

Article first Published online 31 March 2017
Key Words: Asacol, mesalamine, ulcerative colitis, maintenance of remission, once daily administration, 3 times daily administration
Abstract
Background:
The noninferiority of pH-dependent release mesalamine (Asacol) once daily (QD) to 3 times daily (TID) administration was investigated.
Methods:
This was a phase 3, multicenter, randomized, double-blind, parallel-group, active-control study, with dynamic and stochastic allocation using central registration. Patients with ulcerative colitis in remission (a bloody stool score of 0, and an ulcerative colitis disease activity index of ≤2), received the study drug (Asacol 2.4 g/d) for 48 weeks. The primary efficacy endpoint of the nonrecurrence rate was assessed on the full analysis set. The noninferiority margin was 10%.
Results:
Six hundred and four subjects were eligible and were allocated; 603 subjects received the study drug. The full analysis set comprised 602 subjects (QD: 301, TID: 301). Nonrecurrence rates were 88.4% in the QD and 89.6% in the TID. The difference between nonrecurrence rates was −1.3% (95% confidence interval: −6.2, 3.7), confirming noninferiority. No differences in the safety profile were observed between the two treatment groups. On post hoc analysis by integrating the QD and the TID, nonrecurrence rate with a mucosal appearance score of 0 at determination of eligibility was significantly higher than the score of 1. The mean compliance rates were 97.7% in the QD and 98.1% in the TID.
Conclusions:
QD dosing with Asacol is as effective and safe as TID for maintenance of remission in patients with ulcerative colitis. Additionally, this study indicated that maintaining a good mucosal state is the key for longer maintenance of remission.
A basic objective in the management of ulcerative colitis (UC) in the remission phase is to maintain a comfortable daily life for as long as possible while preventing relapse. Oral mesalamine is recommended as basic treatment of UC in the remission phase.1 In most cases, long-term administration is needed to maintain remission. Medication adherence for a long period is known to be a key issue in this patient population.
A previous cohort study indicates that nonadherence is a risk factor for relapse of UC.2 Moreover, it has been reported that administration of multiple daily doses is a factor that influences nonadherence.3 Thus, once daily (QD) administration is a therapeutic option that encourages compliance. In other reports no differences in efficacy and safety between QD and twice or 3 times daily administration in patients with UC in the remission phase have been reported,4,5 and QD dosing is approved in several countries.
In Japan, the noninferiority of QD dosing administration was reported when compared with 3 times daily (TID) dosing administration of a time-dependent release mesalamine formulation,6 and QD dosing is approved. There has, however, not been a report that compares noninferiority of QD with TID use of a pH-dependent release mesalamine formulation. We conducted the current trial to verify that QD administration of a pH-dependent release mesalamine formulation (Asacol) is not inferior to TID administration.
MATERIALS AND METHODS
Informed Consent
Before conducting this clinical study, the investigator's brochure, clinical study protocol, patient information and consent form, other necessary documents, and the conduct of this study were reviewed and approved from ethical and scientific standpoints by the Institutional Review Board.
After thoroughly explaining the study to prospective subjects using the informed consent form, the investigator or subinvestigator obtained their voluntary written consent to participate in the study in person.
The trial was registered in the JapicCTI registry under registration No. JapicCTI-132135.
Study Drugs
A pH-dependent-release mesalamine is coated with Eudragit-S, and contains 400 mg of mesalamine per tablet. Eudragit-S dissolves at a pH ≥7, and the pH-dependent-release mesalamine tablet is designed to release mesalamine in the terminal ileum, where the intraluminal pH exceeds 7. This pH-dependent–release mesalamine tablet is Asacol whose package was changed for use in this study. The placebo did not include mesalamine, and was indistinguishable from the Asacol tablets. The Asacol and placebo tablets used in this study were supplied by Zeria Pharmaceutical Co., Ltd. (Tokyo, Japan).
Inclusion/Exclusion Criteria
Subjects were required to meet all the following inclusion criteria: (1) patients with a bloody stool score of 0, and an UCDAI of ≤2; (2) outpatients aged ≥16–<65 years at the time of informed consent; both males and females were included; (3) individuals who fully understood what was involved in this study and could give documented consent to participate in the study. For patients under the age of 20 at the time of informed consent, the consent of their legal guardian had to be obtained and also that of the patient him/herself.
Patients who met at least one of the following criteria were excluded: (1) the presence of active UC within 83 days before eligibility determination; (2) intestinal stenosis; (3) infectious enteritis or any suspicion of the presence of infectious enteritis; (4) treatment with antidiarrheal drugs or drugs for the treatment of diarrhea within 3 days before eligibility determination; (5) treatment with rectal mesalamine (enema or suppository) within 27 days before eligibility determination; (6) daily dose of oral mesalamine over 2.4 g (if a subject was receiving salazosulfapyridine, the amount equivalent to mesalamine was calculated by multiplying the dose by 0.5), corticosteroids (administration orally, by enema, suppository, or injection), azathioprine or 6-mercaptopurine (oral), cyclosporine or tacrolimus (oral or injection), infliximab or adalimumab (injection), cytapheresis or other study drugs within 83 days before eligibility determination; (7) laparotomy or laparoscopic surgery, surgery for hemorrhoids or perianal abscess, endoscopic mucosal resection or endoscopic intestinal dilatation or endoscopic intestinal dilatation of the colon within 55 days before eligibility determination; (8) patients with a history of resection of the small intestine, cecum, colon, or rectum; (9) patients with moderate or severe hepatic or renal disease; (10) patients with any serious comorbid disease, such as hematological, respiratory, cardiovascular, or neuropsychiatric disease, or metabolic/electrolyte abnormality; (11) patients undergoing treatment for a malignant tumor or patients who had been followed up for under 5 years; (12) patients with hypersensitivity to mesalamine or salicylate drugs; (13) pregnant and nursing women and women suspected of being pregnant; (14) others whom the investigator or subinvestigator considered to be inappropriate for enrollment.
