

Abstract
Objective
To evaluate in normal older adults and preclinical Alzheimer’s disease (AD) the impact of amyloid and regional tauopathy on cerebral glucose metabolism and subsequent memory decline.
Methods
We acquired positron emission tomography using F18 Flortaucipir (tau), C11 Pittsburgh Compound B (amyloid) and F18 Fluorodeoxyglucose in 90 clinically normal elderly of the Harvard Aging Brain Study.
Results
Posterior cingulate metabolism decreased when both amyloid and neocortical tau were high and predicted subsequent memory decline in a larger sample of normal elderly. In contrast, frontal hypometabolism related to the common age-related entorhinal tauopathy, but this dysfunction was independent of amyloid, and did not predict significant memory decline. Neocortical tauopathy was positively associated with metabolism in individuals with sub-threshold amyloid, suggesting that glucose metabolism increases before decreasing in the course of preclinical AD.
Interpretation
Our study identified a synergistic effect of amyloid and tau deposits and demonstrated for the first time in normal elderly its link to AD-like hypometabolism and to AD-like memory decline. The amyloid effect was seen with tau in neocortex, but not with tau in entorhinal cortex, which is the common site of age-related tauopathy. Entorhinal tau was associated with frontal hypometabolism, but this dysfunction was not associated with memory loss.
Alzheimer’s disease (AD) is a neurodegenerative disorder expressed clinically by a progressive memory decline leading to dementia in which brain amyloid-beta (Aβ) plaques and tau neurofibrillary tangles slowly accumulate as the cardinal pathologic features1, 2. Autopsy studies suggest that tau pathology appears in the medial temporal lobe prior to the appearance of Aβ pathology in neocortex3, and that both pathologies appear prior to cognitive decline, defining a preclinical, asymptomatic stage of AD3, 4 that may progress to the symptomatic stage, AD dementia5. Neural dysfunction and cell death are thought to result from the interaction of the two pathologies, but our ability to assess the linkage between early pathology and dysfunction during life has been quite limited. Furthermore, cognitive symptoms correlate best with autopsy evidence of tau deposition that has expanded from medial temporal lobe to neocortex1, but the differential association of medial temporal and neocortical tau depositions with in vivo brain function has not been evaluated.
The development of in vivo imaging of tau pathology followed the introduction of the selective tau ligand 18F Flortaucipir (FTP, also known as AV1451 or T807) used with positron emission tomography (PET)6–11. Tau-PET permits for the first time a regional assessment of tau pathology in relation to age, amyloidosis, and brain functional integrity, measured with 18F Fluorodeoxyglucose (FDG-PET). We sought to determine whether the anatomic pattern of association cortex dysfunction typically associated with AD12–14 was related to brain amyloidosis and tau deposition detected in clinically normal elderly. Second, we aimed to determine whether Aβ- and tau-associated hypometabolism related to subsequent memory decline, using thus Aβ- and tau-PET imaging to identify brain regions in which glucose metabolism predicts future memory decline in older adults with normal memory at baseline. Our results indicate that the interaction of AD pathological hallmarks, Aβ and neocortical tau, relates to posterior cingulate hypometabolism, which in turn predicts memory deterioration.
METHODS
Participants: Table 1
Table 1.
Demographics, Genetics, and Cognitive Data
| Whole Sample | Low-PiB Participants | High-PiB Participants | |
|---|---|---|---|
| Sample 1: Participants with 18F FDG, 11C PiB, and 18F FTP | |||
| N | 90 | 57 | 33 |
| Age, years | 75.6 (6.3) | 74.2 (6.1)** | 77.8 (5.9)** |
| Education, years | 15.8 (3.1) | 15.7 (3.2) | 15.9 (2.9) |
| Female, % (N) | 55.6 (50) | 54.5 (31) | 57.6 (19) |
| E4 carriers, % (N) | 30.0 (27) | 14.0 (8)** | 57.6 (19)** |
| MMSE (/30) | 29.2 (1.0) | 29.3 (0.9) | 29.0 (1.2) |
| Logical Memory (/25) | 15.2 (4.0) | 15.3 (3.9) | 15.0 (4.2) |
| Sample 2: Participants with 18F FDG, 11C PiB, and memory follow-up | |||
| N | 277 | 198 | 79 |
| Age, years | 73.5 (6.1) | 73.0 (6.2)** | 74.9 (5.5)** |
| Education, years | 15.8 (3.1) | 15.6 (3.2) | 16.3 (2.9) |
| Female, % (N) | 59.2 (164) | 57.6 (114) | 64.6 (51) |
| E4 carriers, % (N) | 26.4 (73) | 15.2 (30)** | 54.4 (43)** |
| MMSE (/30) | 29.0 (1.0) | 29.1 (1.0)* | 28.7 (1.0)* |
| Logical Memory (/25) | 13.7 (3.3) | 13.6 (3.4) | 13.9 (3.0) |
Age and cognitive data are given at the closest time of 18F FTP imaging in sample 1 and at baseline in sample 2. Significant differences between low- and high-PiB participants are highlighted: two-sided
p<0.050,
p<0.010.
Participants included in this report were recruited from the Harvard Aging Brain Study (HABS), a longitudinal study of aging and preclinical AD. They were all clinically normal older adults at the time of FTP-imaging: Mini-Mental State Examination (MMSE) was ≥ 26/30, and Logical Memory II subscale (Delayed Paragraph Recall) was normal for education: ≥9/25 if education ≥16 years, ≥6/25 otherwise. Ninety participants with FTP (tau), PiB (Aβ), and FDG (glucose metabolism) PET examinations conducted over the same year were included in a first study sample. Two hundreds and seventy-seven participants with longitudinal memory assessments, PiB, and FDG PET examinations were included in a second sample. All participants underwent a comprehensive medical and neurological evaluation and none had serious medical or neurological conditions, history of alcoholism, drug abuse, or family history of dominant AD. None were clinically depressed when entering the study (Geriatric Depression Scale <11/30) or had other psychiatric illnesses. Participants provided informed consent and were studied under protocols approved by the Partners Human Research Committee at the Massachusetts General Hospital.
Neuropsychological assessment
We evaluated changes in memory performance using the Free and Cued Selective Reminding Test (FCSRT)15. The FCSRT is a 16-item verbal memory test, selectively reminding items that have been forgotten over three successive trials. In the encoding phase, each item is presented with a semantic cue. Every four items, an immediate cued recall controls that encoding has been adequately performed. In the delayed recall phase, participants are first asked to freely recall as many items as possible; next, categorical cueing is provided for those items that were not spontaneously retrieved. We used the total free recall (/48) and the total cued recall (/48) performance over the three trials. HABS participants undergo annual cognitive testing including the FCSRT. All together the 277 HABS participants included in this study had 1230 FCSRT evaluations, on average: 4·4 ± 1·2 per participant (min: 2, max: 6 evaluations). The average follow-up duration was 3·6 ± 1·3 years (min: 0·9, max: 5·6 years).
Brain imaging procedures
MRI was performed on a Siemens 3T Tim Trio and included a 3D structural T1-weighted scan using a repetition time=6400ms, echo time=2.8ms, flip angle=8°, and a voxel size=1×1×1.2mm. Images were processed with Freesurfer v5.1 (http://surfer·nmr·mgh·harvard·edu), to identify gray, white, and pial surfaces to permit region of interest (ROI) parcellation based on the Desikan-Killiany atlas.
