

Abstract
Background and aims
Modern tobacco regulatory science requires an understanding of which biomarkers of cardiovascular injury are most sensitive to cigarette smoking exposure.
Methods
We studied self-reported current smokers from the Multi-Ethnic Study of Atherosclerosis. Smoking intensity was defined by number of cigarettes/day and urinary cotinine levels. Subclinical cardiovascular injury was assessed using markers of inflammation [high-sensitivity C-reactive protein (hsCRP), interleukin 6 & 2 (IL-2 & IL-6), tumor necrosis factor alpha (TNF-α)], thrombosis (fibrinogen, D-dimer, homocysteine), myocardial injury (troponin T; TnT), endothelial damage (albumin: creatinine ratio), and vascular function [aortic & carotid distensibility, flow-mediated dilation (FMD)]. Biomarkers were modeled as absolute and percent change using multivariable-adjusted linear regression models adjusted for cardiovascular risk factors and smoking duration.
Results
Among 843 current smokers, mean age was 58 (9) years, 53% were men, 39% were African American, mean number of cigarettes per day was 13 (10), and median smoking duration was 39 (15) years. Cigarette count was significantly associated with higher hsCRP, IL-6 and fibrinogen (β coefficients: 0.013, 0.011, 0.60 respectively), while In-transformed cotinine was associated with the same biomarkers (β coefficients: 0.12, 0.04, 5.3 respectively) and inversely associated with aortic distensibility (β coefficient: −0.13). There was a limited association between smoking intensity and homocysteine, D-dimer, and albumin:creatinine ratio in partially adjusted models only, while there was no association with IL-2, TNF-α, carotid distensibility, FMD, or TnT in any model. In percent change analyses, relationships were strongest with hsCRP.
Conclusions
Smoking intensity was associated with early biomarkers of CVD, particularly, markers of systemic inflammation. Of these, hsCRP may be the most sensitive.
Introduction
Cigarette smoking remains the leading cause of preventable death with an estimated six million smoking-related deaths per year worldwide.1 Smoking is also a major cause of cardiovascular disease (CVD) and mortality with nearly 10% of worldwide CVD deaths attributable to smoking.2,3
There is a direct correlation between long-term exposure to smoking and cumulative cardiovascular damage, particularly among current smokers.4,5 The precipitous drop in CVD events shortly following smoking bans,6 and the rapid reduction of CVD risk after quitting smoking7,8 suggests that the shorter-term effects of tobacco smoke also may be important.9 Acute exposure to cigarette smoking may be associated with inflammation, thrombosis, endothelial dysfunction, arterial stiffness, and coronary microvascular dysfunction.10–17 Cohort studies have traditionally been used to examine the chronic effects of smoking, but have rarely been used to evaluate immediate inhalational smoking exposure.18 Long-term exposure is commonly assessed using smoking duration (years), or cumulative exposure (pack-years). In contrast, short-term exposure or “smoking intensity”, is measured by the number of cigarettes smoked per day and urinary cotinine, a specific tobacco biomarker with a half-life of 16 hours.19
Knowledge of the CVD biomarkers most sensitive to smoking intensity has implications for tobacco regulatory policy. For example, to formulate timely regulatory policy regarding novel tobacco products, it is critical to establish how their potential CVD risk can be measured. However, there is a long latency period between tobacco exposure and the development of overt symptomatic CVD, therefore necessitating identification of validated biomarkers that can provide data over a shorter time frame. Our prior work has distinguished between biomarkers that allow evaluation of early and late cardiovascular damage in relation to accumulated long-term exposure to smoking.4 The relationship between biomarkers and passive exposure to smoking has also been described.20 There remains a need to define more “proximal” biomarkers that may be abnormal long before distal markers reflecting cumulative damage or dysfunction. Such proximal biomarkers of CVD injury are critical for the Food and Drug Administration (FDA) to determine the potential cardiovascular harms in future epidemiologic studies of e-cigs.
We therefore aimed to study the relationship between smoking intensity and proximal biomarkers of CVD injury in a large, multiethnic and sex-balanced cohort.
Materials and methods
The Multi-Ethnic Study of Atherosclerosis (MESA) is a prospective cohort study aimed at studying the significance of subclinical cardiovascular injury.21 Briefly, 6814 participants aged 45–84 years who identified themselves as White, African-American, Hispanic/Latino, or Chinese-American were recruited from 6 U.S. field centers (Baltimore City and Baltimore County, Maryland; Chicago, Illinois; Forsyth County, North Carolina; Los Angeles County, California; New York, New York and St. Paul, Minnesota) from 2000–2002. Participants were free of clinical CVD at enrollment. The protocols were approved by the institutional review boards of all collaborating institutions and by the National Heart, Lung, and Blood Institute. All participants provided written informed consent.
Inclusion/exclusion criteria
We included self-reported current cigarette smokers at baseline who smoked at least 1 cigarette/day. Participants with missing information on cigarette exposure status (n=22) and number of cigarettes/day (n=41) were excluded. We also excluded those reporting smoking ≥100 cigarettes/day (n=7) in order to minimize the influence of extreme exposure.
Assessment of tobacco exposure
Self-report
Baseline cigarette use was evaluated using questionnaires. Participants were asked about cigarette status (never, former, current), number of cigarettes/day, and duration of smoking. These questions were developed from The National Health and Nutrition Examination Survey (NHANES III), and the Atherosclerosis Risk in Communities Study. Participants were first asked whether they smoked at least 100 cigarettes in their lifetime (the equivalent of 5 packs). Participants who answered “No” were classified as never smokers, whereas those who answered “Yes” were classified as current or former smokers depending on whether they had smoked cigarettes in the past 30 days. Duration of smoking (years) was calculated as present age minus age of smoking onset for current smokers, and age at quitting cigarettes minus age of smoking onset for former smokers. Participants were also asked about the average number of cigarettes that they smoked in a day. Less than 1 cigarette/day was recorded as 0. Number of packs/day was calculated as number of cigarettes/day divided by 20. We calculated pack-years of smoking as number of pack of cigarettes per day multiplied by number of years smoking.
Cotinine
In a subset of participants enrolled in the MESA-Lung Sub-study (3965 of 4484 randomly sampled MESA participants), smoking was also evaluated by urinary cotinine using an immunoassay (Immulite 2000 Nicotine Metabolite Assay; Diagnostic Products Corp., Los Angeles, CA). The intra-assay coefficient of variation was 2.02% and undetectable values of cotinine were assigned to 7.07 ng/mL.22 According to MESA protocol, participants were instructed not to smoke on the morning of the visit and urine was collected mid-to-late morning.
Smoking intensity
Smoking intensity was defined separately by number of cigarettes/day and cotinine levels, which have been shown to correlate with self-reported cigarette count.23,24 For continuous analyses we used number of cigarettes/day and natural logarithm (In)-transformed cotinine. In categorical analyses, number of cigarettes/day was categorized as 1–9, 10–20 and >20,25 while urinary cotinine was divided into tertiles.
Measurement of proximal biomarkers of subclinical cardiovascular injury
Inflammation
Serum high-sensitivity C-reactive protein (hsCRP) was measured using the BNII nephelometer (N High Sensitivity CRP; Dade Behring Inc., Deerfield, IL). The coefficient of variation (CV) was 3.2–9.3% and the lower limit of detection was 0.17 mg/L.26
Serum interleukin-2 soluble receptor alpha (IL-2 sRα) concentration was determined by ultrasensitive ELISA (Quantikine Human IL-2 sRα Immunoassay; R&D Systems, Minneapolis, Minnesota, USA; CV 4.6–7.2%),27 and the assay range was 31.20 – 2,000 pg/mL. IL-2 sRα may regulate IL-2-dependent processes and levels correlate with IL-2.28
Serum Interleukin-6 (IL-6) was also measured using ultra-sensitive sandwich ELISA (Quantikine HS Human IL-6 Immunoassay; R&D Systems, Minneapolis, Minnesota, USA; CV 6.3%). The expected normal range was 0.24–12.5 pg/mL.27
Tumor necrosis factor α soluble receptor 1 (sTNF-R1) concentration was measured using an ultra-sensitive sandwich ELISA (Quantikine Human sTNF R1 Immunoassay; R&D Systems, Minneapolis, Minnesota, USA; CV 5%), and the assay range was 7.80 – 500 pg/mL.29 STNF-R1 modulates TNF-α activity and levels correlate with TNF-α.30
Thrombosis
Serum fibrinogen was measured by immunoprecipitation of fibrinogen antigen using the BNII nephelometer (N Antiserum to Human Fibrinogen; Dade Behring Inc., Deerfield, IL). The intra-assay and inter-assay CV were 2.7% and 2.6%, respectively.31
D-dimer was measured with an immunoturbidimetric assay (Liatest D-DI; Diagnostica Stago) which was used on a Sta-R analyzer (Diagnostica Stago).32 The lower limit of detection of the assay was 0.01 mg/mL.
