

 This work is licensed under a
This work is licensed under a Abstract
Summary
Cholesterol transport into the mitochondria is required for synthesis of the first steroid, pregnenolone. Cholesterol is transported by the steroidogenic acute regulatory protein (STAR), which acts at the outer mitochondrial membrane prior to its import. Mutations in the STAR protein result in lipoid congenital adrenal hyperplasia (CAH). Although the STAR protein consists of seven exons, biochemical analysis in nonsteroidogenic COS-1 cells showed that the first two were not essential for pregnenolone synthesis. Here, we present a patient with ambiguous genitalia, salt-lossing crisis within two weeks after birth and low cortisol levels. Sequence analysis of the STAR, including the exon–intron boundaries, showed the complete deletion of exon 1 as well as more than 50 nucleotides upstream of STAR promoter. Mitochondrial protein import with the translated protein through synthesis cassette of the mutant STAR lacking exon 1 showed protein translation, but it is less likely to have synthesized without a promoter in our patient. Thus, a full-length STAR gene is necessary for physiological mitochondrial cholesterol transport in vivo.
Learning points:
STAR exon 1 deletion caused lipoid CAH.
Exon 1 substitution does not affect biochemical activity.
StAR promoter is responsible for gonadal development.
Background
The activity of a protein depends on multiple factors, including its subcellular localization as well as chaperones that are necessary for appropriate folding. Incorrect folding of metabolic enzymes present in the steroidogenic pathway leads to the reduced production of steroids necessary for mammalian development (1). Specifically, steroidogenic acute regulatory protein (STAR) requires the 78 kDa master chaperone protein, glucose-regulated protein (GRP78) (2). Thus, a mutation in any member of the steroidogenic pathway has the potential to negatively influence the production of cortisol and induce adrenal enlargement, a condition known as lipoid congenital adrenal hyperplasia (CAH) that is characterized by the development of salt-losing crisis and ambiguous genitalia. Although it may be challenging to identify the gene responsible for reduced or low baseline steroid synthesis and subsequently ambiguous genitalia (3), the problem often starts with the reduced availability of the first synthesized steroid, pregnenolone, because the synthesis of all steroids depends on (i) cholesterol flow into the mitochondria and (ii) cleavage of the side chain of cholesterol to generate pregnenolone.
Although a large number of clinical reports from Japan have described patients with large adrenal glands and babies born with abnormal female genitalia, more detailed information is missing (4). The hormonal disorder and levels of the hormonal reduction were first investigated by Prader and coworkers in the mid-1950s although the pathological descriptions of infants dying from lipoid CAH had appeared previously (4). Studies of affected adrenal or testicular tissues or their isolated mitochondria showed an inability to convert cholesterol to pregnenolone due to the severely reduced availability of mitochondrial cholesterol for the synthesis of pregnenolone, which occurs prior to the catalytic activity of the side chain cleavage enzyme, P450scc (4). Patients with lipoid CAH have normal term delivery because placental biosynthesis of progesterone remains unaffected (4).
STAR is expressed in the adrenals and gonads, but not in the placenta, suggesting that mutations of this protein might be responsible for some specific cases of lipoid CAH (4). We proposed a two-hit model to explain the mechanism of action, in which the first hit results from the absence of STAR as in early lipoid CAH. In this scenario, a mechanism, independent of STAR, can transfer few molecules of cholesterol into the mitochondria, resulting in a low level of steroidogenesis, which increases corticotropin secretion and stimulates further production of cholesterol and its accumulation as cholesterol esters in lipid droplets. In the second hit, the accumulated lipid droplets engorge the cell, damaging its cytoarchitecture through both physical displacement and by cholesterol oxidation products, severely reducing all steroidogenic capacity.
Lipoid CAH is a rare disorder in most populations. The spectrum of clinical presentations of patients with STAR mutations is substantially broader than that of classic lipoid CAH, and many affected patients may not be diagnosed correctly (4). An attenuated form of the disease, ‘non-classic lipoid CAH’, is caused by mutations in which 10–25% of normal STAR activity is retained (4). These patients typically experience adrenal insufficiency several years after infancy; the 46,XY individuals may masculinize normally, and mineralocorticoid secretion may be minimally affected. The most common mutation causing this phenotype is StAR p.R188C. After genetic analysis of more than two dozen lipoid CAH patients from six continents, we identified two common STAR mutations: (i) R182L STAR observed in the Arab population and (ii) Q258X STAR identified in the Japanese and Korean populations. We have also identified a common mutation among Japanese, Korean and Palestinian patients that caused lipoid CAH (4). In addition, three groups have reported five Japanese patient alleles carrying StAR p.A218V (4). However, other STAR mutations appear in individual families rather than being widespread in an ethnic group. Although some mutations are more common in some specific ethnicities, STAR mutations are present in any ethnicity independent of the country of origin, and most StAR mutations causing lipoid CAH have been found in exons 5–7 of the StAR gene (4).