Trial Design
This phase 3, multi-center, randomized, double-blind, parallel-group noninferiority trial was conducted at 56 centers in Japan.
The screening period occurred 3 to 14 days after informed consent was obtained. After the screening period, eligibility determination was conducted according to inclusion/exclusion criteria. Eligible subjects were allocated into the QD or TID group, and were instructed to take the study drug (double-dummy) orally after each meal (QD group: 6 pH-dependent-release mesalamine tablets in the morning, 2 placebo tablets at noon, and 2 placebo tablets in the evening; TID group: 2 pH-dependent-release mesalamine tablets and 4 placebo tablets in the morning, 2 pH-dependent-release mesalamine tablets at noon, and 2 pH-dependent-release mesalamine tablets in the evening) for 48 weeks.
At weeks 4, 8, 12, 16, 20, 24, 28, 32, 36, 40, 44, and 48, subjects visited the study site and received relevant tests and examinations of efficacy and safety.
Randomization
The allocation manager, who was independent of this study, prepared a study drug allocation table and randomized study drugs (QD group:TID group = 1:1). After completing the allocation, the allocation manager sealed the study drug allocation table and stored it under strict control until the end of the trial.
The investigator or subinvestigator confirmed whether subjects met the inclusion and exclusion criteria and determined their eligibility. The patient registration center allocated the study drugs randomly to the eligible subjects.
The patients were assigned dynamically and stochastically using central registration. The allocation factors were duration of remission (<2 years and ≥2 years) and the state of the intestinal mucosa (mucosal appearance score [subscore of UCDAI]: 0, 1, and 2). The balance within the study site was also considered.
Blinding
This was a double-blind trial. The subjects, the investigators/subinvestigators, staff of the study site, Zeria Pharmaceutical Co., Ltd., and Kyowa Hakko Kirin Co., Ltd. were blinded to treatment allocation.
Prohibited Concomitant Drugs and Therapies
The following drugs and therapies were prohibited throughout the period from the first day of treatment to the termination of tests (excluding colonoscopy and proctoscopy) and examinations at the week 48 visit (or at discontinuation): (1) mesalamine formulations (oral, enema, and suppository administration, include salazosulfapyridine formulations); (2) corticosteroid formulations (oral, enema, suppository, and injection administration); (3) formulations containing azathioprine or 6-mercaptopurine (oral administration); (4) formulations containing cyclosporine or tacrolimus (oral and injection administration); (5) formulations containing infliximab or adalimumab (injection administration); (6) other study drugs; (7) laparotomy or laparoscopic surgery; (8) surgery for hemorrhoids or perianal abscess; (9) endoscopic mucosal resection, endoscopic submucosal dissection, or endoscopic intestinal dilatation; (10) cytapheresis.
The following drugs and therapies were prohibited during the treatment period throughout the period from the first day of treatment to the termination of tests (excluding colonoscopy and proctoscopy) and examinations at the week 48 visit (or at discontinuation): (1) antidiarrheal drugs or drugs for the treatment of diarrhea (including drugs for irritable bowel syndrome) for 3 days before the scheduled visit; (2) Continuous use of nonsteroidal anti-inflammatory drugs (exclude topical or eye drop forms) for 3 days or more.
Efficacy Variable
The primary efficacy variable was the nonrecurrence rate. Recurrence was defined as a bloody stool score ≥1 and UCDAI ≥3. Four items (stool frequency, bloody stool, mucosal appearance, physician's global assessment) were scored and the sum of all scores was the UCDAI.7 Regardless of an incomplete UCDAI, subjects with a bloody stool score ≥1 and UCDAI ≥3 were handled as recurrence.
The subjects entered into their diary stool frequency, the state of their stool (the extent of bloody stools, if present), and compliance/noncompliance with treatment during the study period, and visited the study site every 4 weeks. The investigator or subinvestigator scored stool frequency score, bloody stool score, and physician's global assessment score based on the diary and the interview with the subject. At the time of screening and final observation (either week 48 visit or visit for discontinuation), the investigator or subinvestigator conducted the colonoscopy and determined the mucosal appearance score by reference to atlases of mucosal appearances based on Mayo endoscopic subscore.
Safety Variables
Safety variables were the incidence of adverse events and adverse drug reactions. Adverse events considered to be related to the study drug were handled as adverse drug reactions.
The investigator or subinvestigator collected information about adverse events by interview with the subjects at each visit. Moreover, to determine the presence or absence of adverse events, laboratory tests (hematological test, biochemical test, urinalysis, and urinary enzyme test) were conducted at the time of screening, at the visits during weeks 12, 24, 36, and 48 (or at discontinuation), and the recording of vital signs were conducted at eligibility determination, at the visits during weeks 12, 24, 36, and 48 (or at discontinuation).
The adverse event terms were coded using MedDRA/J (Ver. 18.1) terms for tabulation and analysis.
Statistical Methods
Noninferiority Margin
In past clinical trials,8,9 the efficacy of Asacol was 20% to 26% higher than that of placebo in patients with UC in the remission phase. Thus, we estimated that the minimum difference between the efficacy of the positive control drug and that of the placebo can be expected to be about 20%; hence the noninferiority margin was set at 10%, which is 1/2 of the minimum value. Moreover, the noninferiority margin was set to −10% in other clinical trials of Asacol in the past.5,7
Sample Size
In the past clinical trials,5,7,10 the efficacy rate of Asacol at final assessment, with efficacy defined as maintenance of remission after 12 months administration, was 65.9% to 85.4%. Thus, we estimated that the nonrecurrence rate in the QD group and TID group would be 75%. The number of cases required for verification of noninferiority is 295 under the following conditions: Significance level = 0.05 (2-tailed), detection power = 80%, and noninferiority margin = 10%. We set the sample size to 300 per group, taking drop-outs from the analysis set into consideration.