Plasma homocysteine was measured using a fluorescence polarization immunoassay with the IMx analyzer (Abbott Diagnostics, Abbott Park, IL; CV 4.5%).33
Endothelial injury
Spot urine samples were collected from participants before venipuncture and then processed and frozen at −70° C. Urine albumin was measured at the Clinical Chemistry Laboratory at Fletcher Allen Health Care (Burlington, VT) using nephelometry (Array 360 CE Protein Analyzer (Beckman Instruments Inc. The lowest detectable level was 0.2 mg/dL.34 Creatinine was measured by the rate Jaffe reaction (Vitros 950IRC instrument (Johnson & Johnson Clinical Diagnostics Inc). The range was 0.05–16.5 mg/dL, with a CV range of 2.5–2.9%.34 Urinary albumin: creatinine ratio was calculated as urinary albumin (mg/dL) divided by creatinine (mg/dL). Estimated glomerular filtration rate (eGFR) was calculated using the CKD-EPI equation.
Vascular function
Aortic distensibility was assessed using gradient echo phase-contrast cine cardiac magnetic resonance imaging (MRI) with electrocardiographic gating. Images of the aorta were obtained in the transverse plane perpendicular to the aortic lumen at the level of the right pulmonary artery. Cross-sectional lumen areas of the aorta were determined using a semi-automated contouring method. Blood pressure was measured immediately before and after aortic measurements using MRI while the participant was in the supine position on the MRI scanner. Pulse pressure was calculated as the difference between the average systolic and diastolic pressure readings. The minimal and maximal aortic areas and the pulse pressure were then used to calculate aortic distensibility.35
Carotid arteries were imaged using B-mode ultrasonography with a GE Logiq 700 machine. All images were interpreted at the MESA ultrasound reading center (Tufts Medical Center). Carotid intima-media thickness (IMT) was measured and reported as the mean of the maximum IMT of the right and left sides for both the near and far walls. Data necessary for calculating carotid distensibility were obtained from a separate 20 second-long acquisition of longitudinal images of the right distal common carotid artery. For each participant, an edge detector was used to process the images and generate carotid arterial diameter curves. Diastolic and systolic diameters were determined as the smallest and largest diameter values during a cardiac cycle. Blood pressure measurements were taken by upper arm sphygmomanometry (DINAMAP system, GE Medical Systems) simultaneously during the carotid artery ultrasound.36
The carotid distensibility was calculated following equations described by Gamble et al:37 Distensibility coefficient = 2ΔD/ΔPDs where ΔD is the change in systolic/diastolic diameter, ΔP is the brachial pulse pressure, Ds is the systolic diameter, D is the average common carotid artery diameter, and h is the, mean wall thickness (IMT) measured 10 mm proximal to the carotid bulb. The intraobserver class correlation coefficient (ICC) was 0.71 and the interobserver ICC was 0.85.
Brachial flow-mediated dilation (FMD) was assessed in a subset of the baseline MESA cohort (N=3013).38 Briefly, a standard blood pressure cuff was placed around the right arm, 2 inches below the antecubital fossa, and the artery was imaged 5 to 9 cm above the antecubital fossa using a linear-array multifrequency transducer operating at 9 MHz (GE Logiq 700 Device). %FMD was computed, as follows: %FMD=[(maximum diameter-baseline diameter)/baseline diameter]×100%. Based on intrareader reproducibility studies, the intraclass correlation coefficients for baseline diameter, maximum diameter, and %FMD were 0.99, 0.99, and 0.93, respectively.
Subclinical myocardial injury
Cardiac troponin T (TnT) was measured in serum using the Elecsys 2010 system (Roche Diagnostics, Indianapolis, IN). All analyses were performed at a core lab (Veteran’s Affairs San Diego Healthcare System, La Jolla, CA; CV 5.6%). The analytical measurement range for TnT was 0.01–25.0 ng/mL39 and values below the limit of detection were recorded as 0.009 ng/mL. The 99th percentile for a healthy population for this assay was <0.01 ng/mL.
Measurement of covariates
Demographic data were self-reported. Body mass index was calculated as measured weight (kg) divided by measured height (m2). Heart rate was measured by electrocardiogram after 5 minutes of rest. Systolic and diastolic blood pressure was measured 3 times using an automated sphygmomanometer (Critikon, Tampa, FL), with final recorded blood pressure considered to be the mean of the last 2 measurements. A central laboratory (University of Vermont, Burlington, VT) measured plasma levels of glucose from blood samples obtained after a 12-hour fast. Diabetes mellitus was defined using the 2003 American Diabetes Association criteria as fasting glucose ≥126 mg/dL or use of hypoglycemic medications or insulin. Alcohol consumption and medication use were self-reported. Intentional exercise was evaluated by the MESA Typical Week Physical Activity Survey (TWPAS), which assesses the amount of time spent in and the frequency of various physical activities during a typical week in the month prior to the baseline study visit. Minutes of activity were summed for each discrete activity type and multiplied by metabolic equivalent (MET) level.40
Statistical analysis
Baseline characteristics of the study population were summarized by category of number of cigarettes smoked per day (1–9, 10–20 and >20) and cotinine tertiles. Continuous variables were reported using means (standard deviation) or medians (interquartile range) and categorical variables using percentages. Differences were tested using a nonparametric test for trend41 for continuous variables and the Chi-square test for categorical variables.
We summarized unadjusted levels of the proximal biomarkers of CVD injury by categories of cigarettes/day and by tertiles of urinary cotinine. Means and medians were reported except for Troponin-T, which was expressed as percentage above the lower limit of detection. p-value for trend was calculated using the equation developed by Cuzick.41
To establish the relation between self-reported smoking and urinary cotinine within MESA, we quantified the association between number of cigarettes/day and In-transformed urinary cotinine levels using spearman’s correlation coefficient and linear regression models adjusting for factors that influence cotinine metabolism (age, sex, race, medication use, kidney function, and duration of smoking).42
Since the relationship between cigarette count and CVD events is proportional but plateaus at higher doses,43,44 we first tested for a non-linear association between smoking intensity and proximal biomarkers of CVD injury in order to inform our choice of modeling strategies for the primary analysis. After excluding non-linearity, we proceeded with linear regression analysis. Sequential models were adjusted for age, sex, race/ethnicity and education (Model 1) and further adjusted for body mass index, systolic blood pressure, anti-hypertensive medication use, statin use, diabetes mellitus, heart rate, exercise, alcohol use and duration of smoking (Model 2). Additional adjustment for glomerular filtration rate (eGFR) was done for TnT and all analyses using cotinine. Biomarkers not normally distributed were In-transformed. Beta coefficients were interpreted as the absolute change in the marker per change in smoking intensity.
To enhance our ability to compare biomarkers on a common unit scale, we In-transformed all biomarkers and using exponentiated beta coefficients from linear regression models expressed the percent change in each marker per change in smoking intensity. Our a priori goal was to identify the most “sensitive” biomarker of subclinical CVD injury to inform future regulatory studies. Therefore, we defined “sensitivity” of a biomarker to the toxic effects of smoking as a function of the beta coefficients from the regression models, considering biomarkers that exhibited largest percent change per unit of smoking intensity as the most sensitive. These relationships were described graphically for the most promising markers using restricted cubic splines with 3 knots placed at the 10th, 50th and 90th percentiles for cigarette count (2, 10, and 25 cigarettes per day) and urinary cotinine levels (5.60, 8.37, and 9.44 ng/mL).
In subsequent analyses, we tested for effect measure modification using multiplicative interaction terms between smoking intensity and sex or race/ethnicity, and stratified analyses if the interaction term was significant.
All reported p-values were two-sided and p<0.05 was considered statistically significant. Analyses were performed using Stata version 13.1 (StataCorp, College Station, Texas), and graphs were generated using R (http://r-project.org), version 2.14.1.
Results
Baseline characteristics
Our final sample size consisted of 843 current smokers who smoked between 1 and 72 cigarettes/day, 450 of whom had measurements of baseline urinary cotinine. The mean age was 58 (9) years, 54% were male, 34% were White, 5% Chinese-American, 39% African American and 22% Hispanic. The mean number of cigarettes/day was 13 (10) and the median duration of smoking was 39 (15) years.
