected by fluorescence microscope (A) and flow cytometry (B) through annexin-V-FITC and propidium iodide (PI) straining. (C) The expression of caspase-1, pro-caspase-3, pro-caspase-9, cleaved caspase-3 and caspase-9 were detected by Western Blot.
PE_PGRS41 suppresses the Autophagy via interfering ATG-8
Autophagy is intensely studied to test its role in host defense against M. tuberculosis29. We interested with if PE_PGRS41 can disturb autophagy. LC3 is a defined marker for autophagy, which plays an essential role in autophagosome formation. We measured LC3 protein expression in THP-1 cells infected with Ms_Vec or Ms_PE_PGRS41 and found that PE_PGRS41 blocks the expression level of LC3B (Fig. 9A). Ms_PE_PGRS41 infected macrophage suppressed the conversion of LC3-I to LC3-II. Hence, the relative ratio of LC3-II to LC3-I was substantially lower in Ms_PE_PGRS41 infected cells than Ms_Vec infected cells (Fig. 9A). Next, the SQSTM1/p62 protein serves as a marker to monitor autophagic flux. SQSTM1 is an ubiquitin-binding scaffold protein that is degraded by autophagy. Thus, decreased levels of SQSTM1 are observed when autophagy is activated30, whereas SQSTM1 accumulates when autophagy is inhibited31. Here, the SQSTM1 protein was detected in Ms_Vec and Ms_PE_PGRS41 infected cells; we found the expression of SQSTM1 significantly increased in cells infected with Ms_PE_PGRS41 by comparison to Ms_Vec infected cells (Fig. 9A). In addition, the expression of ATG8 protein (Fig. 9A) and ATG8 mRNA (Fig. 9B) were both inhibited after Ms_PE_PGRS41 infection, whereas no difference in other autophagy-related proteins such as ATG-5 (Fig. 9A and C) and ATG-12 (Fig. 9A and D), as well as other autophagy related DRAM2H, ATG-10 and ATG-4c (Fig. 5S).
Figure 9. PE_PGRS41 suppresses autophagy of macrophage through interferon with ATG-8.

PMA differentiated THP-1 cells were infected with Ms_Vec and Ms_PE_PGRS41 for indicated time. The cells were washed, collected and subjected to Western Blot for detection the expression of autophagy related protein (A). The expression of ATG-8 (B), ATG-5(C) and ATG-12 (D) mRNA was analyzed by RT-PCR.
Discussion
Deciphering the genome of M. tuberculosis reveals a special PE family protein, which accounts for about 4% of whole genome, with a total of 99 members4. Based on whether C-terminal of PE-encoding proteins bearing Gly-Gly-Ala or Gly-GlyAsn multiple tandem repeat structure, it is divided into two subfamilies: PE_ and PE_PGRS. N-terminal PE domain contains highly conserved amino acid residues, C-terminal PGRS domain characterizes polymorphic GC-rich sequence, which varies in sequence, size and repeat numbers32. The existence and expansion of PE_PGRS genes in certain mycobacteria are associated with the acquisition of new virulence traits3,33. PE_PGRS proteins are major source of antigenic variation6, function as lipase to supply energy for M. tuberculosis persistence and survival34, decrease cytokine production and inhibit phagolysosome maturation of macrophages35,36. The role of most PE_PGRS proteins in M. tuberculosis biology and pathogenesis remains elusive despite sporadic studies33,37.
In this study, we found heterologous expression of M. tuberculosis PE_PGRS41 in M. smegmatis becomes extremely sensitive to multiple antibiotics and in vitro stresses through increasing the cell wall permeability (Figs 2 and 3). M. smegmatis expressing PE_PGRS41 showed enhanced survival both in the RAW264.7 and THP-1 macrophages (Fig. 4). The increased survival within the macrophage cannot be attributed to the increased resistance to any of the tested stresses38, as PE_PGRS41 promoted M. smegmatis is more sensitive to in vitro stress (Fig. 3). Recombinant Ms_PE_PGRS41 suppressed up to ~80% of the cytokine expression at various time points, starting from 24 h after infection, although survival of M. smegmatis did not change substantially until very late time points after infection in murine RAW264.7 macrophage (Fig. 4). Since Ms_Vec and Ms_PE_PGRS41 showed the same growth kinetics (Fig. 1C), the enhanced intracellular survival in macrophages of Ms_PE_PGRS41 might be the effects of PE_PGRS41 on innate immune response.