F18FTP, F18FDG, and C11PiB were prepared and acquired according to previously published8 protocols. The mean ± SD lag time between PET examinations was 11·3 ± 12·3 weeks (max: 50·9 weeks). PET data were acquired on a Siemens ECAT HR+ scanner using a 3D mode; 63 image planes; 15·2cm axial field-of-view; 5·6mm trans-axial resolution; 2·4mm slice interval. T807 was acquired from 80–100 minutes (min) after a 9·0 to 11·0 millicurie (mCi) bolus injection in 4×5-min frames. FDG was acquired from 45–75 min after a 5 to 10mCi bolus injection in 6×5-min frames. PiB was acquired with an 8·5 to 15mCi bolus injection followed immediately by a 60-min dynamic acquisition in 69 frames (12×15 seconds, 57×60 seconds). PET images were co-registered to the corresponding T1 image for each subject using six degrees of freedom, rigid body registration, and structural ROIs, as determined by Freesurfer, were mapped into native PET space in SPM8. FTP and FDG were expressed as standard uptake volume ratios (SUVR), PiB as the distribution volume ratio (DVR) using the Logan graphical method, with slopes extracted from the 40–60min timeframe. All PET data used cerebellum as reference region (gray matter only) and were corrected for partial volume effects using MRI-based methods (geometric transfer matrix16, 17 for ROI and extended Müller-Gärtner17, 18 for surface analyses). The point spread function for partial volume correction was estimated at 6mm.
PiB retention was assessed using a large neocortical aggregate that included superior frontal, rostral middle frontal, rostral anterior cingulate, medial orbitofrontal, inferior and middle temporal, inferior parietal and precuneus (Freesurfer-defined FLR region). Specific analyses were conducted in low- and high-PiB groups: A Gaussian mixture model defined the threshold for PiB positivity (DVR=1·34) using the baseline PiB PET scans obtained from the 277 HABS participants, as previously described19. FDG and FTP retention were assessed using vertex-wise surface mapping of the cortical ribbon (gray matter threshold: 0·25, smoothing kernel: 10mm) and using Freesurfer-defined ROIs.
Statistical analyses
Mean and T-statistics maps were computed using the GLM Flex tools (mrtool.mgh.harvard.edu) implemented in Matlab 8.4 (R2014b). Threshold was selected by applying a correction for multiple comparisons using the false discovery rate (FDR, q<0·050) in the local correlation map between tracers (Fig.1B, left). This correction was obtained for a T=2·65, p<0·010 and used for all subsequent analyses. For ROI analyses, Lilliefors tests confirmed that FDG data was close to normal distribution (K<0·069 for each FDG ROI, p>0·360, criterion for 90 subjects: 0·094). We therefore used linear regressions to assess the correlations between FTP and FDG ROIs, with and without adjusting for covariates. We used linear mixed models with a random intercept per subject to relate FDG ROI data to changes in memory.
Figure 1. Tau deposition locally associated with hypometabolism in the temporal lobe.
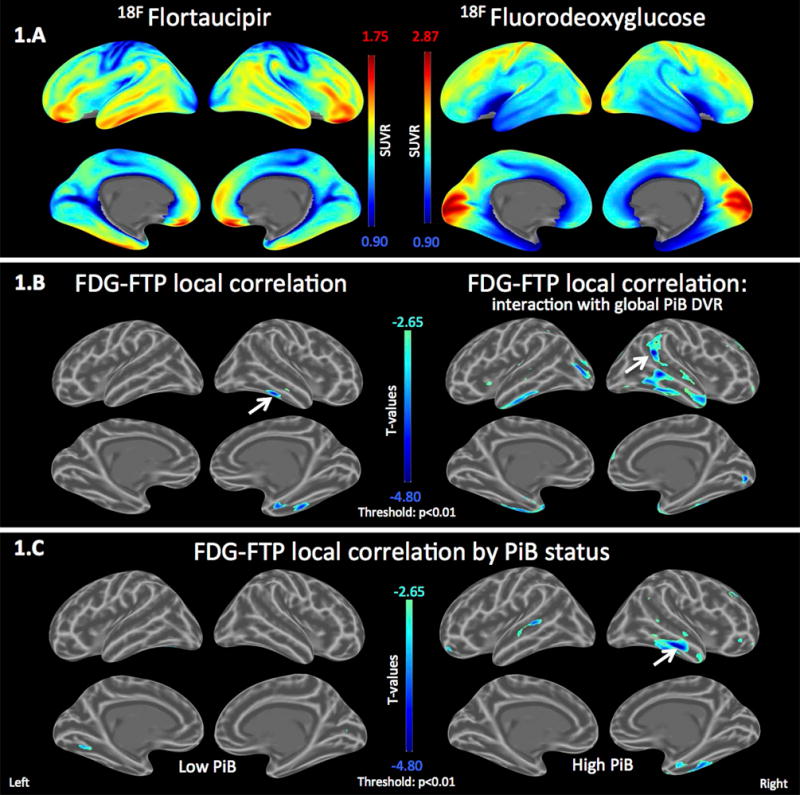
A. Mean SUVR maps for Flortaucipir (FTP) and Fluorodeoxyglucose (FDG) across the 90 study participants.
B. Left: Local correlation map between FTP and FDG, adjusting for age. Temporal tau deposition is locally associated with FDG hypometabolism. Right: Interaction effect of global continuous PiB on the local FTP-FDG association, adjusting for age. FTP-associated temporal hypometabolism is dependent on PiB levels.
C. Local FTP-associated hypometabolism is only observed in the high-PiB participants.
Scale: cold colors represent hypometabolism. Threshold: T<−2·65, p<0·010. Arrows indicate peak statistics.
RESULTS
Temporal tau deposition is associated with local FDG hypometabolism
We first asked whether tau deposits were associated with locally altered glucose metabolism. We computed a mean FTP and a mean FDG map, averaging SUVR values in each vertex across all participants of sample 1. In widespread portions of the temporal and inferior frontal lobes, the average FTP signal was high while the average FDG signal was low (Fig.1A). In contrast, in the primary cortices, the average FTP signal was low and the average FDG signal high. Each FTP vertex was then correlated with its corresponding FDG vertex to evaluate local associations between tracers, adjusting for age. Greater local FTP binding was associated with lower FDG metabolism in the temporal lobe, specifically in the entorhinal and inferior temporal cortices (Fig.1B left: arrow indicates the peak p-value=4*e-5, minimal t-statistic=−4·3). We next inquired whether Aβ burden increased the association between tau and brain dysfunction: We evaluated at each vertex the association between the FDG-PET signal and the statistical interaction between the corresponding local FTP signal and a global continuous measure of PiB DVR binding. The local negative correlation between FTP and FDG increased with increasing levels of PiB binding (Fig.1B right: peak p=9*e-6, t=−4·7). High-PiB participants had significant temporal hypometabolism locally associated with tau-PET signal (Fig.1C right: peak p=4*e-5, t=−4·8) while the association in low-PiB participants was very low or absent (Fig.1C left: peak p=0·001, t=−3·6).
Temporal neocortical tau is associated with distant, Aβ-dependent, FDG hypometabolism
We then asked whether tau deposition in specific regions, where tau was locally associated with hypometabolism, also related to altered metabolism in distant cortical regions. We extracted the FTP signal from two bilateral ROIs: entorhinal and inferior temporal, defined anatomically using Freesurfer, and regressed the FTP signal from these ROIs into the vertex-wise FDG data, adjusting for age. We found that, in addition to the local association, entorhinal FTP was associated with hypometabolism in nearby temporal regions and in inferior frontal, insular, and cingulate gyri (Fig.2A left: peak p=2*e-6, t=−5·1). In contrast, FTP binding in the inferior temporal neocortex was not associated with hypometabolism, except in very small nearby areas (Fig.2A right: peak p=2*e-4, t=−3·9). When correlating global PiB binding to vertex-wise FDG, virtually no effects were observed (peak p>0·001). Thus, neither inferior temporal, i.e., neocortical, tau nor Aβ had major metabolic impact in our sample of normal older adults.
