

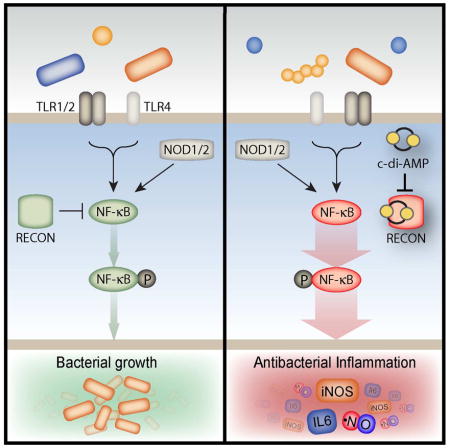
SUMMARY
Bacterial and host cyclic dinucleotides (cdNs) mediate cytosolic immune responses through the STING signaling pathway, though evidence suggests alternative pathways exist. We used cdN-conjugated beads to biochemically isolate host receptors for bacterial cdNs, and identified the oxidoreductase RECON. High-affinity cdN binding inhibited RECON enzyme activity by simultaneously blocking the substrate and co-substrate sites, as revealed by structural analyses. During bacterial infection of macrophages, RECON antagonized STING activation by acting as a molecular sink for cdNs. Bacterial infection of hepatocytes, which do not express STING, revealed that RECON negatively regulates NF-κB activation. Loss of RECON activity, via genetic ablation or inhibition by cdNs, resulted in increased NF-κB activation and reduced bacterial survival, suggesting that cdN inhibition of RECON promotes a proinflammatory, antibacterial state that is distinct from the anti-viral state associated with STING activation. Thus, RECON functions as a cytosolic pattern recognition receptor specific for bacterial cdNs, shaping inflammatory gene activation via its effects on STING and NF-κB.
Keywords: Cyclic dinucleotides, cyclic di-AMP, pattern recognition receptor, STING, aldo-keto reductase, AKR1C13, RECON, NF-κB, Listeria monocytogenes
eTOC Blurb
In this report, McFarland et al. identify a new cytosolic pattern recognition receptor, RECON, that specifically detects bacterial cyclic dinucleotides. Unlike known ligand-PRR interactions, cdN binding to RECON inhibits the enzymatic activity of this sensor. Loss of RECON activity promotes NF-κB activation and shapes infection outcome.
INTRODUCTION
Host cells have evolved a sophisticated arsenal of germ-line encoded pattern recognition receptors (PRRs) that detect a vast array of microorganisms in distinct tissues and cellular compartments. Cytoplasmic sensors monitor for microbes that gain access to the host cell cytosol following breach of the plasma membrane or intracellular invasion. This sensing is achieved through detection of invariant molecular patterns associated with microorganisms or cell stress responses that indicate the presence of a pathogen. Engagement of a PRR by a microbial product triggers a signal transduction pathway that ultimately shapes cellular function to regain sterility. Many of the cellular changes are mediated through activation of transcription factors, such as nuclear factor κB (NF-κB) and/or interferon regulatory factors (IRFs) (Iwasaki and Medzhitov, 2010). Classically, PRR engagement results in recruitment and activation of kinase activity that serves as the driving force of these signaling cascades. For example, retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), which recognize viral and bacterial RNA species in the cytoplasm, activate the IKK kinase complex and TANK-binding kinase 1 (TBK1), leading to NF-κB and IRF3 activation, respectively (Kawai and Akira, 2008). The PRR STING (stimulator of interferon genes) mediates nucleic acid sensing by binding cyclic dinucleotides (Burdette et al., 2011), also resulting in activation of the TBK1-IRF3 signaling axis.
Cyclic dinucleotides (cdNs) have recently emerged as central players in the innate immune response to bacterial and viral infections. Mammals synthesize 2′3′-cGAMP downstream of cytosolic DNA detection of bacterial and viral origin (Cai et al., 2014). Bacteria produce c-di-GMP, c-di-AMP and 3′3′-cGAMP as second messengers that regulate a variety of cellular processes (Davies et al., 2012; Kalia et al., 2013). C-di-GMP regulates motility and biofilm formation while 3′3′-cGAMP regulates metal-reducing activity and host colonization in Geobacter and Vibrio cholera, respectively (Davies et al., 2012; Kellenberger et al., 2015; Mills et al., 2011). C-di-AMP, the only essential cdN, regulates bacterial growth, cell wall homeostasis and central metabolism (Corrigan and Grundling, 2013; Sureka et al., 2014; Witte et al., 2013). C-di-AMP is also implicated in STING-dependent type I interferon (IFN) induction during infection with the intracellular pathogens Listeria monocytogenes (Lm), Chlamydia spp. and Mycobacterium tuberculosis (Mtb) (Barker et al., 2013; Burdette et al., 2011; Dey et al., 2015; Romling, 2008; Sauer et al., 2011; Woodward et al., 2010; Yang et al., 2014). Given the widespread conservation and central physiological function of these metabolites, cdNs are likely to play a role in a diverse array of bacterial-host interactions.
Multiple PRRs exist that sense similar nucleic acid species. For example, several cytosolic PRRs, including DDX41, DHX9, DHX36, IFI16, DAI and cGAS, have been implicated in dsDNA detection, although for some of these receptors, such as IFI16, their ability to bind dsDNA does not necessarily translate into in vivo importance during infection (Cai et al., 2014; Desmet and Ishii, 2012; Gray et al., 2016). Although PRR activation by cdNs is a relatively recent discovery, our understanding of cdN detection by host cells lags significantly behind other nucleic acid species. STING and DDX41 are the only two known PRRs that bind cdNs and downstream responses almost exclusively result in the induction of type I IFN. Interestingly, STING-deficient mice do not exhibit altered bacterial burden or survival during infection with cdN-producing bacteria, such as Lm or Mtb (Collins et al., 2015; Sauer et al., 2011), suggesting that activation of STING does not significantly influence the innate response to infection with these pathogens. However, mice infected with Lm or Mtb producing altered levels of c-di-AMP exhibit profound differences in bacterial loads and survival in an IFN-independent manner, with increased c-di-AMP production correlating with increased host resistance and vice versa (Crimmins et al., 2008; Dey et al., 2015; Yamamoto et al., 2012). The discordant phenotypes between STING-deficient mice and c-di-AMP production suggests the existence of additional host receptors that engage bacterial cdNs.
In this study, we aimed to identify host proteins that interact with bacterial cdNs. We found that the oxidoreductase, aldo-keto reductase family 1, member C13 (AKR1C13), which we refer to as RECON (REductase COntrolling NF-κB) specifically bound to the bacterial nucleotides c-di-AMP and 3′3′-cGAMP, but not mammalian 2′3′-cGAMP. RECON bound c-di-AMP with high affinity, altering STING activation during Lm infection by acting as an intracellular sink for this cdN. In addition, c-di-AMP bound to RECON in the substrate and cosubstrate binding sites, thereby inhibiting its enzyme activity. We found that loss of RECON enzyme activity increased NF-κB activity that resulted in augmented inflammatory gene activation and reduced bacterial survival. Taken together, our data reveal a role for RECON in the shaping of an antibacterial state distinct from the antiviral type I IFN response mediated by the cdN sensing axis comprised of STING and DDX41.
RESULTS
Bacterial cyclic dinucleotides bind and inhibit the oxidoreductase RECON
To identify host proteins that interact with bacterial cdNs, we performed pulldowns from lysates of mouse spleen, liver and lung with c-di-AMP sepharose (Figures 1A and S1A). The oxidoreductase, aldo-keto reductase family 1, member C13 (AKR1C13; protein herein referred to as RECON) was identified by mass spectrometry as the highly abundant protein in c-di-AMP liver pull-downs. Biochemical characterization using purified recombinant protein confirmed that RECON bound to c-di-AMP (Figures 1B and S1B). The affinity of this interaction was significantly higher than that observed between c-di-AMP and STING, with RECON binding to c-di-AMP 16 times more tightly. The binding of c-di-AMP by RECON was comparable to the affinity of STING for 2′3′-cGAMP (Figure 1C), consistent with STING specificity for the endogenously produced 2′3′-cGAMP nucleotide and RECON specificity for exogenously produced bacterial c-di-AMP.