Immunoregulatory molecules, such as TNF-α39, IL-640 and IL-1β41, are essential components of the host immune defense against mycobacterial infection42. M. tuberculosis effectors can suppress the expression of pro-inflammatory cytokines to benefit its survival in macrophages36,43,44. M. tuberculosis Rv0198c (zmp1), required for full virulence of the pathogen in mice, can suppress the IL-1β production by inhibiting inflammasome activation in murine macrophages16. Expression of PE_PGRS62 in M. smegmatis decreases pro-inflammatory cytokines IL-1β and IL-6 mRNA expression in macrophages after infection36. M. tuberculosis Mce3E inhibits TNF-α and IL-6 expression, and the promotion of mycobacterial survival within macrophages43. Consistent with that, we identified that M. tuberculosis PE_PGRS effector PE_PGRS41 down-regulates a specific pool of pro-immunoregulatory molecules, including TNF-α, IL-6 and IL-1β (Fig. 6), while the expression of anti-inflammatory factor IL-10 was increased in Ms_PE_PGRS41 infected macrophage in comparison to Ms_Vec infected cells (Fig. 3S). Studies have shown that IL-10 blocks phagosome maturation in M. tuberculosis infected human macrophages, resulting in the decreasing in pro-inflammatory cytokines production45,46. Thus, M. tuberculosis PE_PGRS41 has the ability to inhibit pro-inflammatory cytokines, resulting in the enhanced intracellular survival of mycobacteria in macrophages.
Recombinant M. smegmatis expressing PE_PGRS41 shows enhanced survival in both murine and human macrophage cell lines indicating a potential role for these proteins in bacterial persistence (Fig. 4). The association of PE_PGRS41 with the M. smegmatis cell wall (Fig. 1C) suggested a role in the mysterious mycobacterium-host interaction47,48. M. tuberculosis can interfere with a number of cellular processes critical for M. tuberculosis intracellular survival, such as phagosome maturation49, phagosome–lysosome fusion, phagosome acidification, apoptosis of infected macrophages50 and induction of autophagy51. PE_PGRS41 suppressed cytokines expression via blocking phagosome maturation, as LPS priming facilitated the expression of these cytokines after infection (Fig. 7).
Apoptosis is a host defence strategy used by the host to eliminate the intracellular bacteria52. We found that M. smegmatis PE_PGRS41 recombinant, instead of the control, inhibits macrophage apoptosis (Fig. 8) via casapse-dependant pathway. Autophagy is another host defence mechanism connected with apoptosis through several possible pathways53. Autophagy involves the formation of a double-membrane cytosolic vesicle, an autophagosome54,55. Central to the formation of the autophagosome is the ubiquitin-like protein autophagy-related LC-3. We found PE_PGRS41 suppresses the conversion of LC3-I to LC3-II, whereas significantly activated the expression of autophagic flux marker SQSTM1/p62 protein (Fig. 9). In addition, ATG-8 controls the expansion of the autophagosome precursor and determines the size of autophagosomes. We demonstrated that PE_PGRS41 expressing M. smegmatis blocks the ATG-8 protein and ATG-8 mRNA expression. Taken together, these data suggest that PE_PGRS41 may suppress autophagy via interaction with autophagy-related ATG-8.
In summary, our results defined a novel role of PE_PGRS41. PE_PGRS41 can decrease pro-inflammatory cytokines expression via blocking phagosome maturation, decreasing apoptosis and autophagy, resulting in the persistence in macrophage. PE_PGRS41 protein seems to be the sole factor mediating abovementioned effect. Hence, PE_PGRS41 represents an important mycobacterial virulence factor in the ongoing battle between host and pathogen.
Methods
Reagents
Middlebrook 7H10 agar and Middlebrook 7H9 broth medium were purchased from BD Difco Laboratories. Annexin V-FITC/PI detection kit was obtained from BD. Anti-caspase-1, anti-caspase-3, anti-caspase-9 (human specific), anti-LC3B, anti-SQSTM/p62, anti-ATG5, anti-ATG12, anti-ATG8 and anti-β-actin antibody were obtained from Cell Signaling Technology. PMA was obtained from Sigma–Aldrich (Sigma), dissolved in DMSO and stored at −20 °C. The final concentration of DMSO in all experiments was less than 0.1% trypsase. pALACE plasmid was a gift from professor Yossef Av-Gay (University of British Columbia).