Figure 2. Tau deposition in the inferior temporal neocortex associated with amyloid-dependent hypometabolism.
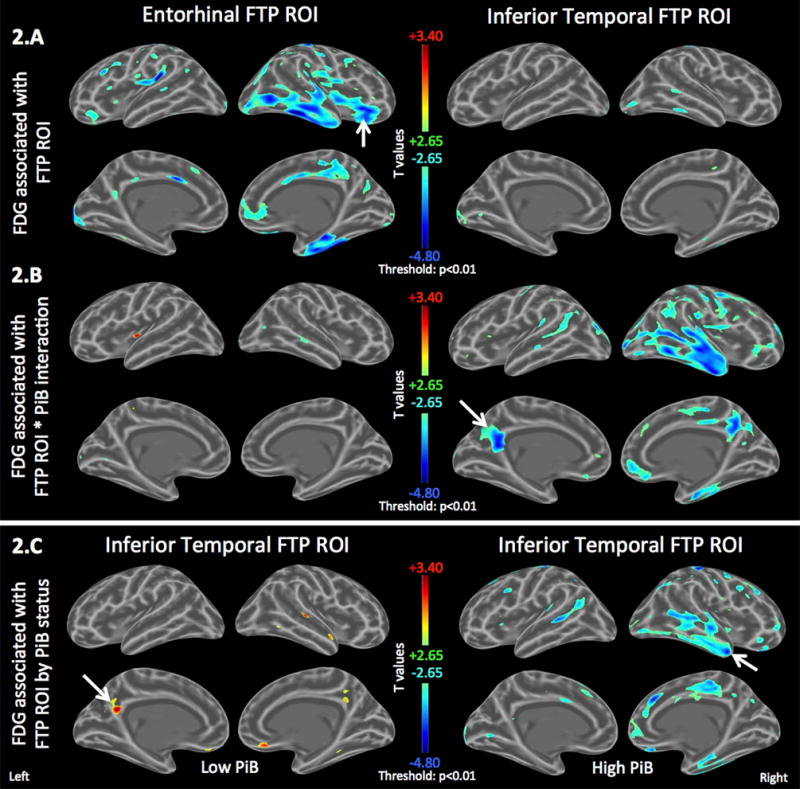
Age-adjusted FDG maps showing the association between the FDG PET signal in each vertex and FTP (tau-PET) signal in two regions of interest (ROI): entorhinal versus inferior temporal.
A. Left: Entorhinal FTP-associated FDG hypometabolism includes inferior frontal, insular, and cingulate gyri. Right: Inferior temporal FTP-associated FDG hypometabolism is restricted to the temporal lobe.
B. Interaction effect of global continuous PiB and FTP ROIs on FDG, adjusting for main effects and age. Entorhinal FTP-associated hypometabolism is not PiB-dependent (left) while inferior temporal FTP is associated with PiB-dependent temporo-parietal FDG hypometabolism (right).
C. Inferior Temporal FTP is associated with FDG hypermetabolism in the subset of low-PiB individuals (left) but FDG hypometabolism in the subset of high-PiB individuals (right). See Figure Sup1 for entorhinal FTP.
Scale: cold colors represent hypometabolism. Threshold: T>±2·65, p<0·010. Arrows indicate peak statistics.
We hypothesized that the observed absence of an effect of either neocortical tau or Aβ on glucose metabolism might be due to differential tau relationships between those with high- and low-Aβ burden. We assessed the joint association of global Aβ burden and regional tau, and found a highly significant interaction between global PiB and inferior temporal FTP predicting FDG metabolism in the bilateral posterior cingulate areas, unilaterally in right anterior temporal, and to a lesser extent, in other temporo-parietal areas (Fig.2B right: peak p=e-5, t=−4·6). High-PiB participants had hypometabolism (Fig.2C right: peak p=9*e-5, t=−4·5) whereas, in contrast, low-PiB participants had increased metabolism associated with higher levels of inferior temporal tau (Fig.2C left: peak p=0·001, t=+3·4). Unlike inferior temporal FTP, the interaction between PiB and entorhinal FTP was not associated with FDG metabolism (Fig.2B left, see Fig.3 for separate PiB subsets).
Figure 3. Tau deposition in the entorhinal cortex associated with amyloid-independent inferior frontal and insular hypometabolism.
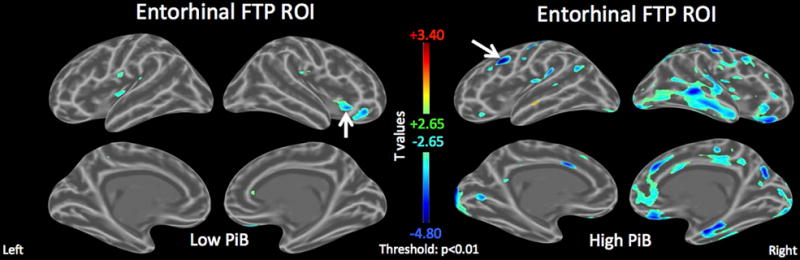
Age-adjusted FDG maps in the subsets of low- and high-PiB individuals showing the association between the FDG PET signal in each vertex and entorhinal FTP (tau-PET) signal.
Left: In the low-PiB individuals, entorhinal FTP is associated with inferior frontal and insular hypometabolism (arrow indicates peak p-value = 3*e-4, minimal t=−3·9).
Right: In the high-PiB individuals, entorhinal FTP is associated with widespread temporo-parietal hypometabolism, including locally in the entorhinal cortex (arrow indicates peak p-value = 4*e-6, minimal t=−5·6).
Scale: cold colors represent hypometabolism. Threshold: T>±2·65, p<0·010. Arrows indicate peak statistics.
In summary, surface analyses indicated that hypometabolism associated with entorhinal tau was not driven by Aβ burden and did not match the regional anatomy typically associated with AD, while FDG associated with inferior temporal tau was driven by Aβ and primarily involved the temporal and parietal lobes, in a set of regions where metabolism is associated with AD, defined clinically14 or pathologically20.
Region-of-interest analyses
We aimed to confirm and further control our vertex-wise findings using FDG ROIs to assess the effects of covariates on the association between tau and metabolism: age, sex, education, Aβ, and APOE e4 status. We investigated the associations between entorhinal and inferior temporal FTP and four bilateral Freesurfer-defined FDG ROIs. Entorhinal and inferior temporal FDG ROIs were selected to assess the local association with the corresponding FTP ROIs. We also selected the regions that included the maximal association with entorhinal or inferior temporal FTP in the vertex-wise maps: Inferior frontal and posterior cingulate respectively, labeled “pars triangularis” and “isthmus cingulate” in Freesurfer. ROI results are summarized in Table 2 and illustrated in Figure 4.
Table 2.