Those who smoked >20 cigarettes/day were more likely to be male, White, physically inactive, have a higher resting heart rate, smoke for a longer duration, and have higher urine cotinine concentrations (p<0.05) (Table 1). There were significantly more African Americans in the highest cotinine tertile, however all other baseline characteristics were not significantly different. (Table 1)
Table 1.
Baseline characteristics of the study population by categories of cigarettes per day and urinary cotinine.
| Cigarette count | Urinary cotinine | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Total
population (N=843) |
1–10
cigs/day (n=299) |
10–20
Cigs/day (n=443) |
>20
Cigs/day (n=101) |
p-value | Total
population (N=450) |
Tertile 1
(7–2421ng/) (n=150) |
Tertile 2
(2422–6436ng/mL) (n=150) |
Tertile 3 (>6437
ng/mL) (n=150) |
p-value | |
| Age, years | 58 (9) | 59 (9) | 58 (9) | 57 (8) | 0.46 | 58 (9) | 58 (9) | 58 (9) | 58 (9) | 0.73 |
| Male, % | 53 | 47 | 55 | 61 | 0.02 | 56 | 53 | 55 | 61 | 0.30 |
| Race, % | <0.001 | <0.001 | ||||||||
| White | 34 | 22 | 34 | 70 | 30 | 28 | 35 | 28 | ||
| Chinese | 5 | 5 | 7 | 1 | 7 | 9 | 7 | 7 | ||
| African American | 39 | 39 | 43 | 22 | 40 | 29 | 38 | 51 | ||
| Hispanic | 22 | 35 | 16 | 7 | 23 | 38 | 21 | 14 | ||
| Body mass index, kg/m2 | 28.0 (5.3) | 28.4 (5.8) | 27.5 (5.8) | 28.5 (5.6) | 0.61 | 28.2 (5.4) | 28.8 (5.5) | 28.0 (6.0) | 27.7 (4.7) | 0.20 |
| Systolic blood pressure, mmHg | 124 (22) | 123 (20) | 124 (22) | 123 (22) | 0.99 | 123.7 (21.0) | 124.6 (21.5) | 124.7 (21.6) | 122.0 (19.8) | 0.45 |
| Antihypertensive medication use, % | 30 | 34 | 28 | 26 | 0.10 | 30 | 33 | 31 | 26 | 0.36 |
| Heart rate, bpm | 63 (10) | 62 (9) | 64 (10) | 67 (10) | <0.001 | 63 (9) | 62 (9) | 63 (9) | 63 (10) | 0.56 |
| Diabetes mellitus, % | 13 | 13 | 12 | 13 | 0.93 | 11 | 13 | 9 | 11 | 0.52 |
| Low-density lipoprotein cholesterol, mg/dL | 116 (33) | 113 (35) | 118 (32) | 115 (34) | 0.16 | 115 (33) | 115 (35) | 114 (29) | 118 (34) | 0.62 |
| High-density lipoprotein cholesterol, mg/dL | 48 (14) | 50 (14) | 48 (14) | 46 (15) | 0.004 | 49 (15) | 49(14) | 50 (16) | 47 (14) | 0.17 |
| Statins, % | 11 | 9 | 12 | 14 | 0.28 | 10 | 9 | 9 | 10 | 0.98 |
| Alcohol use, % | 0.51 | 0.66 | ||||||||
| Never | 9 | 11 | 9 | 5 | 10 | 12 | 7 | 11 | ||
| Former | 24 | 22 | 24 | 26 | 22 | 20 | 21 | 23 | ||
| Current | 67 | 67 | 67 | 69 | 68 | 68 | 71 | 66 | ||
| Physical activity, MET-min/week* | 630 (1890) | 979 (2235) | 593 (1620) | 420 (1103) | <0.001 | 630 (1868) | 630 (1920) | 825 (2070) | 420 (1470) | 0.53 |
| Cotinine (ng/mL)* | 4307 (6328) | 1980 (3581) | 6050 (6780) | 6609 (5878) | <0.001 | 4307 (6328) | 967 (1480) | 4307 (1861) | 10,310 (5385) | <0.001 |
| Duration of smoking, years* | 39 (15) | 38 (14) | 40 (15) | 40 (11) | 0.04 | 38 (14) | 38 (12) | 37 (15) | 38 (16) | 0.71 |
Continuous variables presented as mean (standard deviation) or median (interquartile range)* depending on normality of data while categorical variables were presented as percentages.
p-value for continuous variables was calculated using a nonparametric test for trend and for categorical variables using the Chi-square test Bolded items are significant.
Correlation between self-reported cigarette exposure and urinary cotinine
The Spearman’s correlation coefficient between urinary cotinine and cigarette count was 0.47 and the proportion of variance in urinary cotinine explained by cigarettes was 17%. In multivariable analyses there was a 0.04 (95% CI: 0.03, 0.06) average increase in In-transformed cotinine for every 1 cigarette per day.
Smoking intensity and proximal biomarkers of subclinical cardiovascular injury
Each unit increase in smoking intensity (1 cigarette/day and 1 unit increase in In-transformed cotinine) was positively associated with hsCRP, IL-6, and fibrinogen in unadjusted and multivariable adjusted analyses. In unadjusted models, cotinine was positively associated with D-dimer, but this was not retained after adjustment. Similarly, cigarette count was positively associated with homocysteine in unadjusted models only. There was no association between cigarette count and cotinine with D-dimer and homocysteine respectively (Table 2 and Supplementary Table).
Table 2.
Mean absolute difference (95% CI) in levels of proximal biomarkers of CVD injury by smoking intensity, measured as cigarettes per day or urinary cotinine levels.
| Cigarettes per day | Urinary Cotinine‡ | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unadjusted | p-value | Model 1 | p-value | Model 2 | p-value | Unadjusted | p-value | Model 1** | p-value | Model 2** | p-value | |
| Inflammation | ||||||||||||
| hsCRP‡ | 0.012 (0.004,0.020) | 0.002 | 0.017 (0.009,0.025) | <0.001 | 0.013 (0.005,0.021) | 0.002 | 0.10 (0.04,0.16) | 0.002 | 0.09 (0.03,0.16) | 0.003 | 0.12(0.06,0.18) | <0.001 |
| IL-6‡ | 0.012(0.008,0.168) | <0.001 | 0.014(0.009,0.018) | <0.001 | 0.011(0.006,0.015) | <0.001 | 0.037(0.002,0.073) | 0.04 | 0.03(−0.01,0.07) | 0.11 | 0.04(0.01,0.08) | 0.01 |
| IL-2, pg/mL | 4.1(−0.4,8.5) | 0.07 | 3.2(−1.4,7.8) | 0.17 | 2.1(−2.6,6.7) | 0.38 | 28.6(−2.8,60.1) | 0.07 | 30.9(−0.5,62.3) | 0.054 | 28.6(−3.4,60.6) | 0.08 |
| TNF-α, pg/mL | 4.0(−0.9,8.9) | 0.11 | 2.9(−2.0,7.9) | 0.25 | 2.2(−2.6,7.0) | 0.37 | 25.9(−9.4,61.2) | 0.15 | 23.3(−8.0,54.7) | 0.14 | 24.1(−5.9,54.0) | 0.12 |
| Thrombosis | ||||||||||||
| Fibrinogen, mg/dL | 0.58 (0.05,1.10) | 0.03 | 0.96 (0.42,1.50) | <0.001 | 0.60 (0.05,1.14) | 0.032 | 5.8(1.8,9.8) | 0.005 | 5.1(1.1,9.1) | 0.01 | 5.3 (1.2,9.3) | 0.01 |
| D-dimert‡ | 0.005(−0.001,0.012) | 0.12 | 0.0062 (−0.0003,0.0128) | 0.06 | 0.004(−0.003,0.011) | 0.22 | 0.06 (0.01,0.11) | 0.02 | 0.03(−0.02,0.08) | 0.21 | 0.03(−0.02,0.08) | 0.28 |
| Homocysteine/μmol/L | 0.04 (0.01,0.06) | 0.003 | 0.022 (−0.004,0.049) | 0.10 | 0.02(−0.01,0.05) | 0.215 | 0.04(−0.13,0.22) | 0.62 | 0.02(−0.14,0.18) | 0.82 | 0.02(−0.14,0.19) | 0.78 |
| Endothelial injury | ||||||||||||
| Urinary albumin: Cr‡ | 0.004 (−0.003,0.012) | 0.30 | 0.010(0.002,0.018) | 0.02 | 0.005(−0.003,0.013) | 0.20 | 0.02(−0.04,0.08) | 0.49 | 0.03 (−0.03,0.09) | 0.38 | 0.04(−0.02,0.10) | 0.18 |
| Vascular function | ||||||||||||
| Aortic distensibility, mm/Hg × 10−3 | −0.002(−0.012,0.008) | 0.64 | −0.004 (−0.014,0.006) | 0.44 | −0.001 (−0.011,0.009) | 0.78 | −0.14(−0.25,−0.02) | 0.02 | −0.112 (−0.221,−0.003) | 0.04 | −0.13(−0.25,−0.02) | 0.02 |
| Carotid distensibility coefficient, mm/Hg × 10−3 | −0.002(−0.010,0.006) | 0.65 | −0.008 (−0.016,0.001) | 0.04 | −0.003 (−0.010,0.004) | 0.41 | 0.03(−0.03,0.10) | 0.29 | 0.05 (−0.01,0.11) | 0.099 | 0.02(−0.03,0.08) | 0.41 |
| Flow mediated dilation, % ×10−3 | −5.8(−34.0,22.4) | 0.69 | 8.4(−21.3,38.2) | 0.58 | 9.0(−21.4,39.5) | 0.56 | −27.9(−196.0,140.2) | 0.74 | 69.9 (−101.9,241.8) | 0.42 | 67.3(−111.5,246.1) | 0.46 |
| Subclinical myocardial injury | ||||||||||||
| Troponin T‡* | −0.0003 (−0.0011−0.00005) | 0.43 | −0.0004 (−0.0012,0.0004) | 0.29 | −0.0003 (−0.0012,0.0005) | 0.40 | −0.0006 (−0.0059,0.0047) | 0.83 | −0.001 (−0.006,0.005) | 0.75 | −0.002 (−0.007,0.004) | 0.59 |
Beta coefficients are interpreted as the mean absolute change in levels of proximal biomarkers of CVD injury per 1 cigarette/day or 1 unit of In-transformed cotinine.