Analysis Sets
The safety analysis set (SAF) was the population used for safety assessment and included subjects who took at least one tablet of the study drug, but excluded subjects who were noncompliant in terms of good clinical practice or had no safety data after the start of study treatment.
The full analysis set (FAS) was the population used for efficacy assessment, included all subjects in the SAF, but excluded the subjects who had no efficacy data or were determined not to have UC, after the start of the study treatment.
The per protocol set (PPS) was the population used for efficacy assessment, included all subjects in the FAS, but excluded the subjects who did not meet the inclusion criteria, who fell under the exclusion criteria, used prohibited concomitant drugs and therapies, or had treatment compliance of under 75%.
Statistical Analysis Methods
The statistical analysis plan was finalized after the blind review and before key break. The statistical analyses were performed at Zeria Pharmaceutical Co., Ltd. All statistical calculations were performed with SAS Release 9.3 (SAS Institute Inc., Cary, NC).
We selected a significance level of 0.05 (2-tailed) in statistical tests and a finding was determined to be statistically significant based on the criterion of P < 0.05. In tests for uniformity, a significance level of 0.15 (2-tailed) was used, and P < 0.15 was considered unbalanced. The confidence level of 95% (2-tailed) was used for calculating confidence intervals (CIs) unless otherwise stated.
The primary analysis set was FAS, and the primary efficacy endpoint was the nonrecurrence rate. The lower limit of the 95% CI of the difference in nonrecurrence rate (the nonrecurrence rate in the QD group − the nonrecurrence rate in the TID group) was calculated; based on this calculation, the lower limit of the CI greater than the noninferiority limit value of −10%, we concluded that noninferiority was verified. In addition, we conducted a subgroup analysis (nonrecurrence rate in PPS, nonrecurrence rate by allocation factors), sensitivity analysis, and post hoc analysis after key break. In the sensitivity analysis, we treated as “missing data” a portion of those cases with recurrence, such that determination of recurrence/nonrecurrence depended on the mucosal appearance score. In post hoc analysis after key break, integrating the QD group and TID group, the nonrecurrence rate by each mucosal appearance score at eligibility determination was calculated.