Amyloid- and tau-associated regional hypometabolism
| FDG Regions: (N=90 normal elderly) |
Posterior Cingulate 18F FDG |
Inferior Frontal 18F FDG |
Entorhinal Cortex 18F FDG |
Inferior Temporal 18F FDG |
|---|---|---|---|---|
| 1. Unadjusted | ||||
|
| ||||
| 11C PiB | −.013 (.041) | −.027 (.035) | −.015 (.020) | −.042 (.023) |
| p=.741 | p=.436 | p=.434 | p=.066 | |
|
| ||||
| Entorhinal | −.184 (.074) | −.131 (.064) | −.080 (.036) | −.151 (.040) |
| 18F FTP | p=.014 | p=.042 | p=.028 | p=.0003 |
|
| ||||
| Inferior Temporal | −.173 (.130) | −.109 (.112) | −.135 (.062) | −.190 (.071) |
| 18F FTP | p=.189 | p=.331 | p=.032 | p=.009 |
|
| ||||
| 2. Multiple regressions adjusting for age, sex, education, 11C PiB, and APOE e4 status | ||||
|
| ||||
| Entorhinal | −.197 (.087) | −.139 (.079) | −.068 (.060) | −.133 (.046) |
| 18F FTP | p=.026 | p=.082 | p=.117 | p=.005 |
|
| ||||
| Inferior Temporal | −.053 (.158) | −.069 (.141) | −.089 (.076) | −.105 (.084) |
| 18F FTP | p=.739 | p=.626 | p=.244 | p=.217 |
|
| ||||
| Backward selection: | −.134 (.074) B | −.131 (.064) A | −.080 (.036) A | −.104 (.041)C |
| Entorhinal 18F FTP | p=.073 | p=.042 | p=.028 | p=.012 |
|
| ||||
| 3.A. 18F FTP interactions with PiB status, age-adjusted D | ||||
|
| ||||
| Entorhinal | −.044 (.169) | −.067 (.152) | −.132 (.083) | −.056 (.093) |
| 18F FTP * PiB status | p=.793 | p=.660 | p=.116 | p=.547 |
|
| ||||
| Inferior Temporal | −.866 (.306) | −.285 (.283) | −.206 (.155) | −.443 (.171) |
| 18F FTP * PiB status | p=.006 | p=.318 | p=.188 | p=.011 |
|
| ||||
| 3.B. Low PiB participants, age-adjusted (N=57) | ||||
|
| ||||
| Inferior Temporal | +.524 (.331) | +.058 (.299) | −.016 (.144) | +.196 (.164) |
| 18F FTP | p=.121 | p=.848 | p=.913 | p=.239 |
|
| ||||
| Inferior Temporal | +9.41 (3.38) | −4.89 (3.27) | −1.89 (1.58) | −.590 (1.77) |
| 18F FTP * 11C PiB | p=.007 | p=.141 | p=.239 | p=.740 |
|
| ||||
| 3.C. High PiB participants, age-adjusted (N=33) | ||||
|
| ||||
| Inferior Temporal | −.258 (.108) | −.114 (.111) | −.158 (.083) | −.215 (.087) |
| 18F FTP | p=.023 | p=.311 | p=.067 | p=.020 |
|
| ||||
| Inferior Temporal | −.467 (.262) | −.297 (.266) | −.202 (.210) | −.234 (.219) |
| 18F FTP * 11C PiB | p=.086 | p=.274 | p=.345 | p=.294 |
|
| ||||
| 4. 18F FTP interactions with APOE e4 status, age-adjusted D | ||||
|
| ||||
| Entorhinal | +.115 (.148) | +.106 (.133) | −.035 (.074) | −.011 (.079) |
| 18F FTP * PiB status | p=.442 | p=.428 | p=.641 | p=.885 |
|
| ||||
| Inferior Temporal | +.108 (.270) | +.110 (.241) | −.152 (.131) | −.109 (.144) |
| 18F FTP * e4 status | p=.688 | p=.649 | p=.249 | p=.448 |
Unstandardized estimates (SD) – results significant at two-sided p<0.100 are highlighted in bold
Entorhinal FTP was the only predictor left in the models predicting inferior frontal and entorhinal FDG
Entorhinal FTP and age remained in the model predicting posterior cingulate FDG
Entorhinal FTP, age, and APOE e4 status remained in the model predicting inferior temporal FDG
Estimates are given for the interaction term, adjusting for the main effects and age
Subgroup results are only given if the FTP by PiB interaction is significant at p<0.100 (in gray otherwise).
Figure 4. PiB status only has an effect on hypometabolism associated with inferior temporal tau, not with entorhinal tau.
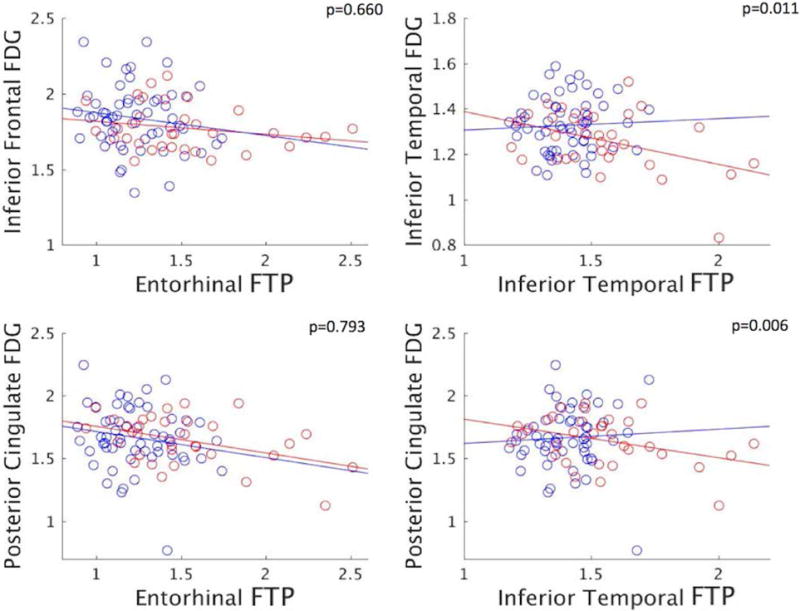
Scatterplots of FTP (tau-PET) against FDG SUVR signals in selected ROIs. P-values in the right-upper corners indicate the significance of the interaction effect between FTP and PiB status on FDG (see Table 2 box 3.A). Dots are categorized by PiB status (blue: low-PiB (n=57), red: high-PiB (n=33)). Entorhinal and inferior temporal FTP have different impact on posterior cingulate FDG in low-PiB, but similar in high-PiB participants.
We first predicted FDG signal in ROIs with PiB or FTP ROIs without adjusting for any covariate (Table 2, box.1): PiB did not significantly predict FDG-PET signal in any ROI. Entorhinal FTP, in contrast, was significantly associated with low metabolism in all FDG ROIs. Inferior temporal FTP was only associated with hypometabolism locally, in the inferior temporal, and in the nearby entorhinal cortex.
We next introduced age, sex, education, PiB, and e4 status as covariates in the FTP-FDG models (box.2): The associations between entorhinal FTP and FDG ROIs remained close to significance after all adjustments, but inferior temporal FTP did not relate to any FDG ROI. To assess the relative strength of these associations we conducted backward selection models for each FDG ROI with FTP ROIs, PiB, and covariates as predictors. Entorhinal FTP consistently remained in the models while inferior temporal FTP and PiB were excluded.
Prediction of signal in FDG ROIs by the interaction between PiB status and FTP ROIs confirmed our vertex-wise findings (box.3A): An interaction between PiB status and inferior temporal FTP, but not entorhinal FTP, was associated with hypometabolism in the posterior cingulate (Fig.4 right). Results were driven by significant hypometabolism in the high-PiB participants, and by a positive association of marginal significance (Table 2, box.3B, p=0·121) in the low-PiB participants.
This latter result suggested the hypothesis that neocortical tau deposition may be associated with increased metabolism at Aβ levels below our PiB-PET threshold. To further explore this possibility we evaluated in the low-PiB participants the association between FDG and the interaction between a continuous PiB measure and inferior temporal FTP. We observed that posterior cingulate FDG increased with the PiB by FTP interaction, demonstrating that the positive FTP-FDG association was driven by individuals with sub-threshold levels of PiB binding. In contrast, in the high-PiB subset, the PiB by FTP interaction related to decreased posterior cingulate FDG, confirming that among high-Aβ individuals, FDG decreases with increasing levels of tau and Aβ burden (box.3C).