STAR requires chaperone, GRP78 (2), for its folding prior to delivery at the outer mitochondrial-associated VDACs (5, 6). STAR entry into mitochondria is not necessary for activity, as deletion of the first 62 amino acids encoding the mitochondrial targeting sequence (N-62 STAR) does not alter the steroidogenic activity of the protein in COS-1 cells or in isolated mitochondria from steroidogenic cells (4). Transfection of full-length amino acid replacement mutant and wild-type STAR followed by Western blotting shows that the proteins are processed similarly and both reach the mitochondrial matrix. N-62 STAR or full-length STAR exhibit maximal activity when permanently affixed to the outer mitochondrial membrane (OMM) (6). Under similar conditions, the 62 amino acid truncated (N-62 STAR) mutant R182L STAR was inactive. Although the biochemical studies suggest that first N-terminal 62 amino acids are not essential for activity, the current clinical analysis indicates that the all the exons are equally important for phenotypic development.
Case presentation
The patient was treated at Memorial Hospital in Savannah, GA. The patient was born by emergency C-section due to non-reassuring fetal heart rate tracing (FHT) at 32 weeks 2 days gestational age with a birth weight of 1410 g (0.17th) percentile-for-gestational age intrauterine growth restriction (IUGR) with multiple complications related to prematurity. The patient also had ambiguous genitalia, including microphallus, bifid scrotum, severe hypospadias and palpable gonads bilateral. There is no change in skin color and chromosomal analysis showed 46 XY. Laboratory analysis revealed 17-OH progesterone normal levels (35.5 ng/dL), low 18-OH corticosterone (<5 ng/dL), low 17-OH pregnenolone (31 ng/dL), normal DHEA (0.078 ng/mL), low anti-Mullerian hormone for a male infant (5.4 ng/mL), normal LH (2.4 IU/mL), normal FSH (0.7 IU/L), normal 5α-dihydrotestosterone (144 pg/mL) and free testosterone (11.3 pg/mL). After two weeks, renin levels were 78 ng/mL/h (1–7 days: 2.0–35.0 ng/mL/h), which was reduced to 36 ng/mL/h after 18 days (1–7 days: 2.0–35.0 ng/mL/h); aldosterone levels were 6.5 ng/dL (1–3 weeks: 6.0–179 ng/dL), ACTH levels were 842 pg/mL (high) and cortisol was 2.8 µg/dL at 15:56 h (am: 6.7–22.6, pm: <10 µg/dL). The patient was treated with 1.25 mg of hydrocortisone three times daily, Florinef (0.2 mg daily) and NaCl supplement (2 mL every 6 h). The patient receives formula feeding through his G tube and showed normal temperature, no excessive crying or vomiting or diarrhea after discharged from the hospital.
After two months of the treatment, the patient’s creatinine levels were below the normal range (0.23 mg/dL), and his chloride was 111 mM, which was a little above the normal range of 98–107 mM. The anion gap was 7.5 (L), which is much lower than the minimum level of 10 (L), indicating the presence of metabolic acidosis. After 6 months of follow-up, 17-OH progesterone androstenedione and DHEA-sulfate levels were all <10 ng/mL.
The patient is currently 16 months old, and he is tolerating glucocorticoid therapy. His recent plasma renin activity (PRA) was low at 1.64 ng/mL/h (normal range, 1.7–11.2 ng/mL/h), and serum sodium was slightly elevated. Thus, his mineralocorticoid therapy was adjusted during the last visit as the patient has not had an adrenal crisis. The patient’s mother decided to raise the patient as a male, and the patient underwent reconstruction of the genitalia in a different hospital. Figure 1 still shows an extremely small penis and an undeveloped scrotum that remains, and labioscrotal fusion with no pendulum.
Figure 1.

Presentation of the genitalia after reconstruction. (A) An overview of the genitalia at 1 year after reconstruction. (B and C) Closer views of the genitalia after reconstruction.