Variables are log-transformed
Model 1 is adjusted for age, sex, race, education.
Model 2 is adjusted for Model 1 plus body mass index, systolic blood pressure, anti-hypertensive medication use, statin use, diabetes mellitus, heart rate, physical activity, alcohol use, duration of smoking.
Model 2 further adjusted for eGFR.
Models 1 and 2 further adjusted for eGFR in all cotinine analyses.
Bolded items are significant.
There was a positive association between cigarette count and urinary albumin: creatinine in partially adjusted models but this did not persist after multivariable adjustment. No association was observed between cotinine and albumin: creatinine. Each 1-unit increase in cotinine was inversely associated with aortic distensibility in both unadjusted and multivariable adjusted analyses, but there was no association with cigarette count.
No association was observed between either measure of smoking intensity with IL-2, TNF-α, carotid distensibility, FMD, or TnT in the unadjusted, partially and multivariable adjusted models. (Table 2 and Supplementary Table)
Biomarker sensitivity to smoking exposure
Defining sensitivity as the slope of percent change in biomarkers, hsCRP was most sensitive by virtue of having the steepest slope and changing the most with the smallest increments of smoking intensity. (Table 3, Fig. 1A and B, 2A and B)
Table 3.
Mean percent change (95% CI) in proximal biomarkers of CVD injury by smoking intensity, measured as cigarettes per day or urinary cotinine levels.
| Cigarette count‡ | Urinary cotinine‡ | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unadjusted | p-value | Model 1 | p-value | Model 2 | p-value | Unadjusted | p-value | Model 1** | p-value | Model 2** | p-value | |
| Inflammation | ||||||||||||
| hsCRP | 20.3(6.9,35.3) | 0.002 | 29.0 (14.2,45.7) | <0.001 | 21.1(7.4,36.6) | 0.002 | 16.9(5.9,29.0) | 0.002 | 16.0(5.0,28.1) | 0.003 | 20.5(9.4,32.8) | <0.001 |
| IL-6 | 20.4(12.6,28.7) | <0.001 | 22.7(14.4,31.6) | <0.001 | 16.4(8.7,24.7) | <0.001 | 6.0 (0.3, 12.1) | 0.04 | 4.8 (−1.0,10.8) | 0.11 | 7.1 (1.4,13.0) | 0.01 |
| IL-2 | 7.2 (1.1,13.8) | 0.02 | 6.0 (−0.2, 12.7) | 0.06 | 4.1 (−2.1,10.7) | 0.20 | 4.4 (0.1,8.9) | 0.04 | 4.7 (0.5,9.2) | 0.03 | 4.38(0.04,8.90) | 0.048 |
| TNF-α | 4.3 (−0.1,8.9) | 0.053 | 3.6 (−0.8,8.1) | 0.11 | 2.7 (−1.4,6.9) | 0.20 | 2.8 (−0.4,6.1) | 0.09 | 2.6 (−0.2,5.5) | 0.07 | 2.9 (0.2,5.6) | 0.03 |
| Thrombosis | ||||||||||||
| Fibrinogen | 2.24(0.05,4.49) | 0.045 | 3.9 (1.6,6.3) | 0.001 | 2.4 (0.1,4.7) | 0.04 | 2.5 (0.8,4.3) | 0.005 | 2.3 (0.5,4.0) | 0.01 | 2.4 (0.7,4.2) | 0.007 |
| D-dimer | 8.2 (−2.0,19.5) | 0.12 | 9.8 (−0.5,21.1) | 0.06 | 6.6 (−3.8,18.0) | 0.22 | 10.2(1.7,19.4) | 0.02 | 5.2 (−2.7,13.7) | 0.21 | 4.5 (−3.6,13.2) | 0.28 |
| Homocysteine | 4.6 (1.3,8.0) | 0.006 | 2.8 (−0.5,6.2) | 0.10 | 1.9 (−1.5,5.4) | 0.28 | 0.6 (−1.9,3.1) | 0.66 | 0.1 (−2.1,2.5) | 0.90 | 0.3 (−2.1,2.6) | 0.83 |
| Endothelial injury | ||||||||||||
| Urinary albumin/Cr | 6.5 (−5.5,20.1) | 0.30 | 16.6 (3.1,32.0) | 0.02 | 8.0 (−3.9,21.4) | 0.20 | 3.4 (−6.0,13.9) | 0.49 | 4.4 (−5.2,15.0) | 0.38 | 6.5 (−2.9,16.7) | 0.18 |
| Vascular function | ||||||||||||
| Aortic distensibility | 1.0 (−6.4,9.0) | 0.79 | −0.2 (−7.1,7.2) | 0.96 | 2.1 (−5.0,9.7) | 0.57 | −7.0 (−13.9,0.5) | 0.07 | −5.5 (−12.0,1.5) | 0.12 | −6.7 (−13.2,0.3) | 0.06 |
| Carotid distensibility coefficient | −0.6 (−4.8,3.7) | 0.78 | −4.1 (−7.8,−0.1) | 0.04 | −1.3 (−4.9,2.5) | 0.50 | 1.4 (−2.1,5.1) | 0.44 | 2.6 (−0.8,6.0) | 0.13 | 1.0 (−2.0,4.2) | 0.51 |
| Flow mediated dilation | −3.2 (−14.6,9.6) | 0.60 | 0.2 (−12.3,14.5) | 0.98 | 0.7 (−12.2,15.4) | 0.92 | −0.2 (−7.7,7.9) | 0.96 | 3.7 (−4.3,12.5) | 0.37 | 3.1 (−5.2,12.1) | 0.48 |
| Subclinical myocardial injury | ||||||||||||
| Troponin T* | −0.5 (−1.6,0.7) | 0.43 | −0.4 (−1.6,0.8) | 0.51 | −0.5 (−1.7,0.7) | 0.40 | −0.1 (−0.9,0.7) | 0.83 | −0.1 (−1.0,0.7) | 0.75 | −0.2 (−1.1,0.6) | 0.59 |
Beta coefficients are interpreted as the mean percent change in levels of proximal biomarkers of CVD injury per interquartile range of cigarette count (15 cigarettes/day) or urinary cotinine levels (1.57 units of log-transformed cotinine).
All proximal biomarkers and cotinine are log-transformed.
Beta coefficients are calculated per interquartile range of cigarettes count (15 cigarettes per day) and urinary cotinine levels (1.57 units of log-transformed cotinine).
Model 1 is adjusted for age, sex, race, education.
Model 2 is adjusted for Model 1 plus body mass index, systolic blood pressure, anti-hypertensive medication use, statin use, diabetes mellitus, heart rate, physical activity, alcohol use, duration of smoking.
Model 2 further adjusted for eGFR.