Bacterial and cell culture
Bacterial culture
M. smegmatis mc2155 and recombinant strains Ms_Vec and Ms_PE_PGRS41 were grown in Middlebrook 7H9 broth medium with or without hygromycin (Hyg), supplemented with 0.5% glycerol at 37 °C, 120 r.p.m. Recombinant strains Ms_Vec and Ms_PE_PGRS41 were grown in 7H9 medium containing 0.05% Tween 80,0.5% glycerol until logarithmic growth phase, the cultures were reinoculated in fresh 7H9 medium supplement with and 1% acetamide at the ratio of 1:1000 dilutions. Cultures were incubated at 37 °C with shaking through the entire growth phase. Samples were collected and the OD600 values were measured at every 4 h interval, the average values were used to plot growth curves.
Cell culture
THP-1 and RAW264.7 cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin, 100 mg/ml streptomycin (GIBCO, Invitrogen), at 37 °C with an atmosphere of 5% CO2.
Construct the recombinant strains
The intact PE_PGRS41 gene was amplified from M.tuberculosis H37Rv genome using gene-specific primers (forward with BamH I site–5′-CGGGATCCATGTCGTTCCTGATTGCTT-3′; reverse with ClaI site-5′-CCATCGATTCGACGTGATCCAGA-3′ (Table 1). The amplified PE_PGRS41 gene was cloned into PGM@19-T vector (Promega, Madison, WI). The BamH I-ClaI digested PCR product was then cloned to plasmid pALACE-His (vector) to generate the plasmid PALACE-PE_PGRS41. The plasmids (PALACE and PALACE-PE_PGRS41) were electroporated into M. smegmatismc2155, a fast-growing non-pathogenic mycobacteria56. The electroporated recombinant M. smegmatis strains were plated on Middlebrook (MB) 7H10 agar containing 100 μg/ml hygromycin after in vitro growth in MB 7H9 liquid medium for 3 h. The constructs harboring PE_PGRS41 gene (Ms_PE_PGRS41) were confirmed by PCR amplification, and the positive recombinant strains were stored with sterile 20% glycerol at −80 °C for further use. Escherichia coli DH5α strains using for gene cloning were grown at 37 °C using Luria–Bertani (LB) broth and LB agar with the addition of appropriate antibiotics. M. smegmatis mc2155 were grown in 7H9 broth medium or on 7H10 agar supplemented with 0.05% (v/v) Tween 80, 0.2% (w/v) glucose and 0.5% (v/v) glycerol.
Detection the expression of PE_PGRS 41 in M. smegmatis
The recombinant M. smegmatis strains Ms_Vec and Ms_PE_PGRS41 were cultured in 7H9 broth medium until the OD600 value between 0.6 and 0.8. Protein expression was induced using 1% (v/v) acetamide (Aladdin, China). M. smegmatis cell fractionation was carried out essentially as described earlier, with minor modifications57. In general, the recombinants including Ms_Vec and Ms_PE_PGRS41were harvested after 16 h acetamide induction using centrifugation at the speed of 3000 × g for 10 min at 4 °C. The collected cells were washed using cold PBS (phosphate-buffered saline). And then sonication was performed in cold PBS supplemented with protease inhibitor P-8849 (Sigma-Aldrich). After sonication, the prepared whole-cell lysate were centrifuged at the speed of 20000 × g for separating the insoluble (pellets in the bottom) and the soluble (supernatant in the upper layer) fractions. The separated fractions were loaded to SDS-PAGE and further detected by Western blot analysis using specific anti-His monoclonal antibody (TIANGEN, China). The blots were formed when incubated with IgG-HRP, anti-mouse IgG antibody labeled with horseradish peroxidase (TIANGEN, China).
Localization of the PE_PGRS41 protein
Recombinant Ms_Vec and Ms_PE_PGRS41 constructs were grown and subjected to cell fractionation using the method previously described. Generally, the acetamide-induced recombinant Ms_Vec and Ms_PE_PGRS41 were subjected to sonication. The whole lysates were centrifuged at the speed of 3,000 × g for 5 min at 4 °C to remove un-lysed cells and cell debris. The supernatants were ultra-centrifuged at the speed of 27,000 × g for 30 min at 4 °C. After ultra-centrifugation, the pellets (cell wall) and the supernatants (cell membrane and cytosol fractions) were collected separately. The pellets were further suspended in PBS. Equal amounts of protein from pellets and supernatants fraction were subjected to Western blotting to determine the expression of PE_PGRS41, using cytosol marker protein GroEL2 as control.