Safety variables were the incidence of adverse events and the incidence of adverse drug reactions.
ETHICAL CONSIDERATIONS
This study was conducted in accordance with the International Conference on Harmonization Good Clinical Practice guidelines and local ethical and legal regulations, in line with the principles of the Declaration of Helsinki, and the “Standards for the Implementation of Clinical Trials on Pharmaceutical Products” (Ordinance).
RESULTS
Subjects
The subjects entered this trial from June 2013 to January 2015. The planned sample size was completed, and follow-up investigation of the final subject was completed on December 5, 2015.
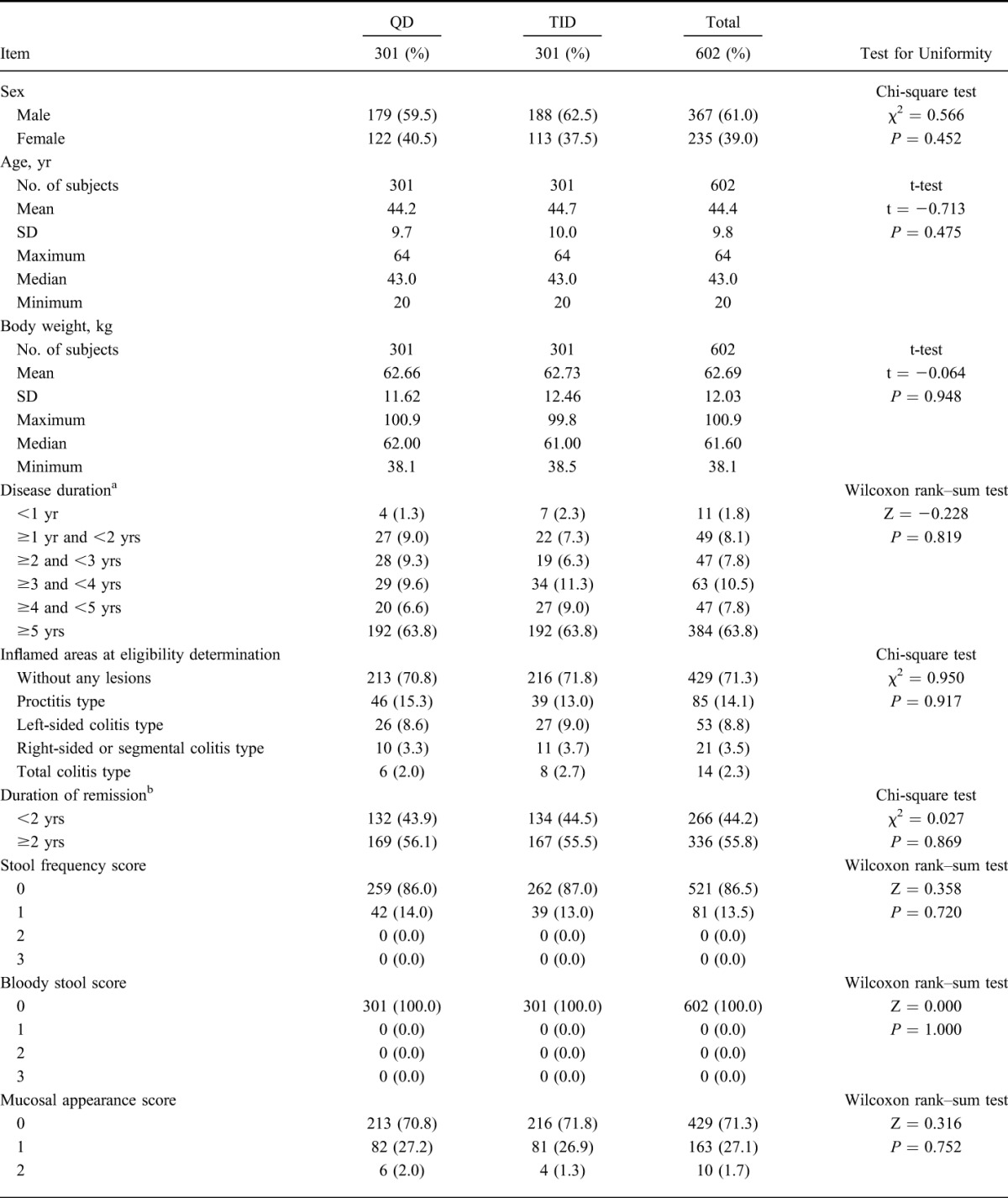
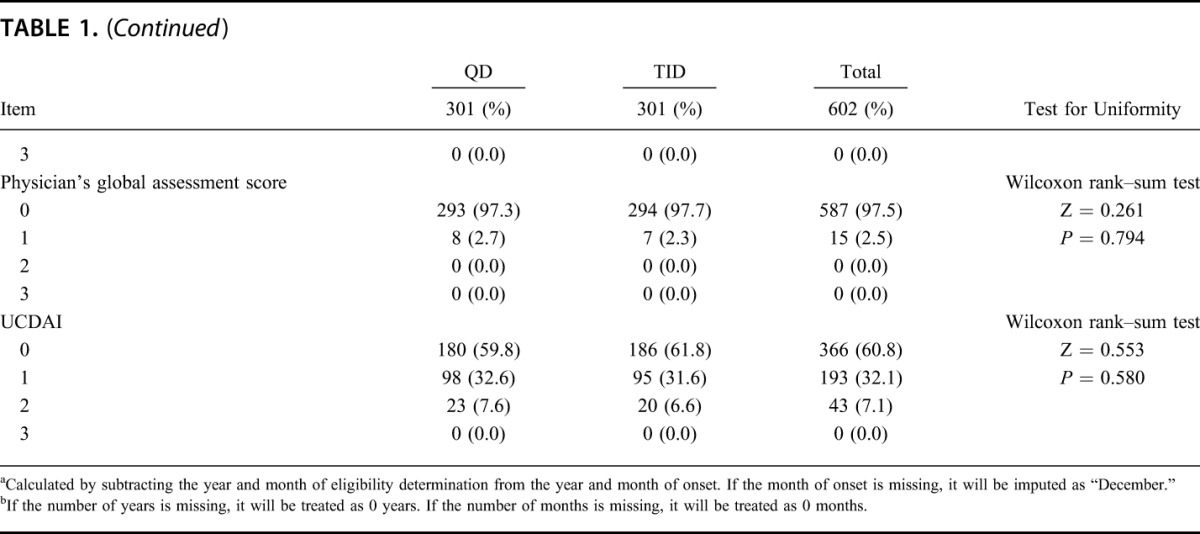
Of 666 patients who provided informed consent, 62 subjects withdrew from study before randomization, 604 subjects were eligible and were allocated into either group (QD group: 303; TID group: 301). One subject withdrew informed consent before starting study drug administration and 603 subjects received the study drug. Five hundred sixteen subjects (QD group: 258, TID group: 258) completed the study and 87 subjects (QD group: 44, TID group: 43) withdrew from the study. The most common reason for withdrawal was aggravation of underlying condition (QD group: 34; TID group: 32). Details of the analysis sets are as follows: 603 subjects (QD group: 302, TID group: 301) were in the SAF; 602 subjects (QD group: 301, TID group: 301) were in the FAS; 588 subjects (QD group: 295, TID group: 293) were in the PPS (Fig. 1). The patient demographics of this study are shown in Table 1 and imbalance (P ≥ 0.15) was not observed.
FIGURE 1.
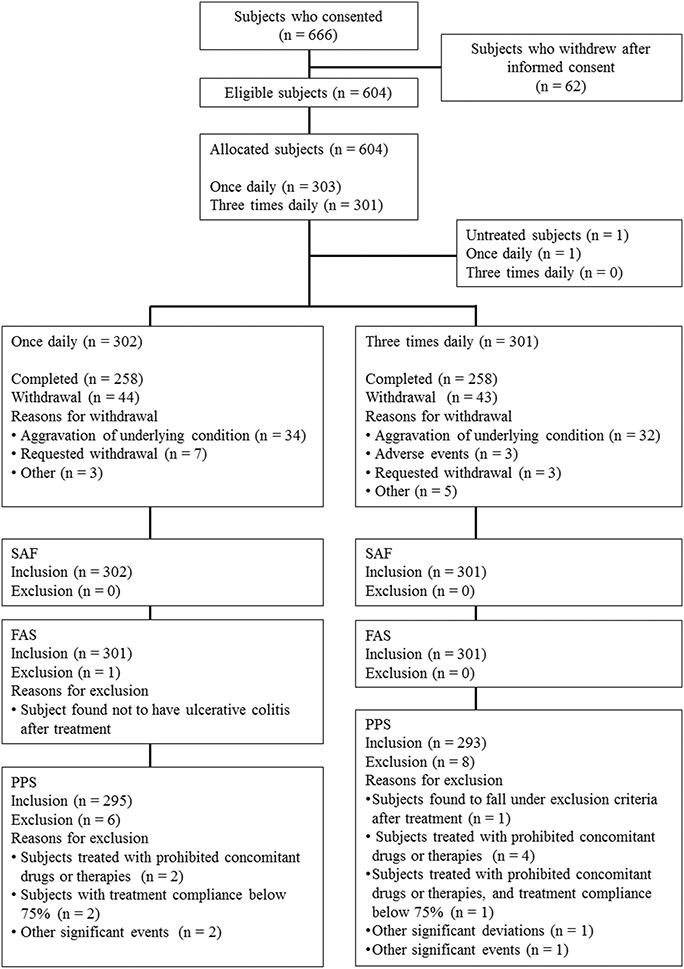
Dispositions of subjects (Subjects who consented). Of 666 patients who provided informed consent, 604 subjects were eligible and were allocated into either group. One subject withdrew informed consent before starting study drug administration and 603 subjects received the study drug. Five hundred sixteen subjects completed the study and 87 subjects withdrew from the study. The most common reason for withdrawal was aggravation of underlying condition.