To test whether the aforementioned impact of PiB status on the FTP-FDG associations was driven by APOE e4 status, we verified that the associations between FTP and FDG were not different according to e4 status (box 4). We also tested these interactions between e4 and FTP in the low- and high-PiB participants separately, to investigate whether e4 status played a role in the positive and negative associations observed between inferior temporal FTP and posterior cingulate FDG. In the low-PiB participants, the positive FTP-FDG association was stronger in the e4 carriers than in the non-carriers (Age-adjusted estimate for the e4 by FTP interaction term: +1·68±0·63, p=0·010, n=8 carriers), although the low-PiB e4 carriers were a small group. In contrast, in the high-PiB participants, the negative FTP-FDG association was not different according to e4 status (Estimate: −0·06±0·26, p=0·825, n=19 carriers).
In summary, ROI analyses confirmed that the interaction, i.e., the multiplicative association, of inferior temporal tauopathy and Aβ burden specifically correlates with temporo-parietal hypometabolism. When Aβ is high, inferior temporal tau is associated with hypometabolism, and the association tends to be more pronounced with increasing levels of Aβ, regardless of e4 status. When Aβ is low, inferior temporal tau is positively associated with glucose metabolism, specifically in those individuals with sub-threshold levels of Aβ or carrying an e4 allele. Our findings suggest that tau-associated hypermetabolism may be an earlier sign of brain dysfunction associated with AD pathology than decreased metabolism.
Associations between FDG metabolism and memory decline
We next tested whether the brain regions in which we identified brain dysfunction associated with tau deposition would also be associated with memory decline. For this purpose, we studied a larger cohort (Sample 2, n=277) of normal older adults including the previously described participants but also others who did not have FTP-PET imaging. We analyzed prospective longitudinal memory performances over a mean four-year period following baseline FDG-PET (one to six years). We used the four FDG ROIs previously described, hypothesizing that posterior cingulate would best predict future memory decline. Our proxy measure of memory decline was the FCSRT15, a highly predictive indicator of progression to AD dementia21 and of AD pathology in the cerebrospinal fluid (CSF)22. Demographics of sample 2 participants are given in Table 1. Memory results are summarized in Table 3 and illustrated in Figure 5.
Table 3.
Associations between longitudinal free and cued recall memory and regional FDG hypometabolism
| FDG Regions: (N=277 normal elderly) |
Posterior Cingulate 18F FDG |
Inferior Frontal 18F FDG |
Entorhinal Cortex 18F FDG |
Inferior Temporal 18F FDG |
|---|---|---|---|---|
| 1. 18F FDG main effectA | ||||
| Longitudinal free recall |
1.08 (0.39) p=.006 |
0.67 (0.39) p=.086 |
0.84 (0.68) p=.217 |
0.90 (0.66) p=.173 |
| Longitudinal cued recall |
0.29 (0.15) p=.045 |
−0.12 (0.15) p=.407 |
0.66 (0.25) p=.010 |
−0.04 (0.25) p=.872 |
| 18F FDG interaction | ||||
|
with PiB status
B Longitudinal free recall |
2.43 (0.85) p=.004 |
0.39 (0.81) p=.630 |
2.97 (1.54) p=.054 |
1.34 (1.82) p=.556 |
| Longitudinal cued recall |
0.73 (0.32) p=.023 |
−0.33 (0.31) p=.282 |
1.57 (0.58) p=.007 |
−0.27 (0.51) p=.600 |
| Low PiB participants (N=198) C | ||||
| Longitudinal free recall | 0.55 (0.42) p=.193 |
0.53 (0.45) p=.238 |
0.21 (0.75) p=.783 |
0.70 (0.77) p=.363 |
| Longitudinal cued recall | 0.11 (0.12) p=.391 |
−0.05 (0.13) p=.732 |
0.35 (0.22) p=.110 |
−0.01 (0.23) p=.986 |
| High PiB participants (N=79) | ||||
| Longitudinal free recall |
2.57 (0.87) p=.004 |
0.51 (0.74) p=.494 |
3.13 (1.47) p=.034 |
0.73 (1.22) p=.547 |
| Longitudinal cued recall |
0.76 (0.43) p=.079 |
−0.55 (0.36) p=.128 |
2.12 (0.71) p=.003 |
−0.46 (0.59) p=.435 |
Results of linear mixed-effects models predicting memory change with FDG, adjusted for age, sex, and education. Separate models were run for the four FDG ROIs and for free and cued recalls resulting in eight models. Unstandardized estimates (SD) and two-sided p-values (bold if significant at p<0.100) are given for the FDG by time interaction term (i.e., how FDG predicts change in memory over time).
Similar models than in A. but including an FDG by PiB status by time interaction term to investigate whether the impact of FDG on memory decline would be different in low and high-PiB participants.
Subgroup results are only given if the FDG by PiB by time interaction is significant at p<0.100 (in gray otherwise).
Figure 5. FDG hypometabolism predicts subsequent memory decline in high-PiB participants only.
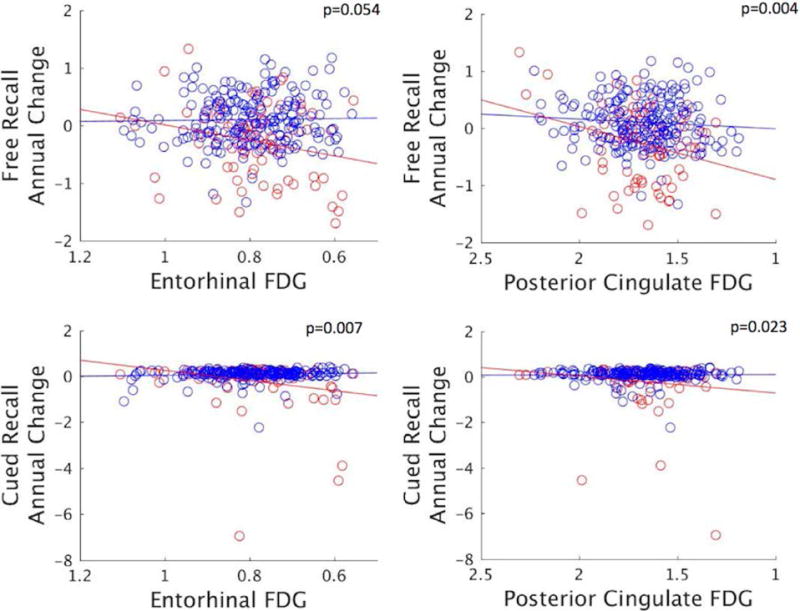
Scatterplots showing annual change in memory scores against FDG metabolism (SUVR) in selected ROIs. Memory and FDG data were residualized for age, sex, and education. P-values indicate the significance of the interaction effect between FDG and PiB status on memory changes (see Table 3). Dots are categorized by PiB status (blue: low-PiB (n=198), red: high-PiB (n=79)).
Posterior cingulate hypometabolism was associated with subsequent decline in both free and cued recall memory, while adjusting for demographics. Adjusting for PiB did not change this result. However, PiB retention strongly interacted with FDG, and when the high and low-PiB subsets were tested separately, posterior cingulate FDG only predicted memory deterioration in the participants with high-PiB (Fig.5 right). Thus, memory decline on a measure highly sensitive to AD was associated with FDG metabolism in the brain area that had the greatest hypometabolism predicted by the tau-Aβ interaction.