In vitro survival under different stresses
To examine their growth patterns, recombinant M. smegmatis strains were cultured in 7H9 medium containing 100 μg/ml hygromycin and grown into an optical density (OD600) of 0.8. The expression of PE_PGRS41 was induced by acetamide. OD plotted growth curves against culture time. Acetamide induced Ms_Vec and Ms_PE_PGRS41 were performed in the presence of surface stress, and acidic pH stress. For surface stress, 0.05% SDS was treated for 1, 2, 3 and 4 h. For acidic pH stress, pH gradient was generated by adding HCl in 7H9 medium and was adjusted down to 3 and 5 filter sterilized by passing through a 2 μm filter. After the treatment with SDS and acid stress, the recombinant strains were diluted and plated into MB 7H10 agar containing hygromycin for bacteria enumeration.
Anti-tuberculosis drug sensitivity assays
Ten antibiotics were used in this study. Acetamide induced Ms_Vec and Ms_PE_PGRS41 strains were prepared for treatment with the ten antibiotics. MIC values of each antibiotic were determined when the bacterial activity was killed at least 99% on liquid medium. After 24 h treatment with these ten antibiotics with different concentration, the bacteria were diluted by 10-fold and plated into 7H10 agar medium and counted the bacterial number after 3 days culture. The medium without any antibiotics serves as the control to confirm the normal growth of bacteria.
In vitro infection with recombinant M. smegmatis
THP-1, the suspension cell line was cultured in RPMI 1640 medium (Invitrogen) supplemented with 10% (v/v) heat inactivated FBS, 2 mM L-glutamine, 100 μg/ml streptomycin and 100 U/ml penicillin (Invitrogen) and cultured in humidified incubator supplemented with 5% CO2 at in 37 °C. Cells were seeded at 2 × 106 cells/well in 6-well tissue culture plates and differentiated by stimulation with 100 ng/ml PMA. The differentiated THP-1 cells were washed and changed the culture without any antibiotics, and then the cells were infected with Ms_Vec and Ms_PE_PGRS41at an MOI of 10. Four hours after infection, the infected cells were washed using PBS and gentamicin (100 μg/ml) was used to kill the bacteria outside the macrophages after four hours infection. For lactate dehydrogenase (LDH) activity assay, the culture supernatants were collected from infected cells after 6, 24, 48 h and 72 h infection. The LDH activities were detected using commercially LDH cytotoxicity kit (Takara Bio) according to standard procedure. For bacteria survival within macrophage, THP-1 cells were infected with Ms_Vec and Ms_PEPGRS41 at an MOI of 10 for 6 h, 24 h, 48 h and 72 h at 37 °C. Then the infected macrophages were washed 3 times using PBS and lysed in 1 ml 0.025% SDS. The cell lysates were diluted and aliquots of each dilution were plated on 7H10 agar plates supplemented with 10% glycerol. After 3 days incubation, colonies on plates were counted. The survival rate was calculated as compared to the control.
RT-PCR and Assay for cytokines
PMA-differentiated THP-1 cells were infected with Ms_Vec and Ms_PEPGRS41 at an MOI of 10. After 6, 24 and 48 h infection, total cellular RNA was extracted from cells was extracted from the infected cells using RNA extraction kit (TIANGEN) according to the manufacturer’s recommendations. cDNA synthesis was performed using the PrimeScript RT reagent kit (Takara, Shiga, Japan). Quantitative real-time RT-PCR reactions were performed using a CFX96 RT-PCR Detection System (Bio-Rad) using SYBR Green Master Mix. Relative mRNA levels were calculated after normalizing to β-actin. The primers of listed in Table 1. Culture supernatants were collected from the infected macrophages, the cytokines production were detected with commercialy available ELISA kits for TNF-α, IL-6, IL-1β and IL-10 (eBioscience).
Apoptosis analysis
2 × 106 THP-1 cells were infected with Ms_Vec and Ms_PE_PGRS41 for 6 h and 48 h, the infected cells were washed with ice-cold PBS and the apoptotic cells were determined by Annexin V-FITC and propidium iodide (PI) straining according to manufacturer’s instructions (Beibo, Shanghai, China). This product detects the externalization of phosphatidylserine in apoptotic cells using recombinant annexin V conjugated to green-fluorescent FITC dye and dead cells using propidium iodide (PI). The cells were subjected to fluorescence microscope analysis and flow cytometer. Untreated cells were taken as negative control.