TABLE 1.
Demographic and Other Baseline Characteristics
Treatment Compliance
The compliance rate (mean ± SD) for the entire period in the FAS was 97.7% ± 5.4% in the QD group and 98.1% ± 4.9% in the TID group.
Efficacy
Nonrecurrence Rate (FAS)
As the primary analysis, the nonrecurrence rates and the 95% CIs were 88.4% (84.3, 91.7) in the QD group and 89.6% (85.7, 92.8) in the TID group, respectively. The difference between nonrecurrence rates and the 95% CI were −1.3% (−6.2, 3.7); as the lower limit of the 95% CI was >−10%, the noninferiority was thus verified (Table 2). The Kaplan–Meier curve is shown in Figure 2. In addition, 2 subjects were handled as “data missing,” because the presence or absence of a recurrence could not be defined. This was to be treated as “data missing” according to the statistical analysis plan.
TABLE 2.
Nonrecurrence Rate (FAS)
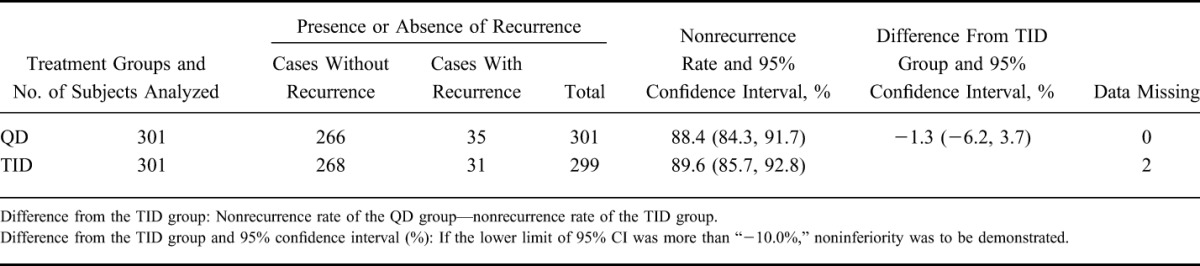
FIGURE 2.
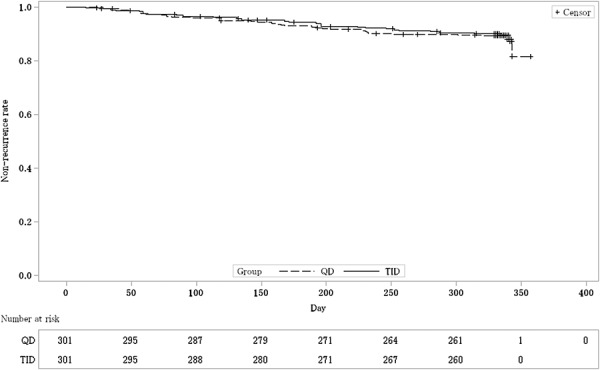
Kaplan–Meier curve estimated nonrecurrence rate (FAS). The dashed line shows the nonrecurrence rate of once-daily (QD) groups. The solid line shows the nonrecurrence rate of 3 times daily (TID) groups.
Subgroup Analysis
Nonrecurrence Rate (PPS)
Using the same procedure used for primary analysis, the nonrecurrence rate in the PPS was analyzed (Table 3). The nonrecurrence rates and 95% CIs were 88.5% (84.3, 91.8) in the QD group and 89.4% (85.3, 92.6) in the TID group, respectively. The difference in nonrecurrence rates and the 95% CIs were −0.9% (−5.9, 4.1). These results are similar to those of the primary analysis.
TABLE 3.
Nonrecurrence Rate (PPS)
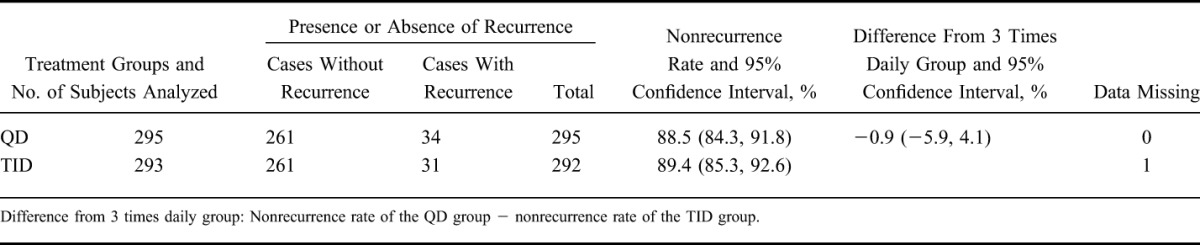
In addition, one subject was handled as data missing, because the presence or absence of a recurrence could not be defined. This was to be treated as “data missing” according to the statistical analysis plan.