Because entorhinal cortex is the earliest site of tau deposition and entorhinal tau was linked to inferior frontal hypometabolism, including in the low-Aβ participants (Fig.3 left), we evaluated the association of inferior frontal FDG and memory decline. We found that inferior frontal hypometabolism was associated with a modest decline in free recall that was independent of Aβ, and marginally significant (Estimate: 0·72±0·38, p=0·061, adjusting for PiB and demographics). There was no interaction with PiB and no effect on cued recall. The result was similar without PiB in the model (Table 3, p=0·086).
We finally tested the association of entorhinal FDG and inferior temporal FDG with memory decline because these were the regions where tau deposition had local effects. Interestingly, entorhinal FDG was also associated with subsequent memory decline, specifically in cued recall performance, and PiB interacted with FDG in this region (Fig.5 left), similar to the finding that tau associates with entorhinal hypometabolism in the high- but not in the low-PiB participants (Fig.1C and 3). In contrast, inferior temporal FDG did not relate to memory decline. APOE e4 status did not affect these associations.
Posterior cingulate FDG mediates the effect of neocortical tau on memory in preclinical AD
Using the 33 high-PiB participants with FTP-data, we tested the hypothesis that memory decline is more closely associated with posterior cingulate FDG than with inferior temporal FTP. When both predictors were simultaneously entered in a mixed-model predicting free recall, FDG (Estimate: 3·51±1·21, p=0·004) was associated with decline but FTP was not (Estimate: −1·11±0·92, p=0·231). A mediation test formally demonstrated that the tauopathy effect on memory decline was mediated by posterior cingulate hypometabolism (Sobel Test: −1·79, p=0·037).
DISCUSSION
We acquired PET biomarker measures of the principal pathologic deposits of AD, Aβ and tau, and related them in normal older persons to FDG-PET, a biomarker of glucose metabolism that indicates synaptic activity. We found that the anatomy of glucose hypometabolism first reported more than 30 years ago in AD, directly correlated with the interaction of both AD pathological hallmarks, Aβ and neocortical tau. In patients with symptomatic AD23, 24, hypometabolic patterns had been previously observed in relation to Aβ and tau measured in the CSF, however the multiplicative effect was not reported. In addition, associations between CSF tau and FDG metabolism have not been found in normal elderly25, 26. The development of tau-PET imaging allowed us to isolate the metabolic signatures of entorhinal and neocortical tauopathy, and to demonstrate that AD-like hypometabolism was specifically associated with neocortical tau. Furthermore, previous studies did not find significant associations between FDG metabolism and memory decline in normal older adults27, 28. Using tau-PET to identify FDG ROIs, we demonstrated that FDG metabolism is predictive of memory decline in normal individuals with various levels of Aβ. More broadly, our finding raises the possibility that AD therapeutic strategies could potentially be successful when either pathology is the target, and that a combination of both strategies and/or therapies targeting the tau-Aβ interaction may be most successful.
Neocortical tau deposition and memory decline point to the same FDG region as AD-vulnerable
We found that tau pathology was better correlated with FDG metabolism than Aβ, in line with previous neuropathology29 and CSF30 studies, but also found that the location of the tau deposition determines the location of tau-related hypometabolism as well as the impact of Aβ on metabolic activity. The strongest predictor of cortical hypometabolism was the neocortical tau-Aβ interaction, and the anatomy of the predicted hypometabolism included predominantly parietal and temporal lobes (Fig.2B right). These limbic and association cortices have long been noted to undergo progressive deterioration in FDG metabolism along the AD trajectory31. Moreover, we found that the area of greatest hypometabolism, the posterior cingulate, predicted subsequent memory decline, which confirmed that the FDG signal we identified related both to AD pathology and to evidence of progressive amnesia. These findings integrate two anatomically distinct phenomena in AD pathophysiology, temporal lobe tauopathy and parietal physiological change, and relate these processes to progressive memory loss in normal elderly.
Tau-associated hypometabolism is both local and distant and both Aβ-dependent and Aβ-independent
To detect signs of a local synaptic toxicity associated with tau deposition, we related FDG and FTP each measured in the same cortical vertex or region. We found local tau effects confined to entorhinal and inferior temporal cortices (Fig.1B left), perhaps reflecting the known association at autopsy of tauopathy and local synaptic loss4. As elaborated below, these local effects were not evident in low-Aβ individuals. We did not observe a local effect of tau in the parietal lobe11, possibly because these normal participants did not have sufficient parietal tauopathy for a local effect to be observed. Additional areas of local negative correlation were evident at increasing Aβ burdens, which is consistent with hypometabolism reported in high-Aβ normal individuals32 or clinical AD12, and with recent work that observed spatial concordance between tau and hypometabolism in AD33.
In contrast, distant associations, i.e., those between FDG and FTP-PET signals each measured in different vertices or regions, were observed in widespread neocortical areas. These fell into two categories, those that were consistently due to an interaction of tau with Aβ, and another set of areas that were associated with tau deposition that was independent of Aβ (Fig.2). The key-differentiating feature was choice of tau-PET region: when neocortical temporal tau deposition predicted hypometabolism, it was due to an interaction with Aβ; whereas when entorhinal tau predicted hypometabolism, there was no Aβ interaction.
Tau deposition in the temporal neocortex was associated with parietal hypometabolism, most strongly involving posterior cingulate. The association in these areas interacted with Aβ, such that FDG only decreased with neocortical tau when Aβ was high. This observation is consistent with the view that Aβ and tau potentiate the physiologic consequences of each other. The mechanism or anatomic connection mediating such an interaction is uncertain; however, a direct disconnection is unlikely to underlie the observed association, because the known connectivity of the inferior temporal gyrus would not predict that the posterior cingulate would be directly affected. A more plausible explanation may simply be that the inferior temporal tau-PET signal is a better tau biomarker because it is amplified among individuals with Aβ-related tauopathy, i.e., it is a proxy measure of potentially more widespread propagating and Aβ-interacting tauopathy11.
Positive associations between tau and FDG metabolism
Among the low-Aβ participants: FDG metabolism was greater with higher levels of inferior temporal FTP and this positive association was greater according to Aβ level within the low-Aβ group. The association was thus driven by sub-threshold Aβ, and was also greater in the small group of low-Aβ e4 carriers. Metabolism did not increase with entorhinal tau. These findings raise the possibility that there is an antecedent period of aberrant tau-related hypermetabolism, consistent with data from preclinical autosomal dominant AD34 and sporadic AD35, 36. Here we provide evidence that tau-associated AD pathophysiology is underway at Aβ levels that are sub-threshold using established cut-points, arguing for an adjustment toward lower cut-points. Above the Aβ threshold, we found tau-associated hypometabolism in regions commonly used as FDG biomarker readouts for AD. Our finding of tau-related increasing metabolism in subjects with sub-threshold Aβ is consistent with postmortem-PET correlations showing that Aβ is evident in a subset of individuals below typical PET thresholds37, 38.
Whether a putative hypermetabolic phase is a cause or consequence of amyloidosis and/or tauopathy remains uncertain. Tau pathology could be driving increased activity either as compensatory or aberrant activity, or it could be increased activity due to incipient amyloidosis. While Aβ has also been linked to hypermetabolism35, 36, there is also support for the notion of increased neuronal activity fostering tau propagation and hyperphosphorylation39. A mechanism related to cognitive reserve could account for higher metabolism in individuals with AD pathology40 and early compensatory responses to AD pathologies are possible, highlighting the need for repeated imaging measures beginning in younger individuals. But the present data do suggest that the link to metabolism may be more evidently tau-associated than Aβ-associated.