Models 1 and 2 further adjusted for eGFR in all cotinine analyses.
Bolded items are significant.
Fig. 1. Multi-variable adjusted percent change in biomarkers as a function of cigarettes per day.
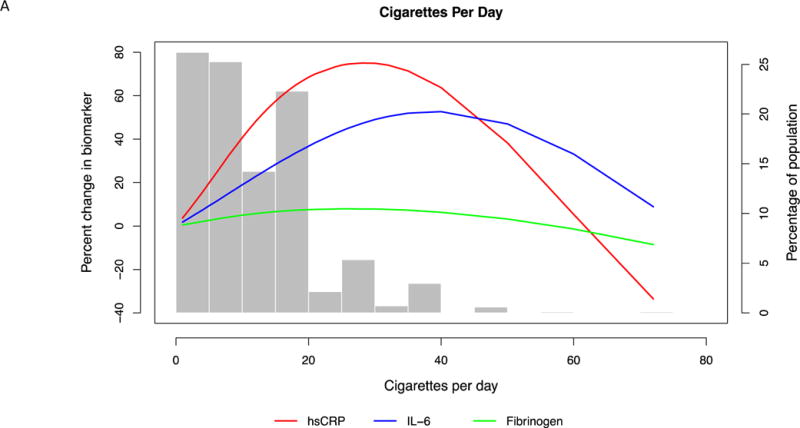
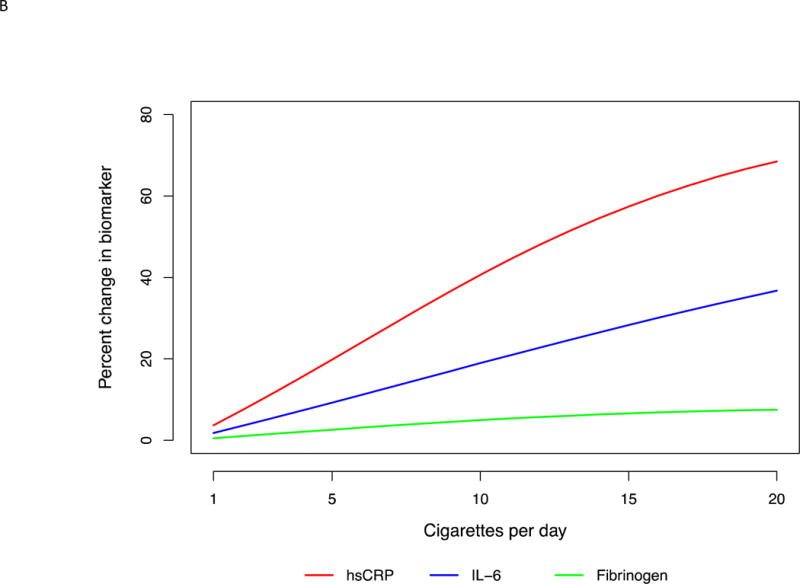
(A) For the entire range of cigarette count. Restricted cubic splines were used with 3 knots placed at 2, 10 and 25 cigarettes per day. Splines were adjusted for age, sex, race, education, body mass index, systolic blood pressure, anti-hypertensive medication use, statin use, diabetes mellitus, heart rate, physical activity, alcohol use, and duration of smoking. (B) In the typical range of smoking intensity. Restricted cubic splines were used with 2 knots placed at 2 and 10 cigarettes per day. Splines were adjusted for age, sex, race, education, body mass index, systolic blood pressure, anti-hypertensive medication use, statin use, diabetes mellitus, heart rate, physical activity, alcohol use, and duration of smoking.
Fig. 2. Multi-variable adjusted percent change in biomarkers as a function of loge -transformed urinary cotinine.
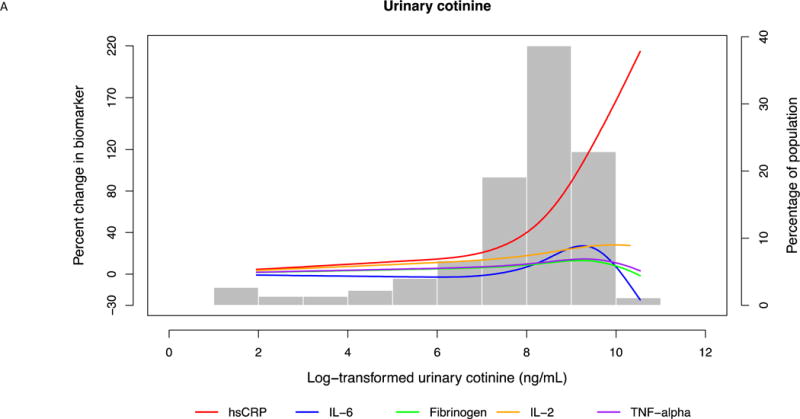
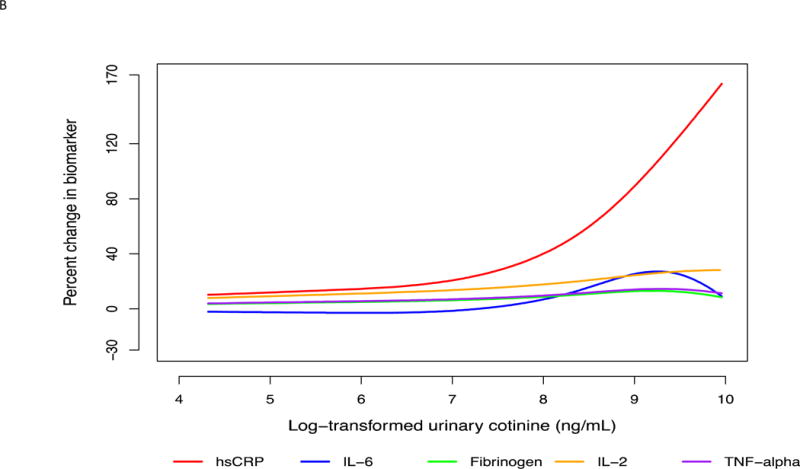
(A) For the entire range of cotinine. Restricted cubic splines were used with 3 knots placed at 5.60, 8.37 and 9.44 ng/mL cotinine. Splines were adjusted for age, sex, race, education, body mass index, systolic blood pressure, anti-hypertensive medication use, statin use, diabetes mellitus, heart rate, physical activity, alcohol use, and duration of smoking. (B) In the typical range of smoking intensity. Restricted cubic splines were used with 3 knots placed at 5.60, 8.37 and 9.44 ng/mL cotinine. Splines were adjusted for age, sex, race, education, body mass index, systolic blood pressure, anti-hypertensive medication use, statin use, diabetes mellitus, heart rate, physical activity, alcohol use, and duration of smoking.
Effect modification
Interaction terms between smoking intensity and sex were non-significant. There was an interaction between cigarette count and race/ethnicity and TNF-α, which was significant (p= 0.03). In race-stratified analyses, Hispanics had the smallest increase in TNF-α compared to Whites.
Discussion
In our cross-sectional study, biomarkers of inflammation were most sensitive to smoking intensity even at the lowest dose of exposure, and this relationship persisted regardless of sex and race/ethnicity. In particular, hsCRP was the marker of subclinical CVD injury most sensitive to smoking intensity. hsCRP has previously been shown to correlate with cumulative exposure to smoking4 and CVD risk,45 which suggests that hsCRP may have great utility for the study of the potential CVD toxicity of new and emerging tobacco products before long-term studies powered for CVD events become available. For example, hsCRP might be a candidate biomarker for the epidemiologic study of novel tobacco products to help inform policy on potential cardiovascular harm long before cardiovascular event data can be generated. Such data is needed to inform regulatory policy regarding the manufacture, marketing and use of novel tobacco products such as waterpipe (hookah) tobacco, electronic cigarettes, nicotine gels and dissolvables not currently under FDA authority.
Our results are best interpreted in light of prior knowledge from mechanistic, acute exposure, and epidemiologic data linking smoking to multiple pathophysiologic steps in atherothrombosis including inflammation, thrombotic risk, and endothelial dysfunction.
Inflammation
The mechanisms by which smoking produces an inflammatory response are well established. Smoking activates the NF-κB pathway, which induces transcription of genes involved in immune regulation.46 As a result, acute smoking exposure results in a systemic inflammatory response indicated by higher levels of leukocytes (neutrophils and macrophages) which release proinflammatory mediators such as TNF-α, IL-6 and hsCRP.18,47–50 The association of smoking and inflammation is also supported by epidemiologic studies which demonstrated a positive association of cigarettes per day with hsCRP, white blood cell count, and fibrinogen in univariate analyses.25 Data from this study indicates that smoking intensity is independently associated with inflammation in a dose dependent manner and that hsCRP is the most sensitive biomarker of this association as compared to IL-2, IL-6, and TNF-alpha.