Figure 1. Bacterial cyclic dinucleotides bind and inhibit the oxidoreductase RECON.

(A) SDS-page analysis of pull-downs from mouse organ lysates with c-di-AMP (+) or control (−) sepharose.
(B) Binding titration of 32P-c-di-AMP with RECON or STING. Kd values are indicated.
(C) Binding titration of 32P-2′3′-cGAMP or 32P-c-di-AMP with STING.
(D) Binding of RECON with 32P-c-di-AMP in the presence of competing unlabeled nucleotides (200 μM).
(E) Binding of RECON or STING with 32P-c-di-AMP in the presence of competing unlabeled cyclic dinucleotides, each at 400 μM concentration.
(F) Michaelis–Menten kinetics of RECON with the substrate geraniol and cosubstrate NAD+ in the presence or absence of 1 μM c-di-AMP.
(G) RECON enzyme activity in the presence of increasing concentrations of the indicated cdNs. Substrate was 9,10-PQ and cosubstrate was NADPH.
Data are plotted as percent of activity without any cdN added. Data are representative of two (A, C, D, E, G) or three (B, F) independent experiments with similar results. In all panels, error bars represent ± SEM of technical replicates. See also Figure S1.
Only the addition of cold c-di-AMP, but not a variey of other cold nucleotides, could interfere with 32P-c-di-AMP binding to RECON (Figure 1D). By competing all of the known cdNs against 32P-c-di-AMP, we found that RECON also bound 3′3′-cGAMP, but not c-di-GMP or host-derived 2′3′-cGAMP, whereas all cold cdNs competed off 32P-c-di-AMP from STING (Figure 1E). Therefore, RECON specifically binds to bacterial cdNs and not the host-derived 2′3′-cGAMP. RECON exhibits both dehydrogenase and reductase activity and can turn over a broad spectrum of substrates (Endo et al., 2007; Ikeda et al., 1999). We found that in the presence of c-di-AMP RECON’s enzyme activity was completely inhibited (Figures 1F and S1C). Consistent with our nucleotide binding studies, bacterial-derived 3′3′-cGAMP, but not host-derived 2′3′-cGAMP, also inhibited RECON activity (Figure 1G). Both c-di-AMP and 3′3′-cGAMP inhibited RECON activity by 50% at concentrations near 300 nM, even in the presence of 200 μM NADPH.
Binding mode of c-di-AMP in RECON
We determined the crystal structure of the c-di-AMP RECON complex at 1.5 Å resolution. The atomic model had good agreement with the crystallographic data and the expected bond lengths, bond angles and other geometric parameters (Table S1). RECON adopts the TIM barrel fold with eight parallel β-strands (β1-β8) surrounded by 8 crossover α-helixes (α1-α8) (Figure 2A) common to aldo-keto reductase superfamily members (Jez et al., 1997). RECON also has a two-stranded anti-parallel β-sheet (β1 and β2) that caps the N-terminal end of the central barrel, and two additional helixes H1 (inserted between β7 and α8) and H2 (after α8) located on the side of the structure (Figure 2A). The active site of the enzyme is located at the top of the structure, near the C-terminal end of the central barrel, and several large loops (loops A, B and C, and loop β1α1) form the NAD+ and substrate binding sites (Figure 2B). Although the structure of the c-di-AMP complex exhibits a very small rms distance of 0.3 Å among equivalent Cα atoms in the NAD+ complex (PDB entry 3LN3) (Figure 2B), important differences between the two structures exist in the active site.
Figure 2. C-di-AMP blocks the NAD+ and substrate binding sites of RECON.

(A) Schematic drawing of structure of RECON (green) in complex with c-di-AMP (magenta, stick models). The view is down the central barrel of the structure. The two Lys to Ala mutation sites are labeled.
(B) Overlay of structure of RECON (green) in complex with c-di-AMP (magenta, stick models) and that of RECON (gray) in complex with NAD+ (cyan, stick models) (PDB entry 3LN3). The view is related to that of panel A by a 90° rotation around the horizontal axis. The bound position of the progesterone substrate (Prog) to AKR1C1 is also shown (orange, stick models) (Couture et al., 2003).
(C) Side views of (A) with two residue-mutations indicated. Omit Fo–Fc electron density for c-di-AMP at 1.5 Å resolution, contoured at 2.5σ.
(D) Molecular surface of RECON near c-di-AMP, colored by electrostatic potential.
(E) Comparison of the binding modes of c-di-AMP (magenta) and NAD+ (cyan) to RECON. All structure figures were produced with PyMOL (www.pymol.org).
(F) Detailed interactions between c-di-AMP and RECON. Hydrogen-bonding interactions are indicated with dashed lines (red). Residues involved in interactions with c-di-AMP are labeled. A water molecule is shown as a red sphere and labeled W.
(G) Overlay of the binding sites of NAD+ (cyan) and c-di-AMP (magenta) near the first nucleotide (AMP1) of c-di-AMP.
(H) Overlay of the binding sites of NAD+ (cyan) and c-di-AMP (magenta) near the second nucleotide (AMP2) of c-di-AMP.
See also Table S1.
Clear electron density was observed for c-di-AMP based on the crystallographic analysis at 1.5 Å resolution (Figure 2C). The compound is an extended conformation where the two adenine bases adopt anti confirmations in nearly the same plane as the phosphodiester ring. The c-di-AMP binding site is in a deep crevice at the top of the central barrel (Figures 2A and 2D). One AMP molecule (AMP1) of c-di-AMP has essentially the same position as the AMP portion of the NAD+ cosubstrate, while the other AMP (AMP2) has small overlap with the nicotinamide ring (Figure 2E) and is positioned mostly in the substrate-binding site (Figure 2B) (Couture et al., 2003).
The N6 and N7 atoms of the AMP1 adenine base are recognized by hydrogen-bonding to the side chain of Asn280 (helix α8), and N6 also interacts with the side chain of Glu279 (α8) (Figure 2F). The adenine base is flanked by the side chain of Glu276 (α8) on one face and those of Leu219 (loop B) and Ala253 (α7) on the other. The phosphate group of AMP1 hydrogen-bonds with the main-chain amides of Leu219 and Thr221 (loop B). The adenine base of AMP2 is π-stacked with the side chain of Tyr24 (β1α1 loop) on one face and is flanked by the side chains of Tyr216 (β7) and Leu306 (loop C) on the other (Figure 2F). This base is pointed toward the catalytic tetrad of the enzyme, composed of Asp50, Tyr55, Lys84, and His117. The 2′ hydroxyl of the ribose is hydrogen-bonded to the main-chain amide of Gly217 (loop B). Several water molecules are in and near the c-di-AMP binding site, including one which mediates hydrogen-bonding interactions between the N1 atom of AMP2 and the catalytic Tyr55 and His117 residues (Figure 2F).
Compared to the structure of the NAD+ complex, most residues in the AMP1 binding site assume the same conformation (Figure 2G). In contrast, substantial conformational differences are observed for residues in the AMP2 binding site (Figure 2H). Tyr24 and the β1α1 loop move closer by ~1 Å to the adenine base, enabling π stacking between the two aromatic moieties. Residues 222-231 of loop B are disordered in the c-di-AMP complex due to clashes of Gln222 and Tyr224 with AMP2. The side chain of Gln270 assumes a different rotamer in the c-di-AMP complex to avoid steric clash with the phosphate group of AMP2 (Figure 2G). Even though c-di-AMP uses the NAD+ binding site and assumes a conformation of similar length as NAD+, only one nucleotide has close overlap with NAD+. The other nucleotide deviates significantly from the NAD+ envelope, exploring a different region of the target protein. This should confer selectivity to the compound, so that it will not inhibit all NAD+/NADP+ binding proteins.