Detection of NO production
THP-1 cells were infected with Ms_PE_PGRS41 vs. control Ms_Vec, the supernatant of each sample was collected at 6, 24, 48 and 72 h after challenge. NO levels were determined by measuring its stable end product, nitrite, using a Griess reagent (Jiancheng Bioengineering Institute, Nanjing). One hundred microliter of supernatant was added to an equal volume of Griess reagent in duplicate on a 96-well plate and incubated at room temperature for 15 min. Absorbance (540 nm) was measured and nitrite concentrations were estimated using a standard nitrite curve. Results were expressed as the mean micromoles of nitrite per sample ± SEM.
Western blot
Western blot was performed as described previously. Cells were washed three times with ice-cold PBS and then lysed in lysis buffer containing 1 mM phenylmethylsulfonyl fluoride, 1% (v/v) protease inhibitor cocktail (Sigma). Lysates were centrifuged at 12,000 × g for 15 min, and the protein concentration was measured using a bicinchoninic acid (BCA) Protein Assay kit (TIANGEN Biotechnology, China). Equal amounts of cell lysates were separated by SDS-PAGE and then transferred to nitrocellulose membranes. Membranes were blocked in 5% non-fat dry milk in TBST and incubated overnight with the respective primary antibodies against caspase-1, caspase-3, caspase-9, LC3B, SQSTM/p62, ATG-5, ATG-12, ATG-8 and His-tag (dilution 1: 1000). β-actin serves as an internal control. The membranes were incubated at room temperature for 1 h with appropriate HRP-conjugated secondary antibodies and X-ray film was developed using Plus-ECL chemiluminescent reagent.
Statistical analysis
Data were expressed as the mean ± SEM of at least three independent experiments. Statistical analysis was performed using GraphPad Prism 6.0. The results from RT-PCR, Griess assay and CFU assay were compared by Student’s t test. Differences were considered statistically significant with *P < 0.05, **P < 0.01, and ***P < 0.001.
Additional Information
How to cite this article: Deng, W. et al. Mycobacterium tuberculosis PE_PGRS41 Enhances the Intracellular Survival of M. smegmatis within Macrophages Via Blocking Innate Immunity and Inhibition of Host Defense. Sci. Rep. 7, 46716; doi: 10.1038/srep46716 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This work was supported by National Natural Science Foundation [grant numbers 81371851, 81071316, 81271882, 81301394, 81172806, 81471563], New Century Excellent Talents in Universities [grant number NCET-11-0703], National Megaprojects for Key Infectious Diseases [grant numbers 2008ZX10003-006, 2008ZX10003-001], Excellent Ph.D. thesis fellowship of Southwest University [grant numbers kb2010017, ky2011003], the Fundamental Research Funds for the Central Universities [grant numbers XDJK2014D040, XDJK2016D025], Graduate research and innovation project of graduate in Chongqing (CYS14044), The Chongqing Municipal Committee of Education for postgraduates excellence program [grant numbers YJG123104], and The undergraduates teaching reform program [grant numbers 2013JY201].
Footnotes
The authors declare no competing financial interests.
Author Contributions These experiments were conceived and designed by W.D., Q.L., X.C., J.X. Most experiments performed by W.D. and Q.L., they contributed equally to this paper. J.Z., P.L., and W.Y. conducted several experiments. All authors have read and approved the manuscript.