Investigation
Sequence analysis
Blood samples of the patient and the mother collected in ethylenediaminetetraacetate (EDTA) were used to prepare genomic DNA using a genomic DNA isolation kit (DNeasy Blood and Tissue Kit, Qiagen). Because the patient was considered to have lipoid CAH, seven exons of StAR gene (Gene ID# NG_011827.1) were analyzed following a standard procedure (3). Analysis of the StAR gene was also undertaken by first amplifying the genomic DNA using a variety of primer combinations. We first amplified exons 5–7, resulting in a 2.1 kbp fragment that contained most of the identified mutations. Next, we amplified exons 1–4 using identical PCR conditions as previously described. To clearly identify the deleted region of exon 1, we amplified a 575 bp fragment containing StAR promoter region with sense Ex-1S (5′TAACACAGGTTTCTGAGCCTCAAT3′) and antisense Ex-1AS (5′TAGTCTTAACCCACCGGACT CGGA3′) primers. PCR products were sequenced directly using commercial resources (MC Lab, South San Francisco, CA, USA).
Mitochondrial translocation assay
A cell-free transcription/translation kit (Promega), which contains rabbit reticulocyte lysate (RRL) for protein expression following our modified procedure (7), was used to express wild-type and mutant STAR proteins after subcloning into the SP6 vector. Briefly, a mixture of all amino acids without methionine was incubated with 35S-methionine (MP Biomedicals, Santa Ana, CA, USA) and SP6 polymerase at 26°C for 2 h. Expression of the radiolabeled proteins was verified using SDS-PAGE. For mitochondrial protein import analysis, mitochondria isolated from MA-10 cells were suspended in 20 mM HEPES, pH 7.5, following a previously described procedure (7). After addition of the freshly prepared cell-free synthesized (CFS) protein of interest, mitochondria were incubated at 26°C.
Treatment
The patient tolerated hydrocortisone sodium succinate therapy for daily physiological glucocorticoid replacement and supra-physiological replacement as needed as well as daily 9 alpha-fludrocortisone therapy for mineralocorticoid replacement. No adverse effects were reported by the mother.
Outcome and follow-up
We predict that the patient will require the current treatment for rest of his life and will be monitored by follow-up at regular intervals.
Results
We first considered that the ambiguous female genitalia and adrenal insufficiency were possibly due to a mutation in the 7.5 kb StAR gene, which has seven exons with exons 5–7 comprising 40% of the protein. Because we did not find any mutation in exons 5–7, we sequenced the complete gene, including the exon–intron boundaries. We found that the nucleotide sequence comprising exons 2–7 is completely protected, including the 50-nucleotide exon–intron boundaries (Fig. 2). Although no mutations were identified in the 5′ nucleotide sequence from 1 to 4786, the region spanning nucleotides 4786–5404 was replaced with a completely unknown nucleotide sequence. Exon 1 is present from nucleotides 5152 to 5328 and exon 1 was completely altered. However, the remaining sequence from nucleotide 5404 (gene sequence) matched wild-type StAR.
Figure 2.
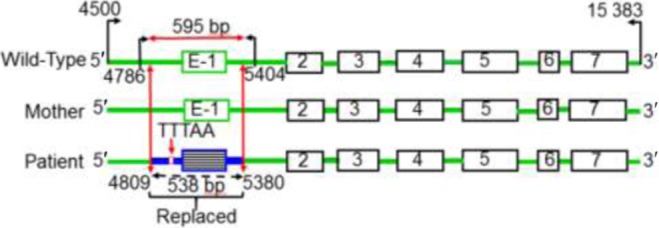
Sequencing analysis of the STAR gene. The seven exons are indicated in boxes, and the PCR amplification of exon 1 including the promoter region and the detailed analysis of the PCR-amplified region are shown by the directional arrows in black. The primers used to amplify exon 1 are shown by black arrows, and the gene sequence is indicated by the specific number. The STAR gene from the mother and the wild-type control are also shown. The replaced region of the patient containing the exon, exon–intron boundary and the extended promoter region is shown in blue. These sequences (indicated in green) were completely identical, suggesting that a de novo exon 1 deletion in the patient.
We next examined the StAR gene of the mother and found that the sequence was similar to the wild-type protein, suggesting that the mother is not affected. As this was a full-term pregnancy, we also analyzed the sequence of the P450 side chain cleavage enzyme. However, after sequencing all nine exons, including the exon–intron boundaries, we found no change in coding sequence. Thus, our genetic analysis suggested that a de novo deletion of exon-1 of StAR resulted in lipoid CAH.