Loss of RECON during L. monocytogenes infection or PRR stimulation augments inflammatory gene expression
To interrogate the effects of RECON on host innate immune responses during infection, we characterized the transcriptional response to Lm, an organism that secretes c-di-AMP and triggers STING-dependent type I IFN expression during infection of macrophages (Figure S2A) (Burdette et al., 2011; Sauer et al., 2011; Woodward et al., 2010). We measured the expression of IFN-β (gene Ifnb1) 4 hours post-infection (hpi) in immortalized bone marrow-derived macrophages (iBMDMs) that overexpressed Akr1c13 (oe-Akr1c13), had reduced levels of Akr1c13 expression via RNAi using a short hairpin RNA (sh-Akr1c13), or were transduced with control scrambled short hairpin RNA (sh-Control) and observed an inverse correlation with Akr1c13 expression, whereby high amounts of Ifnb1 transcripts correlated with low amounts of Akr1c13 transcripts and vice versa (Figure 3A and 3B and S2B). Given that RECON binds c-di-AMP with higher affinity than STING, these observations suggest that RECON antagonizes STING activation by decreasing the bioavailability of c-di-AMP during infection.
Figure 3. Loss of RECON during L. monocytogenes infection or PRR stimulation augments inflammatory gene expression.

(A) Expression of Akr1c13 mRNA in iBMDM cell lines with altered Akr1c13 expression.
(B) Expression of Ifnb1 mRNA in iBMDM cell lines with altered Akr1c13 expression infected with Lm for 4 h.
(C) TaqMan array expression of Ccl5, Cxcl10, Cxcl11, Il1b and Nos2 mRNA in iBMDM cell lines with altered Akr1c13 expression infected with Lm for 4 h.
(D) Expression of Nos2 mRNA at 4 hpi with Lm.
(E) Nitrite levels in iBMDM supernatants 20 hpi with Lm.
(F) Expression of Isg15, Ifnb1 and Nos2 mRNA in sh-Control (grey bars) or sh-Akr1c13 (black bars) iBMDM cell lines infected with Lm for 4 h in the presence (+) or absence (−) of an IFNAR1 blocking antibody (Ab).
(G, H) Expression of Ifnb1 and Nos2 in iBMDMs treated with Pam3CSK4 (G) or Tri-DAP (H) for 4 h.
Data are representative of two or more independent experiments with similar results. qRT-PCR data were performed in technical duplicates or triplicates and were normalized to uninfected/unstimulated sh-Control iBMDM cells. In all panels, error bars represent ± SEM of technical replicates. See also Figure S2 and S7.
We ran a focused TaqMan mouse immune panel on sh-Control, sh-Akr1c13 and oe-Akr1c13 iBMDMs infected with Lm for 4 hours. Ccl5, Cxcl10, Cxcl11, Il1b and Nos2 were all induced during Lm infection, and, similar to Ifnb1, there was a strong inverse correlation between their expression and Akr1c13 levels (Figure 3C). Thus, loss of RECON is associated with increased inflammatory gene expression. Clc5 (RANTES), Cxcl10 (IP-10) and Cxcl11 (I-TAC) are potent chemoattractants for activated T cells, IL-1β is a proinflammatory cytokine that activates innate and adaptive cellular responses, while Nos2 encodes for inducible nitric oxide synthase (iNOS) (Jacobs and Ignarro, 2003; Murphy et al., 2000). Akr1c13 expression itself was not significantly altered during infection, therefore transcriptional regulation of Akr1c13 does not appear to be involved in the response to Lm in iBMDMs (Figure S2C).
The RECON-dependent expression of Nos2 was of interest, considering the important role of nitric oxide (NO) in host defense against pathogenic bacteria (Fang, 1997). The effect of RECON on Nos2 transcription resulted in differences in NO production, as measured by Griess assay (Figures 3D and 3E). Since feedback from IFN-β is known to enhance induction of Toll-like receptor (TLR)-stimulated genes, including iNOS (Jacobs and Ignarro, 2003), we also examined inflammatory gene expression during Lm infection in the presence of an IFNAR1 blocking antibody (Figures 3F and S2D) and following treatment with the TLR2 ligand Pam3CSK4 or NOD1 ligand Tri-DAP (Figures 3G, 3H and S2E). In all cases, we observed that loss of RECON augmented Nos2 expression, even when Ifnb1 expression was similar across the cell lines. Therefore, RECON affects expression of Nos2 and other inflammatory genes through a STING- and IFN-independent pathway.
Bacterial cyclic dinucleotide responses are controlled by RECON and elicit STING-independent host responses
Engagement of TLRs by microbe-associated molecular patterns (MAMPs) leads to MyD88-dependent gene expression. We studied the contribution of this pathway as well as STING activation in primary BMDMs. Infection of WT, Sting−/−, or Myd88/Tlr3−/− primary BMDMs resulted in expression of Ccl5, Cxcl10, Cxcl11 and Ifnb1 that was almost entirely STING-dependent (Figure 4A). We assessed the effect of RECON on the direct ability of cdNs to activate the STING-dependent genes in macrophages. Our biochemical data demonstrated that RECON only binds to bacterial cdNs, c-di-AMP and 3′3′-cGAMP, and not the host-derived 2′3′-cGAMP (Figures 1E). We did not observe a significant difference between the effects of c-di-AMP and 2′3′-cGAMP in sh-Control iBMDMs. However, oe-Akr1c13 iBMDMs treated with c-di-AMP had a significantly impaired ability to induce Ifnb1, Ccl5 and Cxcl11 compared with 2′3′-cGAMP (Figure 4B and 4C). The opposite trend was observed in the sh-Akr1c13 iBMDMs, where c-di-AMP elicited increased gene expression over 2′3′-cGAMP (Figure 4D). These data strengthen a model in which RECON acts as a sink for c-di-AMP and dampens the STING response.
Figure 4. Bacterial cyclic dinucleotide responses are controlled by RECON and elicit STING-independent host responses.

(A) Expression of Ccl5, Cxcl10, Cxcl11, and Ifnb1 mRNA in WT, Sting−/−, or Myd88/Tlr3−/− primary BMDMs infected with Lm for 4 h and 8 h.
(B–D) Expression of Ifnb1, Ccl5, and Cxcl11 in sh-Control (B), oe-Akr1c13 (C) or sh-Akr1c13 (D) iBMDMs treated with c-di-AMP or 2′3′-cGAMP for 4 h.
(E) Expression of Nos2 mRNA in WT, Sting−/−, or Myd88/Tlr3−/− primary BMDMs infected with Lm for 4 h and 8 h.
(F) Primary BMDMs were co-stimulated with c-di-AMP and Pam3CSK4 or Tri-DAP for 20 h. Supernatants were used to determine the ISRE response and nitrite levels.
(G, H) WT or Sting−/− primary BMDMs were co-stimulated with the indicated cdNs and Tri-DAP for 20 h. Supernatants were used to determine the ISRE response (G) and nitrite levels (H).
ISRE luciferase data is plotted as the fold change in relative luciferase units (RLU) with TLR or NOD stimulation in the absence of cdNs set at 1 (N=6). (A, E–H) Data are representative of two or more independent experiments with similar results. (B–D) Data are the means of five biological replicates pooled. qRT-PCR data were performed in technical duplicates or triplicates and were normalized to uninfected WT BMDMs. In all panels, error bars represent ± SEM of technical replicates.
We found that in primary BMDMs infected with Lm, Nos2 expression kinetics significantly differed from Ifnb1 and the chemokines. Early Nos2 induction was MyD88-dependent and STING-independent, whereas late expression was partially restored in the MyD88-deficient cells but was stunted in STING-deficient cells (Figure 4E). These data are consistent with the idea that IFN feedback promotes Nos2 expression during Lm infection. To study the effect of cdNs on iNOS directly, and in the absence of IFN feedback, we co-stimulated primary BMDMs with Pam3CSK4 or Tri-DAP and c-di-AMP. Although c-di-AMP induced an IFN response during co-stimulation with both Pam3CSK4 and Tri-DAP, we only observed increased NO production during co-stimulation with Tri-DAP (Figure 4F). There was no measureable nitrite production in cells treated only with cdNs without TLR or NOD ligand. We studied this effect further by repeating the co-stimulation using Sting−/− primary BMDMs as well as the host-derived 2′3′-cGAMP. Neither 2′3′-cGAMP nor c-di-AMP induced an IFN response in STING-deficient cells, as expected; however, in the absence of STING, c-di-AMP but not 2′3′-cGAMP increased NO production during co-stimulation with Tri-DAP (Figure 4G and 4H). The ability of c-di-AMP but not 2′3′-cGAMP to increase NO in the absence of STING establishes that c-di-AMP can affect antimicrobial responses through other pathways. The synergy of c-di-AMP with NOD1 but not TLR2 stimulation correlates with the in vivo importance of these pathways to host bacterial clearance, whereby NOD1 deficient mice are highly susceptible to Lm infection while TLR2 deficient mice show equivalent resistance to WT mice (Edelson and Unanue, 2002; Mosa et al., 2009).