References
- Barry C. E. 3rd. Interpreting cell wall ‘virulence factors’ of Mycobacterium tuberculosis. Trends in microbiology 9, 237–241 (2001). [DOI] [PubMed] [Google Scholar]
- Kleinnijenhuis J., Oosting M., Joosten L. A., Netea M. G. & Van Crevel R. Innate immune recognition of Mycobacterium tuberculosis. Clinical & developmental immunology 2011, 405310, doi: 10.1155/2011/405310 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole S. T. et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544, doi: 10.1038/31159 (1998). [DOI] [PubMed] [Google Scholar]
- Brennan M. J. & Delogu G. The PE multigene family: a ‘molecular mantra’ for mycobacteria. Trends in microbiology 10, 246–249 (2002). [DOI] [PubMed] [Google Scholar]
- Brennan M. J. et al. Evidence that mycobacterial PE_PGRS proteins are cell surface constituents that influence interactions with other cells. Infection and immunity 69, 7326–7333, doi: 10.1128/IAI.69.12.7326-7333.2001 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banu S. et al. Are the PE-PGRS proteins of Mycobacterium tuberculosis variable surface antigens? Molecular microbiology 44, 9–19 (2002). [DOI] [PubMed] [Google Scholar]
- Delogu G. & Brennan M. J. Comparative immune response to PE and PE_PGRS antigens of Mycobacterium tuberculosis. Infection and immunity 69, 5606–5611 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramovitch R. B., Rohde K. H., Hsu F. F. & Russell D. G. aprABC: a Mycobacterium tuberculosis complex-specific locus that modulates pH-driven adaptation to the macrophage phagosome. Molecular microbiology 80, 678–694, doi: 10.1111/j.1365-2958.2011.07601.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan P. J. & Nikaido H. The envelope of mycobacteria. Annual review of biochemistry 64, 29–63, doi: 10.1146/annurev.bi.64.070195.000333 (1995). [DOI] [PubMed] [Google Scholar]
- Chuang Y. M. et al. Deficiency of the novel exopolyphosphatase Rv1026/PPX2 leads to metabolic downshift and altered cell wall permeability in Mycobacterium tuberculosis. mBio 6, e02428, doi: 10.1128/mBio.02428-14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues L., Machado D., Couto I., Amaral L. & Viveiros M. Contribution of efflux activity to isoniazid resistance in the Mycobacterium tuberculosis complex. Infection, genetics and evolution: journal of molecular epidemiology and evolutionary genetics in infectious diseases 12, 695–700, doi: 10.1016/j.meegid.2011.08.009 (2012). [DOI] [PubMed] [Google Scholar]
- Bottai D. et al. Disruption of the ESX-5 system of Mycobacterium tuberculosis causes loss of PPE protein secretion, reduction of cell wall integrity and strong attenuation. Molecular microbiology 83, 1195–1209, doi: 10.1111/j.1365-2958.2012.08001.x (2012). [DOI] [PubMed] [Google Scholar]
- Tiwari B. M., Kannan N., Vemu L. & Raghunand T. R. The Mycobacterium tuberculosis PE proteins Rv0285 and Rv1386 modulate innate immunity and mediate bacillary survival in macrophages. PloS one 7, e51686, doi: 10.1371/journal.pone.0051686 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S. K., Tripathi D. K., Singh P. K., Sharma S. & Srivastava K. K. Protective and survival efficacies of Rv0160c protein in murine model of Mycobacterium tuberculosis. Applied microbiology and biotechnology 97, 5825–5837, doi: 10.1007/s00253-012-4493-2 (2013). [DOI] [PubMed] [Google Scholar]
- Gil D. P. et al. Differential induction of apoptosis and necrosis in monocytes from patients with tuberculosis and healthy control subjects. The Journal of infectious diseases 189, 2120–2128, doi: 10.1086/386369 (2004). [DOI] [PubMed] [Google Scholar]
- Zhang J., Jiang R., Takayama H. & Tanaka Y. Survival of virulent Mycobacterium tuberculosis involves preventing apoptosis induced by Bcl-2 upregulation and release resulting from necrosis in J774 macrophages. Microbiology and immunology 49, 845–852 (2005). [DOI] [PubMed] [Google Scholar]
- Flynn J. L. & Chan J. Immunology of tuberculosis. Annual review of immunology 19, 93–129, doi: 10.1146/annurev.immunol.19.1.93 (2001). [DOI] [PubMed] [Google Scholar]
- Redpath S., Ghazal P. & Gascoigne N. R. Hijacking and exploitation of IL-10 by intracellular pathogens. Trends in microbiology 9, 86–92 (2001). [DOI] [PubMed] [Google Scholar]
- Murray P. J. & Young R. A. Increased antimycobacterial immunity in interleukin-10-deficient mice. Infection and immunity 67, 3087–3095 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyns H., Plessers E., De Backer P., Meyer E. & Croubels S. In vivo porcine lipopolysaccharide inflammation models to study immunomodulation of drugs. Veterinary immunology and immunopathology 166, 58–69, doi: 10.1016/j.vetimm.2015.06.001 (2015). [DOI] [PubMed] [Google Scholar]