Nonrecurrence Rate (FAS) by Allocation Factors
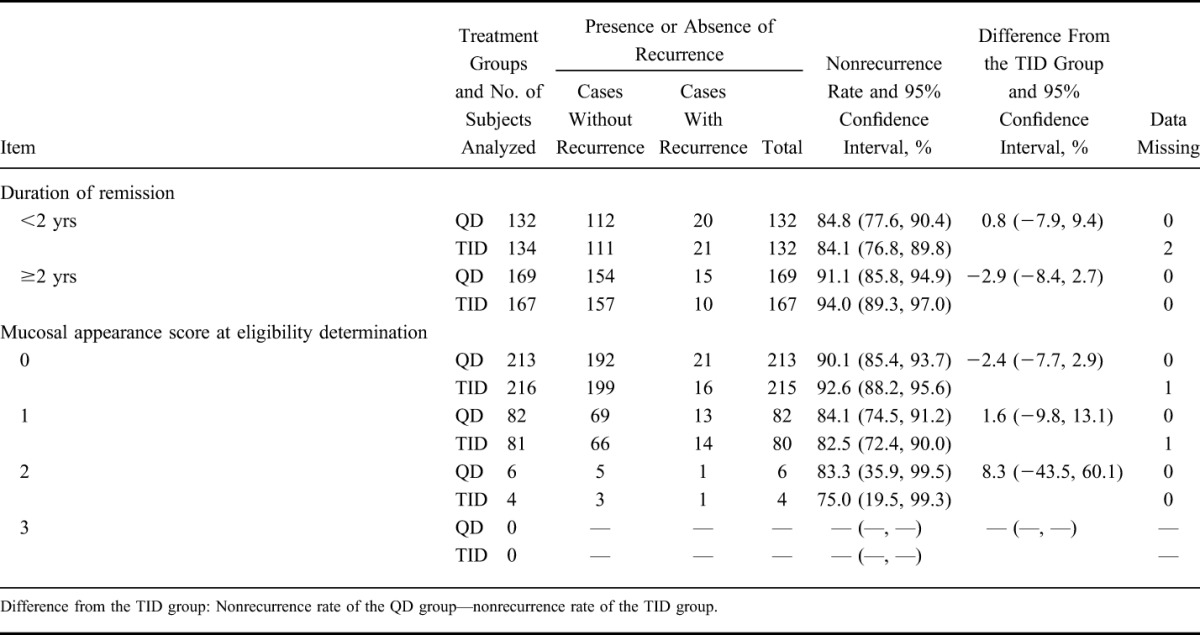
Using the same procedure used in the primary analysis, the nonrecurrence rate was analyzed by allocation factors (Table 4). Nonrecurrence rates in the group with a duration of remission of ≥2 years were higher than the group with a value of <2 years. In addition, nonrecurrence rates in the group with a mucosal appearance score of 0 were higher than in the group with a score of 1. There was no significant difference between the QD group and the TID group.
TABLE 4.
Nonrecurrence Rate by Allocation Factors (FAS)
Sensitivity Analysis
The frequency distribution of partial UCDAI (UCDAI without a mucosal appearance score at the final assessment) in cases with recurrence is shown at Table 5. There were just 7 cases in which determination of recurrence/nonrecurrence depended on the mucosal appearance score (partial UCDAI is ≤2; QD group: 6, TID group: 1). The results of analysis when these 7 subjects were handled as “data missing” are shown in Table 6.
TABLE 5.
Frequency Distribution of UCDAI Without Mucosal Appearance Score (FAS [Only Cases With Recurrence])

TABLE 6.
Nonrecurrence Rate (Sensitivity Analysis) (FAS)
Post-hoc Analysis
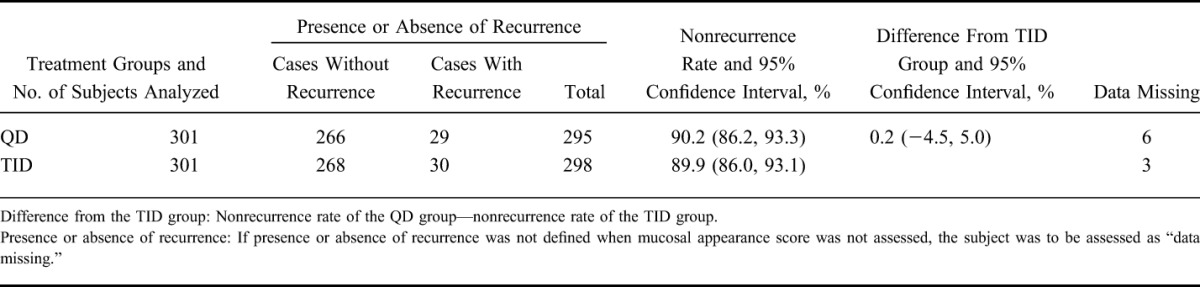
Integrating the QD group and the TID group, the nonrecurrence rate by each mucosal appearance score at eligibility determination is shown in Table 7, and Kaplan–Meier curve is shown in Figure 3. The nonrecurrence rate and the 95% CI for each mucosal appearance score were 91.4% (88.3, 93.8) in the score 0 group, and 83.3% (76.7, 88.7) in the score 1 group. The difference in nonrecurrence rates and the 95% CI were 8.0% (1.7, 14.3). As the lower limit of the 95% CI was >0, the difference between the 2 groups was statistically significant.
TABLE 7.
Nonrecurrence Rate by Mucosal Appearance Score at Eligibility Determination (FAS)
FIGURE 3.
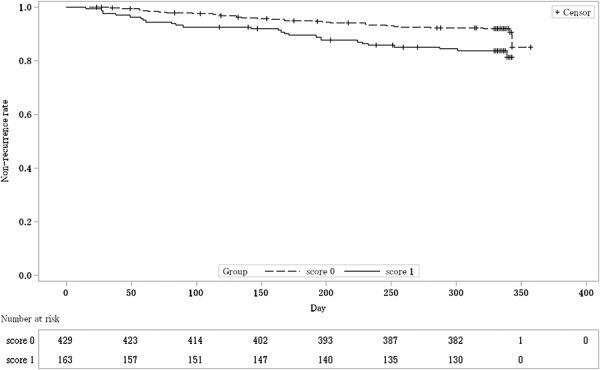
Kaplan–Meier curve estimated nonrecurrence rate by mucosal appearance score at eligibility determination (FAS). The dashed line shows the nonrecurrence rate of the group with mucosal appearance score 0 at eligibility determination. The solid line shows the nonrecurrence rate of the group with mucosal appearance score 1 at eligibility determination.