RECON is a negative regulator of NF-κB activation
Our investigations in macrophages revealed that RECON augmented Nos2 expression in a STING- and IFN-independent manner. Induction of Nos2 is dependent on NF-κB activation, so we further explored the potential link between RECON and NF-κB. We used the embryonically-derived murine hepatocyte cell line, TIB73, because RECON is highly expressed in hepatocytes, and mice with obstructed NF-κB activation specifically in hepatocytes are unable to clear Lm, even in the presence of NF-κB-competent immune cells (Lavon et al., 2000). Additionally, a recent study has shown that primary human and murine hepatocytes do not express STING (Thomsen et al., 2016), therefore the use of this cell type would allow us to study NF-κB activation without complications from IFN feedback.
Using CRISPR/Cas9-based site-directed mutagenesis, we introduced mutations in the Akr1c13 gene in TIB73 hepatocytes. Although there is no commercially available RECON-specific antibody, the resulting clonal Akr1c13−/− cell line had significantly reduced levels of Akr1c13 mRNA (<5% of WT), likely due to nonsense-mediated decay, and contained two mutated compound heterozygous alleles (one with a 3 base pair insertion, one with a single base pair deletion), both of which encoded premature stop codons (Figures 5A and S3A–C). We also confirmed the absence of STING signaling in this cell line, as we were unable to detect Ifnb1 or Isg15 expression in either WT or Akr1c13−/− TIB73 hepatocytes infected with Lm. Likewise, supernatants from infected cells elicited no ISRE response compared with supernatants from iBMDMs (Figure S3D).
Figure 5. RECON is a negative regulator of NF-κB activation.

(A) Steady-state expression of Akr1c13 mRNA in WT and Akr1c13−/− TIB73 cell lines.
(B, C) Expression of Il6 (B) or Nos2 (C) mRNA at 2 h post-treatment with the indicated ligands or Lm infection of WT or Akr1c13−/− TIB73 hepatocytes. T&P = Tri-DAP + Pam3CSK4 co-stimulation.
(D) Expression of Nos2 mRNA in TIB73 cells pre-treated for 1 h with DMSO (ctrl) or the NF-κB inhibitor Celastrol (0.1 and 1 μM).
(E) NF-κB luciferase activity measured from WT or Akr1c13−/− TIB73 hepatocytes treated with the indicated ligands for 6 h. The data are presented as luciferase units divided by the eGFP transfection control fluorescence (LUC) (N=6).
(F) Microscopy analysis of WT (top) or Akr1c13−/− (bottom) TIB73 hepatocytes infected with Lm for 1 h. DAPI (green), NF-κB p65 (red), and F-actin (blue).
(G) Quantification of the nuclear/cytoplasmic ratio of NF-κB p65 fluorescence of data presented in (I) and (K). N=100–120 cells for each treatment. Error bars represent ± SD of biological replicates.
(H) Western blot analyses of proteins involved in the NF-κB pathway; WT or Akr1c13−/− TIB73 hepatocytes were co-stimulated with Tri-DAP + Pam3CSK4 for the indicated times (5–120 min). β-actin is shown as a loading control.
Data are representative of two or more independent experiments with similar results. qRT-PCR data were performed in technical duplicates or triplicates and were normalized to untreated (mock) WT TIB73 cells. Unless otherwise notes, error bars represent ± SEM of technical replicates (*P < 0.0001, unpaired Student’s t test). See also Figures S3–5.
Infection of TIB73 hepatocytes with Lm induced expression of Il6, Nos2, Ccl5 and Cxcl11, and, similar to the results in macrophages, loss of RECON augmented their expression (Figures 5B, 5C and S4A). Similarly, the expression of these genes in Akr1c13−/− hepatocytes was elevated in response to TLR2, TLR4 and NOD1 stimulation. Multiple NF-κB inhibitors (Celastrol, BAY 11-7082 and MG-132) blocked Nos2 expression during Lm infection in both WT and Akr1c13−/− cells without affecting bacterial replication (Figures 5D and S4B–D). Consistent with the gene expression data, the activity of an NF-κB luciferase reporter was increased in the absence of RECON following stimulation with bacterial TLR ligands or Lm infection (Figure 5E). The absence of RECON did not affect Il6 gene expression or luciferase activation following stimulation with the NF-κB activator TNF-α (Figures S4E and S4F). Together, these data demonstrate that loss of RECON augments NF-κB activity and that this effect might only occur alongside detection of other bacterial MAMPs.
We next examined the influence of RECON on components of the NF-κB signaling pathway. We observed an increased ratio of nuclear to cytoplasmic p65 NF-κB in Akr1c13−/− hepatocytes compared to WT cells at 1 hpi as assessed by microscopy, and this translocation was blocked by the addition of an NF-κB inhibitor (Figures 5F, 5G, S5A and S4B). We observed increased phosphorylation of p65 by Western blot in Akr1c13−/− cells co-stimulated with Tri-DAP + Pam3CSK4, which was consistent with the increased nuclear localization we observed by microscopy (Figure 5H). Additionally, the peak of p65 phosphorylation occurred earlier in the absence of RECON (20 versus 40 min). Blots for the inhibitory protein IκBα revealed that after transient degradation (between 20–40 min), its abundance increased over basal levels in WT cells (60–120 min), while they did not rise above the basal state in Akr1c13−/− cells, indicating that the loss of RECON promoted sustained activation of the pathway. We also observed increased phosphorylation of IKKα occurring at 20–40 min in the Akr1c13−/− cells. Thus, the augmentation of NF-κB signaling in the absence of RECON appears to occur at two points – at the level of phospho-IKKα, which would result in increased initiation of the signaling cascade, and phospho-p65, which would result in increased p65 nuclear translocation and DNA binding activity.
Loss of RECON’s enzymatic activity promotes inflammation
To segregate the role of RECON’s enzyme activity from its effects as a nucleotide sink, we mutated the invariant H117 residue that functions as a general acid/base in a proton relay network during oxidative/reductive catalysis by AKRs (Di Costanzo et al., 2009). The H117 residue is more than 5 Å away from c-di-AMP, and thus it is unlikely that it would play an important role in c-di-AMP binding (Figure 6A). Accordingly, mutation of H117 to alanine (H117A mutant) abolished RECON’s enzymatic activity while maintaining high affinity c-di-AMP binding (Figures 6B–D). We stably expressed H117A or WT RECON in Akr1c13−/− hepatocytes and determined that their in vivo c-di-AMP binding capacity was equivalent using a whole cell lysate DRaCALA assay (Figure 6E and 6F). Complementation of the Akr1c13−/− cells with WT RECON reversed the augmentation of NF-κB-driven luciferase activity following co-stimulation with Tri-DAP and Pam3CSK4 while complementation with the H117A mutant did not (Figure 6G). From these results, we can conclude that it is the loss of RECON enzyme activity that is driving increased NF-κB activation. These data support our hypothesis that the accumulation of a RECON substrate or substrates may be responsible for augmenting the activation of the NF-κB pathway.
Figure 6. Loss of RECON activity promotes NF-κB activation.

(A) The conserved residues of the catalytic tetrad of RECON are shown with the measured proximity between H117 and bound c-di-AMP.