- Rich E. A. et al. Mycobacterium tuberculosis (MTB)-stimulated production of nitric oxide by human alveolar macrophages and relationship of nitric oxide production to growth inhibition of MTB. Tubercle and lung disease: the official journal of the International Union against Tuberculosis and Lung Disease 78, 247–255 (1997). [DOI] [PubMed] [Google Scholar]
- Gupta D. et al. Suppression of TLR2-induced IL-12, reactive oxygen species, and inducible nitric oxide synthase expression by Mycobacterium tuberculosis antigens expressed inside macrophages during the course of infection. Journal of immunology 184, 5444–5455, doi: 10.4049/jimmunol.0903283 (2010). [DOI] [PubMed] [Google Scholar]
- Voskuil M. I. et al. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. The Journal of experimental medicine 198, 705–713, doi: 10.1084/jem.20030205 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar S. M. et al. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal immunology 4, 279–287, doi: 10.1038/mi.2011.3 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratazzi C. et al. Macrophage apoptosis in mycobacterial infections. Journal of leukocyte biology 66, 763–764 (1999). [DOI] [PubMed] [Google Scholar]
- Fairbairn I. P. Macrophage apoptosis in host immunity to mycobacterial infections. Biochemical Society transactions 32, 496–498, doi: 10.1042/BST0320496 (2004). [DOI] [PubMed] [Google Scholar]
- Spira A. et al. Apoptosis genes in human alveolar macrophages infected with virulent or attenuated Mycobacterium tuberculosis: a pivotal role for tumor necrosis factor. American journal of respiratory cell and molecular biology 29, 545–551, doi: 10.1165/rcmb.2002-0310OC (2003). [DOI] [PubMed] [Google Scholar]
- Tiwari B., Ramakrishnan U. M. & Raghunand T. R. The Mycobacterium tuberculosis protein pair PE9 (Rv1088)-PE10 (Rv1089) forms heterodimers and induces macrophage apoptosis through Toll-like receptor 4. Cellular microbiology 17, 1653–1669, doi: 10.1111/cmi.12462 (2015). [DOI] [PubMed] [Google Scholar]
- Deretic V. et al. Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cellular microbiology 8, 719–727, doi: 10.1111/j.1462-5822.2006.00705.x (2006). [DOI] [PubMed] [Google Scholar]
- Pursiheimo J. P., Rantanen K., Heikkinen P. T., Johansen T. & Jaakkola P. M. Hypoxia-activated autophagy accelerates degradation of SQSTM1/p62. Oncogene 28, 334–344, doi: 10.1038/onc.2008.392 (2009). [DOI] [PubMed] [Google Scholar]
- Bjorkoy G. et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. The Journal of cell biology 171, 603–614, doi: 10.1083/jcb.200507002 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottai D. & Brosch R. Mycobacterial PE, PPE and ESX clusters: novel insights into the secretion of these most unusual protein families. Molecular microbiology 73, 325–328, doi: 10.1111/j.1365-2958.2009.06784.x (2009). [DOI] [PubMed] [Google Scholar]
- Fishbein S., van Wyk N., Warren R. M. & Sampson S. L. Phylogeny to function: PE/PPE protein evolution and impact on Mycobacterium tuberculosis pathogenicity. Molecular microbiology 96, 901–916, doi: 10.1111/mmi.12981 (2015). [DOI] [PubMed] [Google Scholar]
- Deb C. et al. A novel lipase belonging to the hormone-sensitive lipase family induced under starvation to utilize stored triacylglycerol in Mycobacterium tuberculosis. The Journal of biological chemistry 281, 3866–3875, doi: 10.1074/jbc.M505556200 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y. et al. Phagolysosome maturation of macrophages was reduced by PE_PGRS 62 protein expressing in Mycobacterium smegmatis and induced in IFN-gamma priming. Veterinary microbiology 160, 117–125, doi: 10.1016/j.vetmic.2012.05.011 (2012). [DOI] [PubMed] [Google Scholar]
- Huang Y. et al. Expression of PE_PGRS 62 protein in Mycobacterium smegmatis decrease mRNA expression of proinflammatory cytokines IL-1beta, IL-6 in macrophages. Molecular and cellular biochemistry 340, 223–229, doi: 10.1007/s11010-010-0421-x (2010). [DOI] [PubMed] [Google Scholar]
- Ahmed A., Das A. & Mukhopadhyay S. Immunoregulatory functions and expression patterns of PE/PPE family members: Roles in pathogenicity and impact on anti-tuberculosis vaccine and drug design. IUBMB life 67, 414–427, doi: 10.1002/iub.1387 (2015). [DOI] [PubMed] [Google Scholar]