Thrombosis
Cigarette smoking promotes thrombotic changes by platelet activation and enhancing the effects of clotting factors both playing a prominent role in formation of thrombi. Acute exposure to cigarette smoking increases levels of platelet activating factor and inhibits nitric oxide formation (due to oxidative stress), which normally acts to inhibit platelet activation.51 Smoking also increases formation of thromboxane A2 which promotes platelet aggregation and inhibits release of prostacyclin which reduces platelet aggregation.52 Moreover, smoking increases production of von Willebrand factor, thrombin and fibrinogen, decreases levels of activated protein C, and impairs fibrinolysis, all of which promote thrombosis.53 Smoking also stimulates thrombopoiesis and alters platelet structure, which further contribute to thrombosis.54
Prior observational studies have found a positive association between cigarette smoking, fibrinogen,25 and homocysteine.55 In our study, fibrinogen was mildly associated with both measures of smoking intensity (cigarettes/day or urinary cotinine), however it was not as sensitive as the inflammatory biomarkers. Furthermore, we did not observe a significant increase in homocysteine with either measure of smoking intensity after adjustment for multiple confounding factors.
Vascular function
Cigarette smoking leads to endothelial dysfunction secondary to oxidants and free radicals in cigarette smoke which are generated by activation of the enzyme nicotinamide adenine dinucleotide phosphate oxidase and the NF-κB pathway.56 FMD is a well-established test of endothelial function. In acute inhalational exposure studies, smoking was demonstrated to cause endothelial dysfunction as assessed by FMD and maximum vasodilatation.16 In observational studies, intensity of cigarette smoking was associated with a dose-dependent impairment of FMD related to both duration and number of cigarettes smoked per day.57 In contrast we found no association between smoking intensity and FMD. Compared to the study to Poredo et al, MESA participants were older, which might explain the lack of association in our study given the age-related decline in FMD.58
Prior studies have demonstrated an association between acute smoking and increased arterial stiffness (or decreased aortic distensibility).59 Urinary cotinine is a metabolite of cigarette smoking and may better approximate immediate inhalational exposure to smoking compared to intensity, which is more reflective of shorter-term smoking exposure. Our findings of an association between urinary cotinine, but not cigarette count, and aortic distensibility further support this concept. Similar to our study, there was no association between self-reported smoking intensity and aortic distensibility among a group chronic smokers, while acute exposure to smoking resulted in decreased aortic distensibility.
Subclinical myocardial injury
We were unable to find any study that examined acute exposure to smoking and troponin levels. Smoking is known to cause temporary coronary vasospasm,12 and microvascular ischemia,60 which may result in troponin elevation. The lack of association between smoking intensity and subclinical myocardial injury in our study could be explained by the fact that high-sensitivity troponin assays were not performed in MESA,61 and participants were instructed to abstain from smoking on the morning of the MESA visit. Therefore it is possible that micro elevations in troponin related to acute smoking exposure may have dissipated by the time of the assessment in this study.
Strengths and limitations
We used self-reported exposure to smoking, which may be prone to underreporting.4 However, these self-reported values are consistently correlated with urinary cotinine levels. Certain prior studies reported better correlation between self-reported exposure to smoking and cotinine,23 which may explain why our cotinine concentrations appeared to plateau at the highest selfreported cigarette count. The moderate correlation in our study may also be explained by sex and race differences between categories of reported cigarette count which are known to influence cotinine metabolism.42 Furthermore, MESA participants were instructed to abstain from smoking on the day of the visit, which may impact cotinine levels (including regression of the highest values) and levels of some biomarkers, which have a shorter half-life (IL-6 (<6 hours),62 homocysteine (3–4 hours),63 troponin (2 hours)64 once generated in the body). MESA did not include information on nitric oxide, which is a more specific marker of vascular damage compared to other biomarkers.
There is the possibility for selection bias as participants were required to be free of CVD at baseline and thus our reported associations are most applicable to individuals without prior CVD. Information on type of cigarettes and precise time since last cigarette was not collected in MESA, thus exposure on an hourly scale could not be evaluated. This was a cross-sectional study and therefore data are reported as associations without certain causality. While our sample size consisted of 843 current smokers, our power still remained highly limited to study effect modification by sex and race/ethnicity. Analyses of cotinine were further underpowered as measurements were available in half of self-reported current smokers (n=450). Despite adjustment for multiple confounders in the association of smoking and biomarkers, there remains the possibility of residual confounding that is inherent to observational studies.
Strengths of our study included the large sample size, multiethnic makeup, and the breadth of CVD biomarkers available for analysis.
Conclusion
In this multi-ethnic study smoking intensity, modeled as cigarettes/day and urinary cotinine independent of smoking duration, was associated with early biomarkers of CVD, particularly markers of systemic inflammation. Of these, hsCRP may be the most sensitive. Knowledge of the sensitivity of biomarkers to smoking intensity may guide future epidemiologic studies of the potential harm of existing and novel tobacco products (e.g. electronic cigarettes) prior to longterm outcome studies.
Implications
This study sought to inform the Food and Drug Administration about biomarkers of cardiovascular disease risk that can be used to regulate novel tobacco products (e.g. electronic cigarettes). We demonstrated that biomarkers of inflammation (particularly high-sensitivity C-reactive protein; hsCRP) were most sensitive to smoking intensity. Based on our findings and those of other studies showing a positive association between e-cigarettes and markers of oxidative stress, it may be reasonable to measure hsCRP in future epidemiologic studies of e-cigarette users, and compare these levels to a matched control group consisting of users of combustible cigarette products and never users of tobacco products. Such data can help evaluate the potential cardiovascular toxicity of electronic cigarettes prior to the availability of data on long-term outcomes such as CVD events.
Supplementary Material
Smoking intensity was associated with hsCRP, IL-6, fibrinogen, and aortic distensibility.
hsCRP was the most sensitive biomarker of tobacco-related cardiovascular injury of 12 biomarkers tested in this study.
hsCRP may help in the study of potential cardiovascular effects of novel tobacco products.
Acknowledgments
The authors thank the other investigators, the staff, and the participants of the MESA study for their valuable contributions. A full list of participating MESA investigators and institutions can be found at http://www.mesa-nhlbi.org. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the Food and Drug Administration.
Financial support
This research was supported by contracts N01-HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168 and N01-HC-95169 from the National Heart, Lung, and Blood Institute and by grants UL1-TR-000040 and UL1-TR-001079 from NCRR.
Exam 1 and 5 Urinary Cotinine measurements are available to the MESA study courtesy of MESA Lung contract HL077612.
Research reported in this work was supported by grant number 5P50HL120163 from the National Heart, Lung and Blood Institute (NHLBI) and FDA Center for Tobacco Products (CTP).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declared they do not have anything to disclose regarding no conflict of interest with respect to this manuscript.
References
- 1.WHO. WHO report on the global tobacco epidemic. 2011 http://www.who.int/tobacco/global_report/2011/en/. Accessed November 14, 2015.
- 2.The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General. http://www-ncbi-nlm-nih-gov.ezproxy.welch.jhmi.edu/pubmed/?term=.The+Health+Consequences+of+Smoking%25E2%2580%259450+Years+of+Progress%253A+A+Report+of+the+Surgeon+General. Accessed May 7, 2014.