(B, C) Percent enzyme activity of WT or the H117A mutant RECON. Substrates used were geraniol with the cosubstrate NAD+ (B) or 9,10-Phenanthrenequinone (PQ) with the cosubstrate NADPH (C).
(D) Binding titration of c-di-AMP with the H117A mutant RECON using 32P-c-di-AMP. Kd value is indicated.
(E) Steady-state expression of Akr1c13 mRNA in WT, Akr1c13−/−, or Akr1c13−/− complemented with WT (WT-comp) or H117A (H117A-comp) RECON.
(F) Binding of 32P-c-di-AMP to whole cell lysates from the indicated TIB73 cell lines or to recombinant (rec) RECON as a control.
(G) NF-κB luciferase activity measured from WT or Akr1c13−/− TIB73 hepatocytes co-stimulated with Tri-DAP + Pam3CSK4 for the indicated times. The data are presented as luciferase units divided by the eGFP transfection control fluorescence (LUC) (N=6). Error bars represent ± SD of biological replicates.
Data are from one of two (B–F) or three (G) independent experiments with similar results. Unless otherwise noted, error bars represent ± SEM of technical replicates.
RECON alters bacterial virulence in phagocytic and non-phagocytic cells
We tested whether modulation of RECON levels would influence bacterial virulence. We did not observe any difference in Lm bacterial growth in naïve iBMDMs expressing different levels of RECON (Figure S6A). This was not entirely unexpected considering that neither loss of STING nor MyD88 affects Lm virulence in cell culture (Figure S6B), even though MyD88-deficient mice are highly susceptible to Lm infection (Edelson and Unanue, 2002). However, we observed a significant increase in Lm intracellular replication in oe-Akr1c13 iBMDMs that were activated with IFN-γ (Figure 7A). These data suggest that suppression of RECON drives a cell-intrinsic antibacterial state in activated iBMDMs. It has previously been reported that IFN-γ-mediated inhibition of Lm growth and NO production in BMDMs is dependent on NOD1 (Mosa et al., 2009), and earlier results showed a NOD1-specific synergy of c-di-AMP on NO production (Figure 4F).
Figure 7. RECON-mediated inflammation alters bacterial growth.

(A) iBMDM cell lines with altered Akr1c13 expression were pre-treated with IFN-γ for 20 h and then infected with Lm. CFU were enumerated at the indicated times (0.5 h to 10 h) (N=4, statistics given for 10 h data).
(B) WT or Akr1c13−/− TIB73 cells lines were infected with Lm and CFU were enumerated at the indicated times (N=3).
(C) WT, Akr1c13−/−, or Akr1c13−/− complemented with WT (WT-comp) or H117A (H117A-comp) RECON TIB73 hepatocytes were infected with C. pneumoniae or C. muridarum and inclusion forming units (IFU) were quantified after 24 h (10–20 fields were analyzed across two biological replicates per experiment).
Data are representative of two or more independent experiments with similar results. In (A) and (B) error bars represent ± SD of biological replicates. In (C) all points are shown with min to max indicated. In (A) *P < 0.05, ***P < 0.0005, unpaired Student’s t test of 10 hpi and in (C) ns = not significant, *P < 0.05, ** P < 0.001, ***P <0.0001, ordinary one-way ANOVA. See also Figure S6.
We performed similar experiments in WT or Akr1c13−/− TIB73 hepatocytes but did not observe any difference in Lm intracellular growth (Figure 7B). In the host, Lm can multiply to high numbers in the liver – therefore we reasoned that perhaps Lm has evolved mechanisms to counteract the effects of RECON suppression in this non-immune cell type. For example, Lm is resistant to the antimicrobial effects of nitric oxide (Cole et al., 2012; Shiloh et al., 1999), which is elevated downstream of RECON inhibition. To determine if hepatocyte cell-intrinsic, antibacterial responses are influenced by RECON, we infected TIB73 hepatocytes with Chlamydia spp., another genus of c-di-AMP-producing, intracellular pathogens (Corrigan and Grundling, 2013; Romling, 2008). Both C. pneumoniae and C. muridarum can infect hepatocytes but do not naturally invade this cell type during infection of the host. Infection of Akr1c13−/− TIB73 hepatocytes significantly reduced the growth and infectivity of both Chlamydia spp (Figures 7C). This effect was complemented by re-introduction of WT but not H117A RECON in the Akr1c13−/− cells. These data demonstrate that RECON’s enzyme activity influences intracellular bacterial growth and highlights the importance of this cdN sensor in cell-intrinsic host responses.
DISCUSSION
Microbial nucleic acid recognition by host cell cytosolic PRRs is an important facet of innate immune responses. In this study, we characterized an oxidoreductase PRR that specifically detects canonical bacterial cdNs. Complementation studies presented here support a model whereby cdN binding modulates RECON enzymatic activity to shape downstream inflammation. These observations suggest the presence of an inflammatory mediator that is actively metabolized by RECON. The identity of the substrate or mechanism by which this mediator acts to modulate NF-κB activity remains to be revealed. AKR family proteins have been shown to metabolize nearly all biological lipophilic metabolites, including sterols, isoprenoids, retinoids, and eicosanoids, while many AKRs, RECON included, show promiscuous activity toward a variety of substrates. Thus, the integration of sensor function with enzyme activity may provide plasticity in the host response that is dependent upon the RECON substrates available within the cell or tissue type during infection. Studies to reveal the identity and individual effects of RECON substrates on specific cellular processes will likely expand the repertoire of immunomodulatory effects of this unique enzymatic PRR.
STING has higher affinity for the non-canonical mammalian-derived 2′3′-cGAMP than for bacterial cdNs (Zhang et al., 2013). Interestingly, some human STING alleles have lost the ability to bind bacterial cdNs (Diner et al., 2013; Yi et al., 2013), suggesting that this pathway may not be geared towards recognizing bacterial pathogens but rather towards DNA sensing of viruses. It is clear that STING-dependent induction of type I IFN elicits a robust antiviral response. However, the role of type I IFN in bacterial infection is variable and in some cases detrimental to host clearance of c-di-AMP-producing pathogens such as Lm, Mtb, and C. muridarum, (reviewed in (Monroe et al., 2010)). We found that the binding of c-di-AMP by RECON dampens type I IFN induction during bacterial infection. Therefore, the inability of mammalian-derived 2′3′-cGAMP to bind RECON may maintain the cellular response geared towards type I IFN production during viral infection.
One immediate and important implication of this work is the interpretation of data relating to immune activation by c-di-AMP or bacteria that produce c-di-AMP in mouse models. Recent studies on Mtb have found that STING activation during infection of mouse BMDMs occurs mainly via 2′3′-cGAMP derived from cGAS, with minimal activation contributed by c-di-AMP (Dey et al., 2015; Watson et al., 2015). This finding has raised questions as to whether c-di-AMP produced by Mtb has any role to play in infection. Given that these studies were performed in BMDMs, which express RECON, and that RECON binds c-di-AMP significantly tighter than STING, it is feasible that c-di-AMP is mediating responses independent of STING during infection with this pathogen, as we have found occurs for Lm. Future work to assess the effects of RECON targeting c-di-AMP during infection with Mtb, as well as the relative contribution of c-di-AMP or cGAS-derived 2′3′-cGAMP in activating STING in the absence of RECON will help to shed some light on this active area of investigation.
Another important consideration relates to the translational capacity of Listeria and cdN immunomodulatory studies performed in mice. The intracellular niche and immunostimulatory activity of Lm have prompted the development of live, attenuated Lm vaccine vectors that retain the ability to enter the cytoplasm (Bruhn et al., 2007). Similarly, the use of cdNs as adjuvants or in immunotherapeutic formulations has shown significant promise in the therapeutic protection against infectious and malignant disease (Skrnjug et al., 2014). Vaccine and immunotherapeutic formulations that contain c-di-AMP and/or 3′3′-cGAMP may elicit RECON-mediated responses that may shape the outcome and interpretation of these pre-clinical studies.