- Li W. et al. Mycobacterium tuberculosis Rv3402c enhances mycobacterial survival within macrophages and modulates the host pro-inflammatory cytokines production via NF-kappa B/ERK/p38 signaling. PloS one 9, e94418, doi: 10.1371/journal.pone.0094418 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bopst M. et al. Differential effects of TNF and LTalpha in the host defense against M. bovis BCG. European journal of immunology 31, 1935–1943 (2001). [DOI] [PubMed] [Google Scholar]
- Jang S., Uematsu S., Akira S. & Salgame P. IL-6 and IL-10 induction from dendritic cells in response to Mycobacterium tuberculosis is predominantly dependent on TLR2-mediated recognition. Journal of immunology 173, 3392–3397 (2004). [DOI] [PubMed] [Google Scholar]
- Fremond C. M. et al. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. Journal of immunology 179, 1178–1189 (2007). [DOI] [PubMed] [Google Scholar]
- Cooper A. M., Mayer-Barber K. D. & Sher A. Role of innate cytokines in mycobacterial infection. Mucosal immunology 4, 252–260, doi: 10.1038/mi.2011.13 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. et al. Mycobacterium tuberculosis Mce3E suppresses host innate immune responses by targeting ERK1/2 signaling. Journal of immunology 194, 3756–3767, doi: 10.4049/jimmunol.1402679 (2015). [DOI] [PubMed] [Google Scholar]
- Muttucumaru D. G. et al. Mycobacterium tuberculosis Rv0198c, a putative matrix metalloprotease is involved in pathogenicity. Tuberculosis 91, 111–116, doi: 10.1016/j.tube.2010.11.010 (2011). [DOI] [PubMed] [Google Scholar]
- O’Leary S., O’Sullivan M. P. & Keane J. IL-10 blocks phagosome maturation in mycobacterium tuberculosis-infected human macrophages. American journal of respiratory cell and molecular biology 45, 172–180, doi: 10.1165/rcmb.2010-0319OC (2011). [DOI] [PubMed] [Google Scholar]
- Redford P. S. et al. Enhanced protection to Mycobacterium tuberculosis infection in IL-10-deficient mice is accompanied by early and enhanced Th1 responses in the lung. European journal of immunology 40, 2200–2210, doi: 10.1002/eji.201040433 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G., Wang J., Gao G. F. & Liu C. H. Insights into battles between Mycobacterium tuberculosis and macrophages. Protein & cell 5, 728–736, doi: 10.1007/s13238-014-0077-5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guirado E., Schlesinger L. S. & Kaplan G. Macrophages in tuberculosis: friend or foe. Seminars in immunopathology 35, 563–583, doi: 10.1007/s00281-013-0388-2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welin A., Raffetseder J., Eklund D., Stendahl O. & Lerm M. Importance of phagosomal functionality for growth restriction of Mycobacterium tuberculosis in primary human macrophages. Journal of innate immunity 3, 508–518, doi: 10.1159/000325297 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane J., Remold H. G. & Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. Journal of immunology 164, 2016–2020 (2000). [DOI] [PubMed] [Google Scholar]
- Seto S., Tsujimura K. & Koide Y. Coronin-1a inhibits autophagosome formation around Mycobacterium tuberculosis-containing phagosomes and assists mycobacterial survival in macrophages. Cellular microbiology 14, 710–727, doi: 10.1111/j.1462-5822.2012.01754.x (2012). [DOI] [PubMed] [Google Scholar]
- Duan L., Gan H., Golan D. E. & Remold H. G. Critical role of mitochondrial damage in determining outcome of macrophage infection with Mycobacterium tuberculosis. Journal of immunology 169, 5181–5187 (2002). [DOI] [PubMed] [Google Scholar]
- Sanjurjo L. et al. The scavenger protein apoptosis inhibitor of macrophages (AIM) potentiates the antimicrobial response against Mycobacterium tuberculosis by enhancing autophagy. PloS one 8, e79670, doi: 10.1371/journal.pone.0079670 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D. J. et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 7, 1273–1294, doi: 10.4161/auto.7.11.17661 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D. J. et al. A comprehensive glossary of autophagy-related molecules and processes. Autophagy 6, 438–448, doi: 10.4161/auto.6.4.12244 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goude R. & Parish T. Electroporation of mycobacteria. J Vis Exp, doi: 761 [pii].10.3791/761 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W. et al. Mycobacterium tuberculosis PPE family protein Rv1808 manipulates cytokines profile via co-activation of MAPK and NF-kappaB signaling pathways. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology 33, 273–288, doi: 10.1159/000356668 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.