- 3.Ezzati M, Henley SJ, Thun MJ, Lopez AD. Role of smoking in global and regional cardiovascular mortality. Circulation. 2005;112(4):489–497. doi: 10.1161/CIRCULATIONAHA.104.521708. [DOI] [PubMed] [Google Scholar]
- 4.McEvoy JW, Nasir K, DeFilippis AP, et al. Relationship of Cigarette Smoking With Inflammation and Subclinical Vascular Disease: The Multi-Ethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2015 Mar; doi: 10.1161/ATVBAHA.114.304960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McEvoy JW, Blaha MJ, DeFilippis AP, et al. Cigarette Smoking and Cardiovascular Events: Role of Inflammation and Subclinical Atherosclerosis: The Multiethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2015 Jan; doi: 10.1161/ATVBAHA.114.304562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pell JP, Haw S, Cobbe S, et al. Smoke-free legislation and hospitalizations for acute coronary syndrome. N Engl J Med. 2008;359(5):482–491. doi: 10.1056/NEJMsa0706740. [DOI] [PubMed] [Google Scholar]
- 7.Rosenberg L, Kaufman DW, Helmrich SP, Shapiro S. The risk of myocardial infarction after quitting smoking in men under 55 years of age. N Engl J Med. 1985;313(24):1511–1514. doi: 10.1056/NEJM198512123132404. [DOI] [PubMed] [Google Scholar]
- 8.Dobson AJ, Alexander HM, Heller RF, Lloyd DM. How soon after quitting smoking does risk of heart attack decline? J Clin Epidemiol. 1991;44(11):1247–1253. doi: 10.1016/0895-4356(91)90157-5. [DOI] [PubMed] [Google Scholar]
- 9.Nance R, Delaney J, McEvoy JW, et al. Smoking intensity [pack/day] is a better measure than pack-years or smoking status for modeling cardiovascular disease outcomes. J Clin Epidemiol. 2016 Oct; doi: 10.1016/j.jclinepi.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koczulla A-R, Noeske S, Herr C, et al. Acute and chronic effects of smoking on inflammation markers in exhaled breath condensate in current smokers. Respiration. 2010;79(1):61–67. doi: 10.1159/000245325. [DOI] [PubMed] [Google Scholar]
- 11.van der Vaart H, Postma DS, Timens W, et al. Acute effects of cigarette smoking on inflammation in healthy intermittent smokers. Respir Res. 2005;6:22. doi: 10.1186/1465-9921-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciftci O, Caliskan M, Gullu H, et al. Acute effects of smoking light cigarettes on coronary microvascular functions. Clin Cardiol. 2009;32(4):210–214. doi: 10.1002/clc.20343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otsuka R. Acute Effects of Passive Smoking on the Coronary Circulation in Healthy Young Adults. JAMA. 2001;286(4):436. doi: 10.1001/jama.286.4.436. [DOI] [PubMed] [Google Scholar]
- 14.Kubozono T, Miyata M, Ueyama K, et al. Acute and chronic effects of smoking on arterial stiffness. Circ J. 2011;75(3):698–702. doi: 10.1253/circj.cj-10-0552. http://www.ncbi.nlm.nih.gov/pubmed/21187657. Accessed October 28, 2015. [DOI] [PubMed] [Google Scholar]
- 15.LEVINE PH. An Acute Effect of Cigarette Smoking on Platelet Function: A Possible Link Between Smoking and Arterial Thrombosis. Circulation. 1973;48(3):619–623. doi: 10.1161/01.CIR.48.3.619. [DOI] [PubMed] [Google Scholar]
- 16.Taha NM, Abdel Wahab MA, Amin AS. Acute effects of cigarette smoking in habitual smokers, a focus on endothelial function. Egypt Hear J. 2013;65(4):275–279. doi: 10.1016/j.ehj.2012.09.003. [DOI] [Google Scholar]
- 17.Sassalos K, Vlachopoulos C, Alexopoulos N, Gialernios T, Aznaouridis K, Stefanadis C. The acute and chronic effect of cigarette smoking on the elastic properties of the ascending aorta in healthy male subjects. Hellenic J Cardiol. 47(5):263–268. http://www.ncbi.nlm.nih.gov/pubmed/17134061. Accessed October 29, 2015. [PubMed] [Google Scholar]
- 18.Levitzky YS, Guo C-Y, Rong J, et al. Relation of smoking status to a panel of inflammatory markers: The Framingham offspring. Atherosclerosis. 2008;201(1):217–224. doi: 10.1016/j.atherosclerosis.2007.12.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Avila-Tang E, Al-Delaimy WK, Ashley DL, et al. Assessing secondhand smoke using biological markers. Tob Control. 2013;22(3):164–171. doi: 10.1136/tobaccocontrol-2011-050298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones MR, Magid HS, Al-Rifai M, et al. Secondhand Smoke Exposure and Subclinical Cardiovascular Disease: The Multi-Ethnic Study of Atherosclerosis. J Am Heart Assoc. 2016;5(12):e002965. doi: 10.1161/JAHA.115.002965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bild DE, Bluemke DA, Burke GL, et al. Multi-ethnic study of atherosclerosis: objectives and design. Am J Epidemiol. 2002;156(9):871–881. doi: 10.1093/aje/kwf113. http://www.ncbi.nlm.nih.gov/pubmed/12397006. Accessed August 19, 2014. [DOI] [PubMed] [Google Scholar]
- 22.Donohue KM, Hoffman EA, Baumhauer H, et al. Asthma and lung structure on computed tomography: the Multi-Ethnic Study of Atherosclerosis Lung Study. J Allergy Clin Immunol. 2013;131(2):361-8–11. doi: 10.1016/j.jaci.2012.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDonald SP, Maguire GP, Hoy WE. Validation of self-reported cigarette smoking in a remote Australian Aboriginal community. Aust N Z J Public Health. 2003;27(1):57–60. doi: 10.1111/j.1467-842X.2003.tb00380.x. [DOI] [PubMed] [Google Scholar]
- 24.Etter JF, Perneger TV. Measurement of self reported active exposure to cigarette smoke. J Epidemiol Community Health. 2001;55(9):674–680. doi: 10.1136/jech.55.9.674. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1731967&tool=pmcentrez&rendertype=abstract. Accessed December 29, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bakhru A, Erlinger TP. Smoking cessation and cardiovascular disease risk factors: results from the Third National Health and Nutrition Examination Survey. PLoS Med. 2005;2(6):e160. doi: 10.1371/journal.pmed.0020160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blaha MJ, Budoff MJ, DeFilippis AP, et al. Associations between C-reactive protein, coronary artery calcium, and cardiovascular events: implications for the JUPITER population from MESA, a population-based cohort study. Lancet. 2011;378(9792):684–692. doi: 10.1016/S0140-6736(11)60784-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Damluji AA, Ramireddy A, Al-Damluji MS, et al. Association between anti-human heat shock protein-60 and interleukin-2 with coronary artery calcium score. Heart. 2015;101(6):436–441. doi: 10.1136/heartjnl-2014-306649. [DOI] [PubMed] [Google Scholar]
- 28.Caruso C, Candore G, Cigna D, Colucci AT, Modica MA. Biological significance of soluble IL-2 receptor. Mediators Inflamm. 1993;2(1):3–21. doi: 10.1155/S0962935193000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawut SM, Barr RG, Johnson WC, et al. Matrix metalloproteinase-9 and plasminogen activator inhibitor-1 are associated with right ventricular structure and function: the MESA-RV Study. Biomarkers. 2010;15(8):731–738. doi: 10.3109/1354750X.2010.516455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferrari R, Bachetti T, Confortini R, et al. Tumor necrosis factor soluble receptors in patients with various degrees of congestive heart failure. Circulation. 1995;92(6):1479–1486. doi: 10.1161/01.cir.92.6.1479. http://www.ncbi.nlm.nih.gov/pubmed/7664430. Accessed December 9, 2015. [DOI] [PubMed] [Google Scholar]