Analogous to what has been reported in other PRR families (i.e. Nod-like receptor family, apoptosis inhibitory proteins - NAIPs), mice encode an expanded number of AKR family members, specifically those in the AKR1C subfamily to which RECON belongs. Within this subfamily, mice encode 8 AKRs, while humans encode 4 (http://www.med.upenn.edu/akr). There has been significant interest in the members of the AKR1C subfamily because they are implicated in the progression of several human cancers, including prostate and breast cancer (Penning and Byrns, 2009). To determine whether any human AKR proteins bind c-di-AMP, we performed pulldowns using c-di-AMP sepharose and lysates from the human hepatoma cell line HepG2. The AKR family member AKR1C1 was reproducibly and statistically significantly enriched in c-di-AMP pulldowns over the control (Figure S7A). AKR1C1, also known as 20-alpha-hydroxysteroid dehydrogenase, is in the same subfamily as RECON and catalyzes the reaction of progesterone to the inactive form 20-alpha-hydroxyprogesterone. It has previously been shown to be important during pregnancy and in the development of some human cancers (Yun et al., 2015). It has ~70% identify with RECON and, similar to RECON, it is highly expressed in the liver, stomach and small intestine. However, we were not able to confirm the pulldown results with recombinant AKR1C1 and further examination of the protein revealed that it did not exhibit any enzyme activity (Figure S7B–D). Previous work with other human AKRs showed recombinant proteins expressed in E. coli had up to 20 fold lower activity compared to native enzymes purified from human livers (Martin et al., 2006). Furthermore, some AKR family members require post-translational modifications for their cellular activities (Shimizu et al., 2016), so it possible that AKR1C1 expressed in E. coli lacked the necessary modifications for c-di-AMP binding.
We also stably overexpressed AKR1C1 in iBMDMs and observed a slight increase in c-di-AMP binding over sh-Control iBMDMs, however we observed no effect of AKR1C1 overexpression on Ifnb1 expression in response to c-di-AMP (Figure S7E–G). It is possible that the level of AKR1C1 overexpression achieved in the iBMDMs was not enough to affect c-di-AMP intracellular pools or the use of the reference AKR1C1 sequence (NCBI NM_001353.5) may not resolve the potential role for isoforms or different AKR1C1 alleles that may be important for c-di-AMP binding. AKR1C1 is a complex locus with six transcript isoforms, evidence of exon skipping, over 300 single nucleotide polymorphisms, and varying 5′ and 3′ UTR lengths (GTExPortal). Further work will be necessary to determine if AKR1C1 does interact with bacterial cdNs and whether their interaction shapes the immune response in an orthologous way to RECON. Additionally, given that many AKR members are inducibly expressed and are found in cell types other than hepatocytes, it remains to be seen whether more mouse and human AKR family members are capable of interacting with bacterial cdNs.
In this study, we explored the effect of the oxidoreductase RECON during bacterial infection of macrophages and hepatocytes. RECON protein was abundantly isolated from mouse liver using c-di-AMP sepharose and the liver (specifically hepatocytes) is a principal site for Lm replication following systemic dissemination (Cousens and Wing, 2000). Previous work characterizing tissue-specific expression of RECON has shown that it is most highly expressed in the gastrointestinal tract and is almost exclusively restricted to this site and the liver (Vergnes et al., 2003). Data from FANTOM5, a comprehensive RNA expression analysis database, indicates that GP2+ M cells and enterocytes have the highest steady-state expression of Akr1c13 (EntrezGene: 27384). Stellate cells, hepatocytes and bone marrow-derived macrophages were also among the top seven highest Akr1c13-expressing cells. Given the high expression of Akr1c13 in the gastrointestinal tract, there are several possible effects it may have in these organs. Both Lm and Vibrio cholerae produce RECON-targeted cdNs, while Firmicutes and Bacteroidetes, the two major bacterial phyla in the gastrointestinal tract (>95% in humans), all produce c-di-AMP (Moreno-Indias et al., 2014; Yang et al., 2009). Considering this, RECON-mediated surveillance of bacterial cdNs may have widespread implications for the host immune response to bacteria, both pathogens and commensals alike.
EXPERIMENTAL PROCEDURES
Nucleotide-Binding and Enzyme Assays
Differential radial capillary action of ligand assays (DRaCALA) were performed with recombinant proteins or whole cell lysates as described previously (Roelofs et al., 2011). For competition assays, unlabeled nucleotides or cyclic dinucleotides were added prior to the addition of 32P-c-di-AMP. To measure RECON enzyme activity, geraniol or 9,10-Phenanthrenequinone were supplied as substrates with NAD+ or NADPH as cosubstrates, respectively (see Extended Experimental Procedures – E.E.P.).
Tissue Culture and Infection/Biological Assays
TIB73 and ISRE-L929 cells were grown at 37°C in 5% CO2 in phenol red-free DMEM medium (GIBCO) with 10% heat-inactivated FBS (HyClone) and supplemented with 2 mM sodium pyruvate and 1 mM L-Glutamine (Thermo Fisher). J2 immortalized BMDMs (iBMDMs) and primary BMDMs were grown in the same media as above except with additional supplementation of 55 μM 2-mercaptoethanol and M-CSF harvested from L929 conditioned medium (final volume 10%). Primary BMDMs were generated as previously described (Jones and Portnoy, 1994). Bone marrow from WT, Sting−/− or MyD88/Tlr3−/− mice was generously provided by Daniel Stetson and Michael Gale, Jr. at the University of Washington. All protocols were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Washington.
Wild-type 10403s Lm was used to infect iBMDMs as previously described (Sauer et al., 2010) with an MOI of 0.1. TIB73 cells were infected with an MOI of 0.001. For Chlamydia infections, C. pneumoniae TW73, and C. muridarum were infected onto TIB73 cells and 24 hpi an inclusion forming units (IFU) assay was performed (see E.E.P. for full infection details). Cell-free supernatants were used to quantify nitrite levels using a colorimetric assay kit (Cayman Chemical) and IFN levels using a bioassay involving ISRE-L929 reporter cells. For full details regarding cdN and MAMP stimulation of iBMDM and primary BMDMs see E.E.P. For NF-κB luciferase assays, TIB73 hepatocytes were co-transfected with pNiFty-Luc plasmid (InvivoGen) and eGFP plasmid (internal control) using TransIT-LT1 transfection reagent (Mirus Bio). Cells were stimulated 20 h post-transfection. For NF-κB Western blots, whole cell lysates were transferred to nitrocellulose membranes, and blocked and probed in 5% non-fat dry milk with NF-κB antibodies or β-actin (Cell Signaling NF-κB pathway kit #9936) (see E.E.P. for full IFN bioassay, NF-κB luciferase assay, NF-κB p65 microscopy and Western details).
Generation of Cell Lines with Modified RECON Expression
Overexpression of RECON was achieved via retroviral transduction and lentivirus-transduced scrambled control and Akr1c13 RNAi (sh-Akr1c13) iBMDMs were generated using the pLKO.1 vector system (see E.E.P.). Cas9 guide sequences specifically targeting Akr1c13 were determined using http://crispr.mit.edu/. TIB73 cells were transfected with pX330-U6-Chimeric_BB-CBh-hSpCas9 (a gift from Feng Zhang (Addgene plasmid # 42230)) with Akr1c13 guide RNA sequences cloned in (see E.E.P. and Figure S3). Complementation of Akr1c13−/− TIB73 hepatocytes with WT or H117A RECON was accomplished by transduction with MSCV virus (see E.E.P. for H117A cloning and virus generation details).
RT-qPCR Assays and Inhibitors
RNA was extracted with the RNAqueous Total RNA Isolation Kit, DNase treated using the TURBO DNA-free Kit (Thermo Fisher) and reverse-transcribed with the iScript cDNA synthesis kit (Bio-Rad). A TaqMan Mouse Immune Panel (Thermo Fisher 4367786) was used for data presented in Figure 3C and TaqMan RT-qPCR assays were used for quantification of mouse Akr1c13, Ccl5, Cxcl10, Cxcl11, Ifnb1, Il1b, Il6, Isg15, and Nos2. Hprt served as an endogenous control. All RT-qPCR data are plotted as the mean ± SEM of technical duplicates or triplicates. Purified anti-mouse IFNAR-1 antibody (BioLegend) (1 μg/ml) was added to cells 1 hpi with Lm. NF-κB inhibitors Celastrol, BAY 11-7082, MG-132 (InvivoGen) were added to cells for 1 h prior to infection, maintained in the presence of the inhibitors during infection and then again after bacteria were washed off out to 4 hpi. All inhibitors were reconstituted in DMSO, which by itself was used as a control treatment.