Safety
SAF was the primary analysis set for the safety analysis.
The incidence of adverse events was 71.2% in the QD group and 76.1% in the TID group, and the incidence of adverse drug reactions was 4.3% in the QD group and 5.3% in the TID group; thus, there was no clear difference in incidence between the 2 groups (shown in Table 8).
TABLE 8.
Incidence of Adverse Events and Adverse Drug Reactions
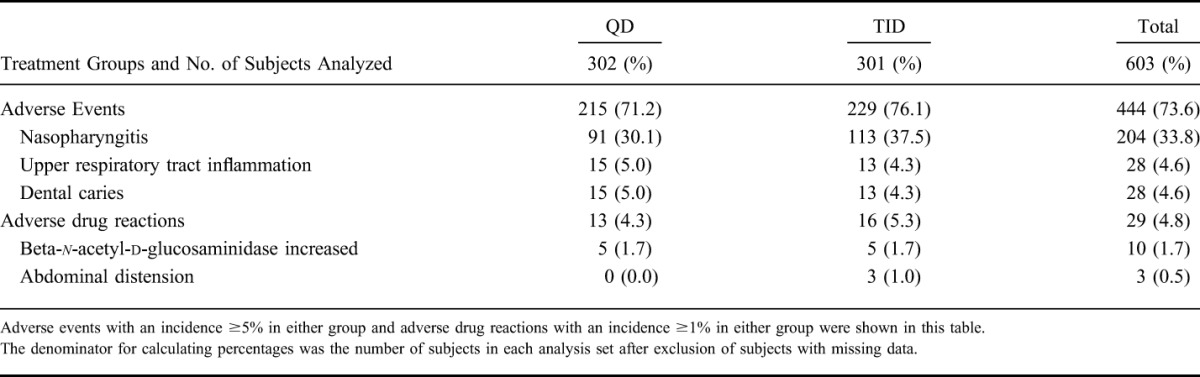
Of the adverse event that occurred, events with an incidence ≥5% in either group were nasopharyngitis (30.1% in the QD group and 37.5% in the TID group), upper respiratory tract inflammation (5.0% in the QD group and 4.3% in the TID group), and dental caries (5.0% in the QD group and 4.3% in the TID group). All episodes of nasopharyngitis, upper respiratory tract inflammation, and dental caries were considered unrelated to the study drug.
Of the adverse drug reactions that occurred, events with an incidence ≥1% in either group were beta-n-acetyl-d-glucosaminidase increased (1.7% in the QD group and 1.7% in the TID group) and abdominal distension (did not occurred in the QD group and 1.0% in the TID group).
Five serious adverse events occurred in 5 subjects in the QD group (detachment of retina (left), fracture of the right radius, a right tibial diaphysis fracture, a left thighbone trochanteric fracture, and choledocholithiasis), and 5 serious events occurred in 5 subjects in the TID group (a fissured fracture of the left radius, Hunt syndrome, hypersensitivity pneumonitis related to 5-ASA, an inguinal hernia, aggravation of an anal fistula). Only the hypersensitivity pneumonitis due to 5-ASA was considered related to the study drug, as this is a known adverse drug reaction. All serious adverse events had recovered or were recovering before the end of this trial.
DISCUSSION
As a result of the primary analysis in UC patients in remission, QD dosing was noninferior to TID dosing in maintenance of remission, using 2.4 g/d of a pH-dependent release mesalamine formulation. Findings in the PPS analysis were equivalent to those of the primary analysis, demonstrating robustness of the result of the primary analysis.
The nonrecurrence rate in this trial was approximately 90% in both groups, and was higher than we expected (75%) based on the result of the past study,7 before the start of this trial. We consider that this finding was a consequence of the greater number of stable patients (e.g., those with a mucosal appearance score of 0 or remission-maintenance period of ≥2 years) led by an increase in the number of therapeutic options and a prevailing emphasis on the importance of mucosal healing, subsequent to the time when the past trials5,9,10 were conducted—trials on which the expected recurrence rate was based.
Besides, the subgroup analyses of the nonrecurrence rate by prior use of mesalamine or corticosteroid were conducted in this trial (these data were not shown). In almost all of these subgroups, significant difference between QD group and TID group was not observed. Only in a subgroup with the patients who used suppository of corticosteroid in the most recent active phase of UC, the significant difference was observed but we thought that there was a large impact of dispersion caused by small number of subjects.
In this trial, central reading of mucosal appearance scores was not used. To investigate the effect of not using central reading, we conducted a sensitivity analysis. There were just 7 cases in which determination of recurrence/nonrecurrence depended on the mucosal appearance score. If these subjects were handled as “data missing,” the results of analysis are equivalent to those of the primary analysis. In this trial, we thus consider the effect of not using central reading to be limited. Besides, we did not conduct a sensitivity analysis in cases with nonrecurrence. This was because, in all cases with nonrecurrence, blood was not apparent in the stool at final assessment; thus, the determination of recurrence/nonrecurrence did not depend on mucosal findings.
Although differences were reported based on the genetic background of Asians (including Japanese) and Caucasians,11 the results of this trial, in which the enrolled subjects were solely Japanese, matched the results of a meta-analysis12—there is no significant statistically difference in efficacy between QD and conventional dosing.
As the double-dummy method was used in this trial, high compliance rate was observed in the QD group and the TID group. There was no difference in treatment compliance and the safety profile between 2 groups. In comparison to the known safety profile, there was no incidence of events that needed special consideration in this trial.