We and others have reported that N-62 STAR, in which exons 1 and 2 as well as the first three amino acids of exon 3 were deleted, had similar activity after overexpression in nonsteroidogenic COS-1 cells along with F2 as compared to wild-type STAR (8). Similarly, expression of N-21 STAR resulted in the similar pregnenolone synthesis (data not shown), suggesting that the results obtained through biochemical analyses may be different from that observed in vivo. Furthermore, given that the structural model of STAR was developed in the absence of the N-terminal 62 amino acids, the proposed mechanism of action based on the crystal structure is likely to be different. Thus, we analyzed the mitochondrial import and processing of the mutant STAR using isolated mitochondria from MA-10 cells as STAR folding is crucial for its activity. As shown in Figure 3A, full-length STAR (37 kDa) was imported into the mitochondria as detected by the 30 kDa mature imported protein that is identified after cleavage of the N-terminal sequence (arrowhead). Separation of the imported fraction from the unimported by centrifugation and then washing with the mitochondrial import buffer showed that the imported fraction was present in the pellet (P) fraction, and the unimported fraction was present in the supernatant (S). Next, extraction with sodium carbonate followed by separation by centrifugation showed that the imported fraction remained in the pellet, confirming the imported fraction was integrated into the membrane because the alkaline pH can break protein–protein interactions but not lipid–protein interactions (8, 9). In contrast, no mitochondrial import was detected with the N-62 or N-30 STAR mutants (Fig. 3B and C).
Figure 3.

Mitochondrial import of the wild-type StAR, N-62 StAR and N-30 StAR. 35S-methionine-labeled (A) wild-type StAR, (B) N-62 StAR and (C) N-30 StAR were synthesized in a cell-free rabbit reticulocyte lysate system (CFS), and then, the newly synthesized protein was imported into mitochondria isolated from MA-10 cells. The imported and unimported fractions were separated by washing (Wash) followed by centrifugation, where the unimported fraction remained in the supernatant (S), and the imported fraction remained in the pellet (P). Sodium carbonate (Carb) determines the lipid integrated from the unintegrated protein.
Discussion
STAR processing from the ribosome to the OMM is crucial for its activity, and GRP78 is necessary for its synthesis, proper folding (2) and delivery at the OMM for interaction with Tom22 (8). We have reported that STAR import is slower than that observed for other mitochondrial steroidogenic proteins, and its import is unique as only its own leader sequence but not that of other mitochondrial proteins competed with its import (7). Computational analysis of this region, which includes the mitochondrial leader sequence, shows a potentially flexible amphiphilic helix and a large number of positively charged amino acids comprising the exon 2 sequence, and thus, N-terminal sequence of exon 1 is possibly influenced by the exon 2 sequence. The minimum length of a mitochondrial leader sequence is 15 amino acids (7), given the induction of lipoid CAH with the exon 1 deletion, we deleted this region and examined STAR import and compared it with full-length STAR and another STAR mutant in which exons 1 and 2 were deleted. Neither mutant protein was imported into the mitochondria, suggesting that exon 1 may play a role for association with the OMM.
Because of the substitution of exon 1 DNA sequence, there was a dramatic failure of STAR transcription in this patient as the STAR (TTTAA) box present at −24 to −20 bp of the transcriptional start site in the human locus. For example, a SNP c > T 9 bp upstream of TTTAA in human patients showed reduction in STAR promoter activity by 35% (10). The disruption of ~50 bp upstream in this patient suggests that this could have been removed/replaced and therefore STAR transcription was possibly disrupted resulting in no STAR protein available for cholesterol transport from the outer to inner mitochondria.
We were the first to measure StAR activity by overexpressing full-length and mutant proteins in nonsteroidogenic cells after providing the electron donating partners, P450scc, adrenodoxin and adrenodoxin reductase. Although the results unveiled important findings, they may not reflect what is occurring physiologically. Specifically, in nonsteroidogenic COS-1 cells, deletion of N-terminal sequence of STAR did not affect the folding of the protein, and the activity of the exon 1 replacement mutant protein was similar to that observed for the wild-type protein. Our in vitro experiment was performed with a promoter present in the synthesis cassette (DNA plasmid vector) but mother nature performed experiment in the patient that has a disruption of its own promoter necessary for STAR protein synthesis. So, it is likely there was no STAR protein present in our patient. In summary, in the absence of promoter, our patient may not have any STAR protein present necessary for survival.