- 31.Harhay MO, Tracy RP, Bagiella E, et al. Relationship of CRP, IL-6, and fibrinogen with right ventricular structure and function: the MESA-Right Ventricle Study. Int J Cardiol. 2013;168(4):3818–3824. doi: 10.1016/j.ijcard.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neuhaus J, Jacobs DR, Baker JV, et al. Markers of inflammation, coagulation, and renal function are elevated in adults with HIV infection. J Infect Dis. 2010;201(12):1788–1795. doi: 10.1086/652749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perng W, Villamor E, Shroff MR, et al. Dietary intake, plasma homocysteine, and repetitive element DNA methylation in the Multi-Ethnic Study of Atherosclerosis (MESA) Nutr Metab Cardiovasc Dis. 2014;24(6):614–622. doi: 10.1016/j.numecd.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zemaitis P, Liu K, Jacobs DR, et al. Cumulative systolic BP and changes in urine albumin-to-creatinine ratios in nondiabetic participants of the multi-ethnic study of atherosclerosis. Clin J Am Soc Nephrol. 2014;9(11):1922–1929. doi: 10.2215/CJN.02450314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Redheuil A, Wu CO, Kachenoura N, et al. Proximal aortic distensibility is an independent predictor of all-cause mortality and incident CV events: the MESA study. J Am Coll Cardiol. 2014;64(24):2619–2629. doi: 10.1016/j.jacc.2014.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blaha MJ, Budoff MJ, Rivera JJ, et al. Relationship of carotid distensibility and thoracic aorta calcification: multi-ethnic study of atherosclerosis. Hypertension. 2009;54(6):1408–1415. doi: 10.1161/HYPERTENSIONAHA.109.138396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gamble G, Zorn J, Sanders G, MacMahon S, Sharpe N. Estimation of arterial stiffness, compliance, and distensibility from M-mode ultrasound measurements of the common carotid artery. Stroke. 1994;25(1):11–16. doi: 10.1161/01.str.25.1.11. http://www.ncbi.nlm.nih.gov/pubmed/8266356. Accessed February 19, 2016. [DOI] [PubMed] [Google Scholar]
- 38.Yeboah J, Folsom AR, Burke GL, et al. Predictive value of brachial flow-mediated dilation for incident cardiovascular events in a population-based study: the multi-ethnic study of atherosclerosis. Circulation. 2009;120(6):502–509. doi: 10.1161/CIRCULATIONAHA.109.864801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daniels LB, Clopton P, deFilippi CR, et al. Serial measurement of N-terminal pro-B-type natriuretic peptide and cardiac troponin T for cardiovascular disease risk assessment in the Multi-Ethnic Study of Atherosclerosis (MESA) Am Heart J. 2015 Sep; doi: 10.1016/j.ahj.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bapat A, Zhang Y, Post WS, et al. Relation of Physical Activity and Incident Atrial Fibrillation (from the Multi-Ethnic Study of Atherosclerosis) Am J Cardiol. 2015;116(6):883–888. doi: 10.1016/j.amjcard.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cuzick J. A Wilcoxon-type test for trend. Stat Med. 4(1):87–90. doi: 10.1002/sim.4780040112. http://www.ncbi.nlm.nih.gov/pubmed/3992076. Accessed November 6, 2015. [DOI] [PubMed] [Google Scholar]
- 42.Benowitz NL, Hukkanen J, Jacob P. Nicotine chemistry, metabolism, kinetics and biomarkers. Handb Exp Pharmacol. 2009;(192):29–60. doi: 10.1007/978-3-540-69248-5_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pope CA, Burnett RT, Krewski D, et al. Cardiovascular mortality and exposure to airborne fine particulate matter and cigarette smoke: shape of the exposure-response relationship. Circulation. 2009;120(11):941–948. doi: 10.1161/CIRCULATIONAHA.109.857888. [DOI] [PubMed] [Google Scholar]
- 44.Schane RE, Ling PM, Glantz SA. Health effects of light and intermittent smoking: a review. Circulation. 2010;121(13):1518–1522. doi: 10.1161/CIRCULATIONAHA.109.904235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaptoge S, Di Angelantonio E, Pennells L, et al. C-reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med. 2012;367(14):1310–1320. doi: 10.1056/NEJMoa1107477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gonçalves RB, Coletta RD, Silvério KG, et al. Impact of smoking on inflammation: overview of molecular mechanisms. Inflamm Res. 2011;60(5):409–424. doi: 10.1007/s00011-011-0308-7. [DOI] [PubMed] [Google Scholar]
- 47.Petrescu F, Voican SC, Silosi I. Tumor necrosis factor-alpha serum levels in healthy smokers and nonsmokers. Int J Chron Obstruct Pulmon Dis. 2010;5:217–222. doi: 10.2147/copd.s8330. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2921689&tool=pmcentrez&rendertype=abstract. Accessed February 24, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barbieri SS, Zacchi E, Amadio P, et al. Cytokines present in smokers’ serum interact with smoke components to enhance endothelial dysfunction. Cardiovasc Res. 2011;90(3):475–483. doi: 10.1093/cvr/cvr032. [DOI] [PubMed] [Google Scholar]
- 49.Drost EM, Selby C, Bridgeman MM, MacNee W. Decreased leukocyte deformability after acute cigarette smoking in humans. Am Rev Respir Dis. 1993;148(5):1277–1283. doi: 10.1164/ajrccm/148.5.1277. [DOI] [PubMed] [Google Scholar]
- 50.Patiar S, Slade D, Kirkpatrick U, McCollum CN. Smoking causes a dose-dependent increase in granulocyte-bound L-selectin. Thromb Res. 2002;106(1):1–6. doi: 10.1016/s0049-3848(02)00082-8. http://www.ncbi.nlm.nih.gov/pubmed/12165281. Accessed October 28, 2015. [DOI] [PubMed] [Google Scholar]
- 51.Caponnetto P, Russo C, Di Maria A, et al. Circulating endothelial-coagulative activation markers after smoking cessation: a 12-month observational study. Eur J Clin Invest. 2011;41(6):616–626. doi: 10.1111/j.1365-2362.2010.02449.x. [DOI] [PubMed] [Google Scholar]
- 52.Nowak J, Murray JJ, Oates JA, FitzGerald GA. Biochemical evidence of a chronic abnormality in platelet and vascular function in healthy individuals who smoke cigarettes. Circulation. 1987;76(1):6–14. doi: 10.1161/01.cir.76.1.6. http://www.ncbi.nlm.nih.gov/pubmed/3297389. Accessed February 24, 2016. [DOI] [PubMed] [Google Scholar]
- 53.Sambola A, Osende J, Hathcock J, et al. Role of risk factors in the modulation of tissue factor activity and blood thrombogenicity. Circulation. 2003;107(7):973–977. doi: 10.1161/01.cir.0000050621.67499.7d. http://www.ncbi.nlm.nih.gov/pubmed/12600909. Accessed February 24, 2016. [DOI] [PubMed] [Google Scholar]
- 54.Lupia E, Bosco O, Goffi A, et al. Thrombopoietin contributes to enhanced platelet activation in cigarette smokers. Atherosclerosis. 2010;210(1):314–319. doi: 10.1016/j.atherosclerosis.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 55.Chen S, Wu P, Zhou L, Shen Y, Li Y, Song H. Relationship between increase of serum homocysteine caused by smoking and oxidative damage in elderly patients with cardiovascular disease. Int J Clin Exp Med. 2015;8(3):4446–4454. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4443202&tool=pmcentrez&rendertype=abstract. Accessed December 9, 2015. [PMC free article] [PubMed] [Google Scholar]
- 56.Takac I, Schröder K, Brandes RP. The Nox family of NADPH oxidases: friend or foe of the vascular system? Curr Hypertens Rep. 2012;14(1):70–78. doi: 10.1007/s11906-011-0238-3. [DOI] [PubMed] [Google Scholar]
- 57.Poredos P, Orehek M, Tratnik E. Smoking is associated with dose-related increase of intima-media thickness and endothelial dysfunction. Angiology. 1999;50(3):201–208. doi: 10.1177/000331979905000304. http://www.ncbi.nlm.nih.gov/pubmed/10088799. Accessed October 29, 2015. [DOI] [PubMed] [Google Scholar]
- 58.Skaug E-A, Aspenes ST, Oldervoll L, et al. Age and gender differences of endothelial function in 4739 healthy adults: the HUNT3 Fitness Study. Eur J Prev Cardiol. 2013;20(4):531–540. doi: 10.1177/2047487312444234. [DOI] [PubMed] [Google Scholar]
- 59.Doonan RJ, Hausvater A, Scallan C, Mikhailidis DP, Pilote L, Daskalopoulou SS. The effect of smoking on arterial stiffness. Hypertens Res. 2010;33(5):398–410. doi: 10.1038/hr.2010.25. [DOI] [PubMed] [Google Scholar]
- 60.Miyazaki T, Ashikaga T, Ohigashi H, Komura M, Kobayashi K, Isobe M. Impact of smoking on coronary microcirculatory resistance in patients with coronary artery disease. Int Heart J. 2015;56(1):29–36. doi: 10.1536/ihj.14-189. [DOI] [PubMed] [Google Scholar]
- 61.Al-Otaiby MA, Al-Amri HS, Al-Moghairi AM. The clinical significance of cardiac troponins in medical practice. J Saudi Hear Assoc. 2011;23(1):3–11. doi: 10.1016/j.jsha.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma Concentration of Interleukin-6 and the Risk of Future Myocardial Infarction Among Apparently Healthy Men. Circulation. 2000;101(15):1767–1772. doi: 10.1161/01.CIR.101.15.1767. [DOI] [PubMed] [Google Scholar]
- 63.Guttormsen AB, Mansoor AM, Fiskerstrand T, Ueland PM, Refsum H. Kinetics of plasma homocysteine in healthy subjects after peroral homocysteine loading. Clin Chem. 1993;39(7):1390–1397. http://www.ncbi.nlm.nih.gov/pubmed/8330395. Accessed February 24, 2016. [PubMed] [Google Scholar]
- 64.White HD. Pathobiology of troponin elevations: do elevations occur with myocardial ischemia as well as necrosis? J Am Coll Cardiol. 2011;57(24):2406–2408. doi: 10.1016/j.jacc.2011.01.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.