Aβ-independent entorhinal tauopathy relates to frontal and insular hypometabolism
In contrast to neocortical, entorhinal tauopathy is very commonly observed in this age range at autopsy, and is thought to begin prior to substantial Aβ and cognitive impairment41, 42. Our entorhinal tau measures did not interact with Aβ to predict metabolism. Rather, in those with low-Aβ, entorhinal tauopathy was associated most prominently with inferior frontal and insular hypometabolism (Fig.3). Glucose metabolism in these regions has been associated with aging17, and while our data were age-adjusted, evaluation of younger individuals will be required. Some entorhinal connectivity to these regions has been reported in rodents43; however medial temporal ablation in primates was not linked to FDG hypometabolism in these regions, but rather to hippocampal and cingulate hypometabolism44. We also observed an Aβ-independent association between entorhinal tauopathy and cingulate metabolism. Nevertheless, the overall pattern of entorhinal tau-associated hypometabolism was distinct from the typical temporo-parietal AD pattern. Longitudinal PET studies will help determine whether entorhinal tauopathy fosters additional pathology, but without PET detectable amyloidosis, our study suggests that older individuals with entorhinal tauopathy do not manifest the AD-metabolic endophenotype.
Associations between glucose metabolism and memory decline
We found that memory progressively declined in individuals with posterior cingulate hypometabolism, the region most strongly linked to the tau-Aβ interaction. Previous studies evaluating regional FDG predicted cognitive decline in MCI45 and in normal subjects reporting subjective impairment46. However, in normal elderly, association between FDG and memory decline had not been observed so far28.
Here we used a novel tau-Aβ interaction assessment to identify a region of FDG hypometabolism, and then demonstrated that FDG in this region predicts memory decline in a group of normal elderly with various levels of Aβ. Previous work showed that posterior cingulate is critically involved in encoding and retrieval memory processes47, likely accounting for the association of metabolism in this region and memory decline. We also found a strong association between memory and entorhinal hypometabolism, similarly driven by the high-Aβ participants. These findings are consistent with the idea that episodic memory specifically decreases with temporo-limbic damage48, 49. In contrast, the inferior temporal FDG region also identified by the tau-Aβ interaction was not associated with memory decline, suggesting that not all hypometabolic regions indicated by the tau-Aβ interaction are associated with memory decline.
In addition, we found a modest effect of inferior frontal FDG on free recall decline that was independent of Aβ, suggesting that retrieval-based memory inefficiencies prevalent in aging and less specific to AD may be more associated with frontal dysfunction50. Our finding that entorhinal tauopathy was strongly associated with inferior frontal hypometabolism, particularly in low-Aβ subjects, raises the possibility that medial temporal tauopathy, highly prevalent in this age range, may affect free recall, mediated by frontal dysfunction. Pursuit of this hypothesis will require longer follow-up among those with earlier stages of tau deposition.
Acknowledgments
This work was made possible thanks to the generous support of the Belgian American Education Foundation, the Belgian Neurological Society, the Saint-Luc Foundation, the National Institute of Health, Fidelity Biosciences Research Initiative, Harvard Neurodiscovery Center, and the Alzheimer’s Association.
Footnotes
AUTHORS CONTRIBUTIONS
Conception and Design of the study: BH, DR, RS, KJ
Data Analyses: MRI and PET preprocessing: ML, JB, DC, AS – Statistics: BH, RB, EM, KP, JC, JS, AS
Drafting of the manuscript and figures: BH, KJ
POTENTIAL CONFLICT OF INTEREST DR, RS, and KJ have been consultants for Eli Lilly/Avid, which owns FTP, the tau tracer used in this study.
References
- 1.Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012 May;71(5):362–81. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006 Oct;112(4):389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Price JL, McKeel DW, Jr, Buckles VD, et al. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging. 2009 Jul;30(7):1026–36. doi: 10.1016/j.neurobiolaging.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012 Jan;8(1):1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011 May;7(3):263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chien DT, Bahri S, Szardenings AK, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis. 2013 Jan 1;34(2):457–68. doi: 10.3233/JAD-122059. [DOI] [PubMed] [Google Scholar]
- 7.Marquie M, Normandin MD, Vanderburg CR, et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol. 2015 Nov;78(5):787–800. doi: 10.1002/ana.24517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson KA, Schultz A, Betensky RA, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol. 2016 Jan;79(1):110–9. doi: 10.1002/ana.24546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scholl M, Lockhart SN, Schonhaut DR, et al. PET Imaging of Tau Deposition in the Aging Human Brain. Neuron. 2016 Mar 02;89(5):971–82. doi: 10.1016/j.neuron.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwarz AJ, Yu P, Miller BB, et al. Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain. 2016 May;139(Pt 5):1539–50. doi: 10.1093/brain/aww023. [DOI] [PubMed] [Google Scholar]
- 11.Sepulcre J, Schultz A, Sabuncu M, Gomez-Isla T, Chhatwal J, Becker A, Sperling R, Johnson K. In vivo Tau, Amyloid and Grey Matter Profiles in the Aging Brain. Journal of Neuroscience. doi: 10.1523/JNEUROSCI.0639-16.2016. in-press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foster NL, Chase TN, Fedio P, Patronas NJ, Brooks RA, Di Chiro G. Alzheimer’s disease: focal cortical changes shown by positron emission tomography. Neurology. 1983 Aug;33(8):961–5. doi: 10.1212/wnl.33.8.961. [DOI] [PubMed] [Google Scholar]
- 13.Friedland RP, Budinger TF, Ganz E, et al. Regional cerebral metabolic alterations in dementia of the Alzheimer type: positron emission tomography with [18F]fluorodeoxyglucose. J Comput Assist Tomogr. 1983 Aug;7(4):590–8. doi: 10.1097/00004728-198308000-00003. [DOI] [PubMed] [Google Scholar]
- 14.Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann Neurol. 1997 Jul;42(1):85–94. doi: 10.1002/ana.410420114. [DOI] [PubMed] [Google Scholar]
- 15.Gröber E, Buschke H. Genuine memory deficits in dementia. Dev Neuropsychol. 1987;3:13–36. [Google Scholar]
- 16.Rousset E, Harel J, Dubreuil JD. Binding characteristics of Escherichia coli enterotoxin b (STb) to the pig jejunum and partial characterization of the molecule involved. Microb Pathog. 1998 May;24(5):277–88. doi: 10.1006/mpat.1997.0193. [DOI] [PubMed] [Google Scholar]
- 17.Greve DN, Salat DH, Bowen SL, et al. Different partial volume correction methods lead to different conclusions: An F-FDG-PET study of aging. Neuroimage. 2016 Feb 23;132:334–43. doi: 10.1016/j.neuroimage.2016.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muller-Gartner HW, Links JM, Prince JL, et al. Measurement of radiotracer concentration in brain gray matter using positron emission tomography: MRI-based correction for partial volume effects. J Cereb Blood Flow Metab. 1992 Jul;12(4):571–83. doi: 10.1038/jcbfm.1992.81. [DOI] [PubMed] [Google Scholar]