Statistical Analyses
Data were analyzed using Prism 6 software. Unpaired Student’s t-tests were used to determined significance of data unless otherwise noted.
Supplementary Material
Highlights.
Bacterial but not eukaryotic cyclic dinucleotides bind RECON with high affinity.
Loss of RECON enzyme activity promotes inflammation by regulating NF-κB activity.
RECON shapes cell-intrinsic, antibacterial responses.
RECON is a cytosolic PRR that detects bacterial cyclic dinucleotides.
Acknowledgments
We would like to Dan Stetson and Michael Gale, Jr. (U. of Washington) for murine bone marrow, Ram Savan (U. of Washington) for help with RT-qPCR experiments, Kamakshi Sureka for helpful discussions, Alex Pollock for feedback on the preparation of the manuscript, and TuAnh Ngoc Huynh for technical help with pulldown experiments. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program (to A.P.M.) under Grant No. DGE-1256082 and by National Institutes of Health grants AI116669 (to J.J.W. and L.T.), AI108698 (to J.J.W.), and the Pew Scholars Program in the Biomedical Sciences (to J.J.W.). The in-house instrument for X-ray diffraction screening was supported by an NIH grant to LT (S10OD012018).
Footnotes
Accession Numbers
The Research Collaboratory for Structural Bioinformatics Protein Data Bank accession number for the new structure reported in this paper is 5UXF.
AUTHOR CONTRIBUTIONS
Conceptualization, A.P.M. and J.J.W.; Methodology, A.P.M., S.L., M.Z., Y.A.G., K.H., L.T., and J.J.W.; Investigation, A.P.M., S.L., F.A.Q., M.Z., E.F.T., K.H. L.T., and J.J.W.; Validation, A.P.M., F.A.Q., M.Z., and J.J.W.; Writing – Original Draft, A.P.M., S.L., L.T., and J.J.W.; Writing – Review & Editing, A.P.M., S.L., K.H., L.T., J.J.W.; Funding Acquisition, A.P.M., L.T., and J.J.W.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barker JR, Koestler BJ, Carpenter VK, Burdette DL, Waters CM, Vance RE, Valdivia RH. STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during Chlamydia trachomatis infection. MBio. 2013;4:e00018–00013. doi: 10.1128/mBio.00018-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruhn KW, Craft N, Miller JF. Listeria as a vaccine vector. Microbes Infect. 2007;9:1226–1235. doi: 10.1016/j.micinf.2007.05.010. [DOI] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell. 2014;54:289–296. doi: 10.1016/j.molcel.2014.03.040. [DOI] [PubMed] [Google Scholar]
- Cole C, Thomas S, Filak H, Henson PM, Lenz LL. Nitric oxide increases susceptibility of Toll-like receptor-activated macrophages to spreading Listeria monocytogenes. Immunity. 2012;36:807–820. doi: 10.1016/j.immuni.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR, Scharn CR, Stamm CE, Levine B, Chen ZJ, et al. Cyclic GMP-AMP Synthase Is an Innate Immune DNA Sensor for Mycobacterium tuberculosis. Cell Host Microbe. 2015;17:820–828. doi: 10.1016/j.chom.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan RM, Grundling A. Cyclic di-AMP: another second messenger enters the fray. Nat Rev Microbiol. 2013;11:513–524. doi: 10.1038/nrmicro3069. [DOI] [PubMed] [Google Scholar]
- Cousens LP, Wing EJ. Innate defenses in the liver during Listeria infection. Immunol Rev. 2000;174:150–159. doi: 10.1034/j.1600-0528.2002.017407.x. [DOI] [PubMed] [Google Scholar]
- Couture JF, Legrand P, Cantin L, Luu-The V, Labrie F, Breton R. Human 20alpha-hydroxysteroid dehydrogenase: crystallographic and site-directed mutagenesis studies lead to the identification of an alternative binding site for C21-steroids. J Mol Biol. 2003;331:593–604. doi: 10.1016/s0022-2836(03)00762-9. [DOI] [PubMed] [Google Scholar]
- Crimmins GT, Herskovits AA, Rehder K, Sivick KE, Lauer P, Dubensky TW, Jr, Portnoy DA. Listeria monocytogenes multidrug resistance transporters activate a cytosolic surveillance pathway of innate immunity. Proc Natl Acad Sci U S A. 2008;105:10191–10196. doi: 10.1073/pnas.0804170105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies BW, Bogard RW, Young TS, Mekalanos JJ. Coordinated regulation of accessory genetic elements produces cyclic di-nucleotides for V. cholerae virulence. Cell. 2012;149:358–370. doi: 10.1016/j.cell.2012.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet CJ, Ishii KJ. Nucleic acid sensing at the interface between innate and adaptive immunity in vaccination. Nat Rev Immunol. 2012;12:479–491. doi: 10.1038/nri3247. [DOI] [PubMed] [Google Scholar]
- Dey B, Dey RJ, Cheung LS, Pokkali S, Guo H, Lee JH, Bishai WR. A bacterial cyclic dinucleotide activates the cytosolic surveillance pathway and mediates innate resistance to tuberculosis. Nat Med. 2015;21:401–406. doi: 10.1038/nm.3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Costanzo L, Penning TM, Christianson DW. Aldo-keto reductases in which the conserved catalytic histidine is substituted. Chem Biol Interact. 2009;178:127–133. doi: 10.1016/j.cbi.2008.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 2013;3:1355–1361. doi: 10.1016/j.celrep.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelson BT, Unanue ER. MyD88-dependent but Toll-like receptor 2-independent innate immunity to Listeria: no role for either in macrophage listericidal activity. J Immunol. 2002;169:3869–3875. doi: 10.4049/jimmunol.169.7.3869. [DOI] [PubMed] [Google Scholar]
- Endo S, Sanai M, Horie K, Matsunaga T, Ishikura S, Tajima K, El-Kabbani O, Hara A. Characterization of rat and mouse NAD+-dependent 3alpha/17beta/20alpha-hydroxysteroid dehydrogenases and identification of substrate specificity determinants by site-directed mutagenesis. Arch Biochem Biophys. 2007;467:76–86. doi: 10.1016/j.abb.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Fang FC. Perspectives series: host/pathogen interactions. Mechanisms of nitric oxide-related antimicrobial activity. J Clin Invest. 1997;99:2818–2825. doi: 10.1172/JCI119473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Winship D, Snyder JM, Child SJ, Geballe AP, Stetson DB. The AIM2-like Receptors Are Dispensable for the Interferon Response to Intracellular DNA. Immunity. 2016;45:255–266. doi: 10.1016/j.immuni.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda S, Okuda-Ashitaka E, Masu Y, Suzuki T, Watanabe K, Nakao M, Shingu K, Ito S. Cloning and characterization of two novel aldo-keto reductases (AKR1C12 and AKR1C13) from mouse stomach. FEBS Lett. 1999;459:433–437. doi: 10.1016/s0014-5793(99)01243-0. [DOI] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs AT, Ignarro LJ. Cell density-enhanced expression of inducible nitric oxide synthase in murine macrophages mediated by interferon-beta. Nitric Oxide. 2003;8:222–230. doi: 10.1016/s1089-8603(03)00027-2. [DOI] [PubMed] [Google Scholar]
- Jez JM, Bennett MJ, Schlegel BP, Lewis M, Penning TM. Comparative anatomy of the aldo-keto reductase superfamily. Biochem J. 1997;326:625–636. doi: 10.1042/bj3260625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Portnoy DA. Characterization of Listeria monocytogenes pathogenesis in a strain expressing perfringolysin O in place of listeriolysin O. Infect Immun. 1994;62:5608–5613. doi: 10.1128/iai.62.12.5608-5613.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia D, Merey G, Nakayama S, Zheng Y, Zhou J, Luo Y, Guo M, Roembke BT, Sintim HO. Nucleotide, c-di-GMP, c-di-AMP, cGMP, cAMP, (p)ppGpp signaling in bacteria and implications in pathogenesis. Chem Soc Rev. 2013;42:305–341. doi: 10.1039/c2cs35206k. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Toll-like receptor and RIG-I-like receptor signaling. Ann N Y Acad Sci. 2008;1143:1–20. doi: 10.1196/annals.1443.020. [DOI] [PubMed] [Google Scholar]