This particular mutant revealed four important points regarding STAR activity. First, it is likely that STAR folding is most crucial prior to reaching the OMM. In the absence of the N-terminal sequence, the protein is likely not folded and is not capable of transporting cholesterol from the OMM to inner mitochondrial membrane (IMM). Second, the N-terminus may be extremely important for STAR association with the outer mitochondrial translocase. In the absence of this sequence, the protein cannot specifically interact, resulting in complete inactivity. Third, STAR needs to be present close to the mitochondria, where it retains a role for continuous association with Tom22 and VDAC2 (6, 8). Fourth, the STAR promoter region is responsible for regulating for STAR synthesis, thus regulating steroidogenesis. Finally, and possibly most importantly, in the absence of STAR promoter, no STAR protein was synthesized having no availability of substrate cholesterol for pregnenolone synthesis transported by STAR. As the disposal mechanism of the mitochondrial imported STAR is unknown, it is possible that it participates in additional, yet unknown mitochondrial activities in steroidogenic cells and requires further investigation.
Taken together, analysis of pregnenolone production after overexpression of wild-type and mutant STAR protein in nonsteroidogenic cells can be used as a measure of protein activity; however, certain differences should be noted from that obtained using in vivo analyses. Thus, sequencing remains the definitive way to determine disease mechanism not only in the exons or exon–intron boundaries but also in the promoter region.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Patient consent
Included previously during the first submission.
Author contribution statement
The patient is seen by A Piya and regularly is in touch with the development in clinic, provided clinical data and recorded the photograph. J Kaur isolated genomic DNA and performed PCR. H Bose performed the protein import experiments; analyzed and interpreted the data and wrote the manuscript. The patient was also seen by A Rice. They are also thankful to Meenakshi Bose (Dipa) for critically reading the manuscript.
References
- 1.Bose HS. 2011. Mechanistic sequence of mitochondrial cholesterol transport by StAR proteins. Journal of Proteins and Proteomics 2 1–9. [Google Scholar]
- 2.Prasad M, Pawlak KJ, Burak WE, Perry EE, Marshall B, Whittal RM, Bose HS. 2017. Mitochondrial metabolic regulation by GRP78. Science Advances 3 e1602038 ( 10.1126/sciadv.1602038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaur J, Casas L, Bose HS. 2016. Lipoid congenital adrenal hyperplasia due to StAR mutations in a Caucasian patient Endocrinology, Diabetes and Metabolism. ( 10.1530/EDM-15-0119) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller WL, Bose HS. 2011. Early steps in steroidogenesis: intracellular cholesterol trafficking. Journal of Lipid Research 52 2111–2135. ( 10.1194/jlr.R016675) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prasad M, Kaur J, Pawlak KJ, Bose M, Whittal RM, Bose HS. 2015. Mitochondria-associated endoplasmic reticulum membrane (MAM) regulates steroidogenic activity via steroidogenic acute regulatory protein (StAR)-voltage-dependent anion channel 2 (VDAC2) interaction. Journal of Biological Chemistry 290 2604–2616. ( 10.1074/jbc.M114.605808) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bose M, Whittal RM, Miller WL, Bose HS. 2008. Steroidogenic activity of StAR requires contact with mitochondrial VDAC1 and phosphate carrier protein. Journal of Biological Chemistry 283 8837–8845. ( 10.1074/jbc.M709221200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rajapaksha M, Kaur J, Bose M, Whittal RM, Bose HS. 2013. Cholesterol-mediated conformational changes in the steroidogenic acute regulatory protein are essential for steroidogenesis. Biochemistry 52 7242–7253. ( 10.1021/bi401125v) [DOI] [PubMed] [Google Scholar]
- 8.Prasad M, Walker AN, Kaur J, Thomas JL, Powell SA, Pandey AV, Whittal RM, Burak WE, Petruzzelli G, Bose HS. 2016. Endoplasmic reticulum stress enhances mitochondrial metabolic activity in mammalian adrenals and gonads. Molecular and Cellular Biology 36 3058–3074. ( 10.1128/MCB.00411-16) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajapaksha M, Kaur J, Prasad M, Pawlak KJ, Marshall B, Perry EW, Whittal RM, Bose HS. 2016. An outer mitochondrial translocase, Tom22, is crucial for inner mitochondrial steroidogenic regulation in adrenal and gonadal tissues. Molecular and Cellular Biology 36 1032–1047. ( 10.1128/MCB.01107-15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casai AJ, Sinclair VJP, Capponi AM, Nicod J, Huynh-Do U, Ferrari P. 2006. A novel mutation in the steroidogenic acute regulatory protein gene promoter leading to reduced promoter activity. Journal of Molecular Endocrinology 37 71–80. ( 10.1677/jme.1.02082) [DOI] [PubMed] [Google Scholar]