The various risk factors for recurrence are known, and the influence of mucosal state on maintenance-remission has been reported.13 On post hoc analysis of trial data, nonrecurrence rate with a mucosal appearance score of 0 at determination of eligibility was significantly higher than the score of 1. Although this analysis is ad hoc, we consider the result of this analysis to be very important because it was prospective and showed the importance of maintaining a good mucosal state in the maintenance–remission period.
In conclusion, we consider QD dosing maintenance–remission therapy to be as effective and safe as TID dosing, and that it can contribute to better adherence by adding a therapeutic option in keeping with the subject's lifestyle. In addition, to prolong remission, it is important to maintain a good mucosal state.
ACKNOWLEDGMENTS
The authors thank all of the study participants, doctors, and staff who supported this study. The authors thank Hikaru Ito (Clinical Research 2, Zeria Pharmaceutical Co., Ltd., Tokyo, Japan) for help in writing this article.
Footnotes
Supported by Zeria Pharmaceutical Co., Ltd. and Kyowa Hakko Kirin Co., Ltd. (Tokyo, Japan).
Y. Suzuki has received speaker fees and manuscript fees from Zeria Pharmaceutical Co., Ltd. M. Iida has received speaker fees from Zeria Pharmaceutical Co., Ltd. H. Ito has received speaker fees and manuscript fees from Kyowa Hakko Kirin Co., Ltd., and consulting fees from Zeria Pharmaceutical Co., Ltd. H. Nishino has received speaker fees and manuscript fees from Zeria Pharmaceutical Co., Ltd. T. Ohmori has received speaker fees from Zeria Pharmaceutical Co., Ltd. T. Yokoyama has received speaker fees and manuscript fees from Zeria Pharmaceutical Co., Ltd. T. Okubo is an employee of Zeria Pharmaceutical Co., Ltd. T. Hibi has received speaker fees, manuscript fees, and grant support from Zeria Pharmaceutical Co., Ltd. T. Arai has no conflict of interest to disclose.
Supply of study drug The Asacol and placebo tablets used in this study were supplied by Zeria Pharmaceutical Co., Ltd. (Tokyo, Japan). Help in writing this article Hikaru Ito (Clinical Research 2, Zeria Pharmaceutical Co., Ltd., Tokyo, Japan).
REFERENCES
- 1.Research Group for Intractable Inflammatory Bowel Disease. Guidelines for the management of ulcerative colitis in Japan-developed through integration of evidence and consensus among experts. January, 2006. Available at: http://minds4.jcqhc.or.jp/minds/kaiyouseida/ucgl+201102.pdf. Accessed August 18, 2016.
- 2.Kane SV. Systematic review: adherence issues in the treatment of ulcerative colitis. Aliment Pharmacol Ther. 2006;23:577–585. [DOI] [PubMed] [Google Scholar]
- 3.Higgins PDR, Rubin DT, Kaulback K, et al. Systematic review: impact of non-adherence to 5-aminosalicylic acid products on the frequency and cost of ulcerative colitis flares. Aliment Pharmacol Ther. 2009;29:247–257. [DOI] [PubMed] [Google Scholar]
- 4.Ford AC, Khan KJ, Sandborn WJ, et al. Once-daily dosing vs. conventional dosing schedule of mesalamine and relapse of quiescent ulcerative colitis: systematic review and meta-analysis. Am J Gastroenterol. 2011;106:2070–2077. [DOI] [PubMed] [Google Scholar]
- 5.Sandborn WJ, Korzenik J, Lashner B, et al. Once-daily dosing of delayed-release oral mesalamine (400-mg tablet) is as effective as twice-daily dosing for maintenance of remission of ulcerative colitis. Gastroenterology. 2010;138:1286–1296. [DOI] [PubMed] [Google Scholar]
- 6.Watanabe M, Hanai H, Nishino H, et al. Comparison of QD and TID oral mesalazine for maintenance of remission in quiescent ulcerative colitis: a double-blind, double-dummy, randomized multicenter study. Inflamm Bowel Dis. 2013;19:1681–1690. [DOI] [PubMed] [Google Scholar]
- 7.Ito H, Iida M, Matsumoto T, et al. Direct comparison of two different mesalamine formulations for the maintenance of remission in patients with ulcerative colitis: a double-blind, randomized study. Inflamm Bowel Dis. 2010;16:1575–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ardizzone S, Petrillo M, Imbesi V, et al. Is maintenance therapy always necessary for patients with ulcerative colitis in remission? Aliment Pharmacol Ther. 1999;13:373–379. [DOI] [PubMed] [Google Scholar]
- 9.Hawkey CJ, Dube LM, Rountree LV, et al. A trial of zileuton versus mesalazine or placebo in the maintenance of remission ulcerative colitis. Gastroenterology. 1997;112:718–724. [DOI] [PubMed] [Google Scholar]
- 10.Prantera C, Kohn A, Campieri M, et al. Clinical trial: ulcerative colitis maintenance treatment with 5-ASA: a 1-year, randomized multicentre study comparing MMX with Asacol. Aliment Pharmacol Ther. 2009;30:908–918. [DOI] [PubMed] [Google Scholar]
- 11.Ng SC, Tsoi KKF, Kamm MA, et al. Genetics of inflammatory bowel disease in Asia: systematic review and meta-analysis. Inflamm Bowel Dis. 2012;18:1164–1176. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Parker CE, Feagan BG, et al. Oral 5-aminosalicylic acid for maintenance of remission in ulcerative colitis. Cochrane Database Syst Rev. 2016;5:CD000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Acosta MB, Vallejo N, Iglesia D, et al. Evaluation of the risk of relapse in ulcerative colitis according to the degree of mucosal healing (mayo score 0 vs 1): a longitudinal cohort study. J Crohns Colitis. 2016;10:13–19. [DOI] [PubMed] [Google Scholar]