- 19.Mormino EC, Betensky RA, Hedden T, et al. Amyloid and APOE epsilon4 interact to influence short-term decline in preclinical Alzheimer disease. Neurology. 2014 May 20;82(20):1760–7. doi: 10.1212/WNL.0000000000000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffman JM, Welsh-Bohmer KA, Hanson M, et al. FDG PET imaging in patients with pathologically verified dementia. J Nucl Med. 2000 Nov;41(11):1920–8. [PubMed] [Google Scholar]
- 21.Sarazin M, Berr C, De Rotrou J, et al. Amnestic syndrome of the medial temporal type identifies prodromal AD: a longitudinal study. Neurology. 2007 Nov 6;69(19):1859–67. doi: 10.1212/01.wnl.0000279336.36610.f7. [DOI] [PubMed] [Google Scholar]
- 22.Wagner M, Wolf S, Reischies FM, et al. Biomarker validation of a cued recall memory deficit in prodromal Alzheimer disease. Neurology. 2012 Feb 7;78(6):379–86. doi: 10.1212/WNL.0b013e318245f447. [DOI] [PubMed] [Google Scholar]
- 23.Chiaravalloti A, Martorana A, Koch G, et al. Functional correlates of t-Tau, p-Tau and Abeta(1)(-)(4)(2) amyloid cerebrospinal fluid levels in Alzheimer’s disease: a (1)(8)F-FDG PET/CT study. Nucl Med Commun. 2015 May;36(5):461–8. doi: 10.1097/MNM.0000000000000272. [DOI] [PubMed] [Google Scholar]
- 24.Dowling NM, Johnson SC, Gleason CE, Jagust WJ. Alzheimer’s Disease Neuroimaging I The mediational effects of FDG hypometabolism on the association between cerebrospinal fluid biomarkers and neurocognitive function. Neuroimage. 2015 Jan 15;105:357–68. doi: 10.1016/j.neuroimage.2014.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alexopoulos P, Kriett L, Haller B, et al. Limited agreement between biomarkers of neuronal injury at different stages of Alzheimer’s disease. Alzheimers Dement. 2014 Nov;10(6):684–9. doi: 10.1016/j.jalz.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 26.Yakushev I, Muller MJ, Buchholz HG, et al. Stage-dependent agreement between cerebrospinal fluid proteins and FDG-PET findings in Alzheimer’s disease. Curr Alzheimer Res. 2012 Feb;9(2):241–7. doi: 10.2174/156720512799361592. [DOI] [PubMed] [Google Scholar]
- 27.Landau SM, Mintun MA, Joshi AD, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol. 2012 Oct;72(4):578–86. doi: 10.1002/ana.23650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jagust WJ, Landau SM, Koeppe RA, et al. The Alzheimer’s Disease Neuroimaging Initiative 2 PET Core: 2015. Alzheimers Dement. 2015 Jul;11(7):757–71. doi: 10.1016/j.jalz.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeCarli CAJ, Ball MJ, et al. Post-mortem neurofibrillary tangle densities but not senile plaque densities are related to regional metabolic rates for glucose during life in Alzheimer’s disease patients. Neurodegeneration. 1991;1(1):113:21. [Google Scholar]
- 30.Petrie EC, Cross DJ, Galasko D, et al. Preclinical evidence of Alzheimer changes: convergent cerebrospinal fluid biomarker and fluorodeoxyglucose positron emission tomography findings. Arch Neurol. 2009 May;66(5):632–7. doi: 10.1001/archneurol.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minoshima S. Imaging Alzheimer’s disease: clinical applications. Neuroimaging Clin N Am. 2003 Nov;13(4):769–80. doi: 10.1016/s1052-5149(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 32.Hanseeuw BJ, Schultz AP, Betensky RA, Sperling R, Johnson KA. Hippocampal metabolism is decreased in high-amyloid mild cognitive impairment. Alzheimers Dement. doi: 10.1016/j.jalz.2016.06.2357. in-press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ossenkoppele R, Schonhaut DR, Scholl M, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain. 2016 Mar 8; doi: 10.1093/brain/aww027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benzinger TL, Blazey T, Jack CR, Jr, et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proc Natl Acad Sci U S A. 2013 Nov 19;110(47):E4502–9. doi: 10.1073/pnas.1317918110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altmann A, Ng B, Landau SM, Jagust WJ, Greicius MD. Alzheimer’s Disease Neuroimaging I. Regional brain hypometabolism is unrelated to regional amyloid plaque burden. Brain. 2015 Dec;138(Pt 12):3734–46. doi: 10.1093/brain/awv278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oh H, Madison C, Bakerm S, Rabinovici G, Jagust W. Dynamic relationships between age, amyloid-beta deposition, and glucose metabolism link to the regional vulnerability to Alzheimer’s disease. Brain. 2016 Aug;139(Pt 8):2275–89. doi: 10.1093/brain/aww108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murray ME, Lowe VJ, Graff-Radford NR, et al. Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer’s disease spectrum. Brain. 2015 May;138(Pt 5):1370–81. doi: 10.1093/brain/awv050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thal DR, Beach TG, Zanette M, et al. [(18)F]flutemetamol amyloid positron emission tomography in preclinical and symptomatic Alzheimer’s disease: specific detection of advanced phases of amyloid-beta pathology. Alzheimers Dement. 2015 Aug;11(8):975–85. doi: 10.1016/j.jalz.2015.05.018. [DOI] [PubMed] [Google Scholar]
- 39.Wu JW, Hussaini SA, Bastille IM, et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. 2016 Jun 20; doi: 10.1038/nn.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cohen AD, Price JC, Weissfeld LA, et al. Basal cerebral metabolism may modulate the cognitive effects of Abeta in mild cognitive impairment: an example of brain reserve. J Neurosci. 2009 Nov 25;29(47):14770–8. doi: 10.1523/JNEUROSCI.3669-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014 Dec;128(6):755–66. doi: 10.1007/s00401-014-1349-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duyckaerts C, Braak H, Brion JP, et al. PART is part of Alzheimer disease. Acta Neuropathol. 2015 May;129(5):749–56. doi: 10.1007/s00401-015-1390-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kondo H, Witter MP. Topographic organization of orbitofrontal projections to the parahippocampal region in rats. J Comp Neurol. 2014 Mar;522(4):772–93. doi: 10.1002/cne.23442. [DOI] [PubMed] [Google Scholar]
- 44.Meguro K, Blaizot X, Kondoh Y, Le Mestric C, Baron JC, Chavoix C. Neocortical and hippocampal glucose hypometabolism following neurotoxic lesions of the entorhinal and perirhinal cortices in the non-human primate as shown by PET. Implications for Alzheimer’s disease. Brain. 1999 Aug;122(Pt 8):1519–31. doi: 10.1093/brain/122.8.1519. [DOI] [PubMed] [Google Scholar]
- 45.Landau SM, Harvey D, Madison CM, et al. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging. 2011 Jul;32(7):1207–18. doi: 10.1016/j.neurobiolaging.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scheef L, Spottke A, Daerr M, et al. Glucose metabolism, gray matter structure, and memory decline in subjective memory impairment. Neurology. 2012 Sep 25;79(13):1332–9. doi: 10.1212/WNL.0b013e31826c1a8d. [DOI] [PubMed] [Google Scholar]
- 47.Huijbers W, Vannini P, Sperling RA, C MP, Cabeza R, Daselaar SM. Explaining the encoding/retrieval flip: memory-related deactivations and activations in the posteromedial cortex. Neuropsychologia. 2012 Dec;50(14):3764–74. doi: 10.1016/j.neuropsychologia.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sarazin M, Chauvire V, Gerardin E, et al. The amnestic syndrome of hippocampal type in Alzheimer’s disease: an MRI study. J Alzheimers Dis. 2010;22(1):285–94. doi: 10.3233/JAD-2010-091150. [DOI] [PubMed] [Google Scholar]
- 49.de Leon MJ, Convit A, Wolf OT, et al. Prediction of cognitive decline in normal elderly subjects with 2-[(18)F]fluoro-2-deoxy-D-glucose/poitron-emission tomography (FDG/PET) Proc Natl Acad Sci U S A. 2001 Sep 11;98(19):10966–71. doi: 10.1073/pnas.191044198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wheeler MA, Stuss DT, Tulving E. Frontal lobe damage produces episodic memory impairment. J Int Neuropsychol Soc. 1995 Nov;1(6):525–36. doi: 10.1017/s1355617700000655. [DOI] [PubMed] [Google Scholar]