- Kellenberger CA, Wilson SC, Hickey SF, Gonzalez TL, Su Y, Hallberg ZF, Brewer TF, Iavarone AT, Carlson HK, Hsieh YF, et al. GEMM-I riboswitches from Geobacter sense the bacterial second messenger cyclic AMP-GMP. Proc Natl Acad Sci U S A. 2015;112:5383–5388. doi: 10.1073/pnas.1419328112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavon I, Goldberg I, Amit S, Landsman L, Jung S, Tsuberi BZ, Barshack I, Kopolovic J, Galun E, Bujard H, et al. High susceptibility to bacterial infection, but no liver dysfunction, in mice compromised for hepatocyte NF-kappaB activation. Nat Med. 2000;6:573–577. doi: 10.1038/75057. [DOI] [PubMed] [Google Scholar]
- Martin HJ, Breyer-Pfaff U, Wsol V, Venz S, Block S, Maser E. Purification and characterization of akr1b10 from human liver: role in carbonyl reduction of xenobiotics. Drug Metab Dispos. 2006;34:464–470. doi: 10.1124/dmd.105.007971. [DOI] [PubMed] [Google Scholar]
- Mills E, Pultz IS, Kulasekara HD, Miller SI. The bacterial second messenger c-di-GMP: mechanisms of signalling. Cell Microbiol. 2011;13:1122–1129. doi: 10.1111/j.1462-5822.2011.01619.x. [DOI] [PubMed] [Google Scholar]
- Monroe KM, McWhirter SM, Vance RE. Induction of type I interferons by bacteria. Cell Microbiol. 2010;12:881–890. doi: 10.1111/j.1462-5822.2010.01478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Indias I, Cardona F, Tinahones FJ, Queipo-Ortuno MI. Impact of the gut microbiota on the development of obesity and type 2 diabetes mellitus. Front Microbiol. 2014;5:190. doi: 10.3389/fmicb.2014.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosa A, Trumstedt C, Eriksson E, Soehnlein O, Heuts F, Janik K, Klos A, Dittrich-Breiholz O, Kracht M, Hidmark A, et al. Nonhematopoietic cells control the outcome of infection with Listeria monocytogenes in a nucleotide oligomerization domain 1-dependent manner. Infect Immun. 2009;77:2908–2918. doi: 10.1128/IAI.01068-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy PM, Baggiolini M, Charo IF, Hebert CA, Horuk R, Matsushima K, Miller LH, Oppenheim JJ, Power CA. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev. 2000;52:145–176. [PubMed] [Google Scholar]
- Penning TM, Byrns MC. Steroid hormone transforming aldo-keto reductases and cancer. Ann N Y Acad Sci. 2009;1155:33–42. doi: 10.1111/j.1749-6632.2009.03700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelofs KG, Wang J, Sintim HO, Lee VT. Differential radial capillary action of ligand assay for high-throughput detection of protein-metabolite interactions. Proc Natl Acad Sci U S A. 2011;108:15528–15533. doi: 10.1073/pnas.1018949108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romling U. Great times for small molecules: c-di-AMP, a second messenger candidate in Bacteria and Archaea. Sci Signal. 2008;1:pe39. doi: 10.1126/scisignal.133pe39. [DOI] [PubMed] [Google Scholar]
- Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, Hyodo M, Hayakawa Y, Woodward JJ, Portnoy DA, et al. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun. 2011;79:688–694. doi: 10.1128/IAI.00999-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer JD, Witte CE, Zemansky J, Hanson B, Lauer P, Portnoy DA. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe. 2010;7:412–419. doi: 10.1016/j.chom.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh MU, MacMicking JD, Nicholson S, Brause JE, Potter S, Marino M, Fang F, Dinauer M, Nathan C. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity. 1999;10:29–38. doi: 10.1016/s1074-7613(00)80004-7. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Tatano Y, Tomioka H. Aldose reductase participates in the downregulation of T cell functions due to suppressor macrophages. Sci Rep. 2016;6:21093. doi: 10.1038/srep21093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrnjug I, Rueckert C, Libanova R, Lienenklaus S, Weiss S, Guzman CA. The mucosal adjuvant cyclic di-AMP exerts immune stimulatory effects on dendritic cells and macrophages. PLoS One. 2014;9:e95728. doi: 10.1371/journal.pone.0095728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureka K, Choi PH, Precit M, Delince M, Pensinger DA, Huynh TN, Jurado AR, Goo YA, Sadilek M, Iavarone AT, et al. The cyclic dinucleotide c-di-AMP is an allosteric regulator of metabolic enzyme function. Cell. 2014;158:1389–1401. doi: 10.1016/j.cell.2014.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen MK, Nandakumar R, Stadler D, Malo A, Valls RM, Wang F, Reinert LS, Dagnaes-Hansen F, Hollensen AK, Mikkelsen JG, et al. Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology. 2016 doi: 10.1002/hep.28685. [DOI] [PubMed] [Google Scholar]
- Vergnes L, Phan J, Stolz A, Reue K. A cluster of eight hydroxysteroid dehydrogenase genes belonging to the aldo-keto reductase supergene family on mouse chromosome 13. J Lipid Res. 2003;44:503–511. doi: 10.1194/jlr.M200399-JLR200. [DOI] [PubMed] [Google Scholar]
- Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, Vance RE, Stallings CL, Virgin HW, Cox JS. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe. 2015;17:811–819. doi: 10.1016/j.chom.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte CE, Whiteley AT, Burke TP, Sauer JD, Portnoy DA, Woodward JJ. Cyclic di-AMP is critical for Listeria monocytogenes growth, cell wall homeostasis, and establishment of infection. MBio. 2013;4:e00282–00213. doi: 10.1128/mBio.00282-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward JJ, Iavarone AT, Portnoy DA. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science. 2010;328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Hara H, Tsuchiya K, Sakai S, Fang R, Matsuura M, Nomura T, Sato F, Mitsuyama M, Kawamura I. Listeria monocytogenes strain-specific impairment of the TetR regulator underlies the drastic increase in cyclic di-AMP secretion and beta interferon-inducing ability. Infect Immun. 2012;80:2323–2332. doi: 10.1128/IAI.06162-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Bai Y, Zhang Y, Gabrielle VD, Jin L, Bai G. Deletion of the cyclic di-AMP phosphodiesterase gene (cnpB) in Mycobacterium tuberculosis leads to reduced virulence in a mouse model of infection. Mol Microbiol. 2014;93:65–79. doi: 10.1111/mmi.12641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Xie L, Li Y, Wei C. More than 9,000,000 unique genes in human gut bacterial community: estimating gene numbers inside a human body. PLoS One. 2009;4:e6074. doi: 10.1371/journal.pone.0006074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi G, Brendel VP, Shu C, Li P, Palanathan S, Cheng Kao C. Single nucleotide polymorphisms of human STING can affect innate immune response to cyclic dinucleotides. PLoS One. 2013;8:e77846. doi: 10.1371/journal.pone.0077846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun H, Xie J, Olumi AF, Ghosh R, Kumar AP. Activation of AKR1C1/ERbeta induces apoptosis by downregulation of c-FLIP in prostate cancer cells: A prospective therapeutic opportunity. Oncotarget. 2015;6:11600–11613. doi: 10.18632/oncotarget.3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen ZJ. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell. 2013;51:226–235. doi: 10.1016/j.molcel.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.